
Abstract
Background
Osteoarthritis (OA) is a common cause of pain and disability. Currently available analgesics are often insufficiently effective or have unacceptable adverse effects. Tricyclic antidepressants may offer a useful centrally-acting analgesic. Nortriptyline is a readily-available, cheap and comparatively well-tolerated tricyclic antidepressant.
Methods/Design
We will conduct a parallel group, two-arm, participant and investigator-blinded, randomised controlled superiority trial comparing nortriptyline with placebo. Two hundred participants with primary knee OA will be enrolled. Participants will take study medication for 14 weeks. The primary outcome is difference between treatment arms in mean pain score measured on the Western Ontario and McMaster Universities (WOMAC) pain scale at 14 weeks.
Discussion
This protocol describes the first randomised controlled trial of a tricyclic antidepressant in the treatment of OA. The results of the study may have significant implications for the management of this common and painful condition.
Trial registration
The trial was registered with the Australian New Zealand Clinical Trials Registry on 27 June 2014. The trial registration number is: ACTRN12614000683639.
Similar content being viewed by others


Background
Osteoarthritis (OA) is the most common joint disease and is a major cause of joint pain and disability [1, 2]. In high-income countries, OA is the fifth leading cause of years lost to disease, [3] and in New Zealand 15 % of 55–64 year-olds and 26 % of 65–74 year-olds have OA [4]. The burden of disease and its associated costs are predicted to rise as the population ages [5]. OA is characterised by pain and loss of function and typically affects the weight-bearing joints of the lower limbs (hip, knee) as well as the hand and spine, but it can affect any joint. OA affecting the knee carries the greatest public health burden [6]. The major pathological features of the disease are degradation of articular cartilage and adjacent bone remodelling. OA is categorised as either primary or secondary: primary OA has no known trigger whereas secondary OA occurs as the result of joint damage from mechanical damage (e.g. fracture), inflammation (e.g. gout and rheumatoid arthritis) or infection. Risk factors for primary OA include advancing age, female sex, and obesity.
OA is predominantly managed in primary care. There is no cure and management is focused on relief of pain and maintenance of function. A range of interventions is available according to the severity of the condition. Initial management consists of patient education, exercise and weight loss, but many patients will also require analgesics [7]. First-line analgesic choices are typically paracetamol and topical non-steroidal anti-inflammatory drugs (NSAIDs); these may be followed by oral NSAIDs or weak opioids [7]. None of these analgesics is ideal: paracetamol, although safe and well-tolerated, may be no more effective than placebo [8, 9]. NSAIDs are effective in reducing pain, [9] but they are nephrotoxic, significantly increase the risk of gastrointestinal ulceration and bleeding, and are associated with cardiovascular events [10]. The long-term use of NSAIDs is, therefore, contraindicated in many patients. Opioids are also used despite poor evidence of their efficacy, and their use is limited by side effects [11]. Ultimately, OA may be treated with joint replacement, but access to this intervention is limited by resource constraints, [12] and patient co-morbidity, and for younger patients delayed joint replacement is desirable due to prostheses’ limited life-spans. Hence, there is a need for more effective and better tolerated pain management for patients with OA.
Central processing of pain in OA
Pain in OA has historically been attributed to the local nociceptive effects of degeneration of cartilage at the affected joint. Functional imaging has recently revealed areas of the brain that are involved in central processing of pain in OA. Functional magnetic resonance imaging (fMRI) has been used to investigate the central response to painful mechanical stimulation of osteoarthritic knees, and has shown an increase in central activation in brain regions associated with pain [13]. Subsequent treatment of the knee pain with topical lignocaine reduces central activity [13]. fMRI studies of patients with hip OA have also revealed greater central activation amongst those patients with higher levels of neuropathic pain [14]. The central effects of OA pain have also been explored using positron emission tomography (PET) and a similar increase in activity in pain-processing centres in participants with OA pain compared to those without pain has been demonstrated [15].
Antidepressants as a novel pain treatment in OA
The discovery of central pain-processing activity in OA has suggested a potential new therapeutic approach for pain control in OA [16]. Recent trials of the serotonin and noradrenalin reuptake inhibitors (SNRIs) venlafaxine [17] and duloxetine [18–20] have shown statistically and clinically significant reductions in pain in patients with OA. The study of venlafaxine, however, was very small (n = 18) and used a single-blind, placebo run-in design rather than a randomised controlled trial (RCT). The duloxetine studies were larger (n = 256 to n = 524), were double-blind RCTs, and demonstrated significantly greater reduction in pain and greater improvement in physical functioning with duloxetine than with placebo; however, duloxetine was also associated with significantly more side effects and higher rates of discontinuation than placebo. Furthermore, whilst duloxetine is licensed in New Zealand, it is not subsidised so is not readily available to many of our patients.
Tricyclic antidepressants (TCAs), which are pharmacologically-related to the SNRIs, are commonly used in other chronic pain conditions: for example, in back pain and fibromyalgia, [21–23] but their potential benefits in treating OA pain have not yet been tested in a well-designed RCT. A very small (n = 24) cross-over study of imipramine in 1969 suggested a reduction in OA pain [24]. However, this study had several limitations in addition to its small size: the study population was mixed and only 7 participants had OA (other participants had rheumatoid arthritis and ankylosing spondylitis), the treatment periods were short (3 weeks), and there was no washout period between treatment periods. Furthermore, imipramine has a high rate of adverse effects [25]. Nortriptyline is a better-tolerated TCA and is readily available at low cost. TCAs have a range of adverse effects, particularly dry mouth, constipation and drowsiness but nortriptyline is amongst the best tolerated [25, 26]. If nortriptyline proves to have a useful analgesic effect and a tolerable side effect burden it will be a valuable addition to the analgesic options for patients with OA.
Study aim
To measure the effectiveness, safety and tolerability of nortriptyline compared to placebo in reducing pain and improving function in patients with knee OA.
Hypotheses
In patients with knee OA, a 14-week course of nortriptyline delivered in addition to usual care will lead to:
-
1.
A significant reduction in OA pain compared to placebo.
-
2.
A significant improvement in physical function greater than with placebo.
Methods/Design
We propose a parallel group, two-arm, participant and investigator-blinded, randomised controlled superiority trial comparing nortriptyline with placebo.
The study population consists of adult patients living in the Canterbury region of New Zealand with primary knee OA (defined according to American College of Rheumatology (ACR) criteria) causing moderate to severe pain [27]. The ACR clinical criteria will be used as these require clinical findings without x-ray confirmation to confirm the diagnosis of OA. These criteria have slightly lower specificity than the clinical plus x-ray criteria, but their use more closely matches current practice and management guidelines for primary care management of OA. Recruitment will be from the Canterbury District Health Board’s (CDHB’s) orthopaedic service, from primary care, and from public advertisements.
Inclusion criteria
-
1.
Primary knee OA defined according to ACR classification criteria (knee pain plus 3 of: age > 50 years, morning stiffness lasting < 30 minutes, crepitus, bony tenderness, no palpable warmth) [27].
-
2.
Pain severity of ≥ 20 points on the Western Ontario and McMaster Universities (WOMAC) numerical rating scale (range 0 to 50 points) at the study knee [28].
-
3.
Stable analgesic regime for 2 months before entering the study.
Exclusion criteria
-
1.
Prior joint replacement surgery on the study knee
-
2.
Intra-articular steroid injection within the previous 3 months
-
3.
Secondary OA (OA due to inflammatory arthritis (e.g. gout, rheumatoid arthritis, juvenile arthritis), septic arthritis or trauma (articular fracture))
-
4.
Known hypersensitivity to nortriptyline or history of adverse reaction to any TCA
-
5.
Current use of nortriptyline or other antidepressants, amiodarone or domperidone
-
6.
Myocardial infarction within 6 months before study entry
-
7.
Heart block
-
8.
Postural hypotension
-
9.
Pregnancy
-
10.
Hyperthyroidism or phaeochromocytoma under current investigation or treatment
-
11.
History of epilepsy or other seizure
-
12.
History of bipolar disorder or manic episode
-
13.
History of increased intra-ocular pressure or history of angle-closure glaucoma
-
14.
Chronic constipation
-
15.
Urinary retention
Participant recruitment
Potential participants will be identified from the CDHB orthopaedic department and from local general practitioners’ (GPs’) enrolled patient lists. Patients identified from the orthopaedic department will have been assessed as being below threshold for operative treatment for OA. Public advertising will be undertaken in venues typically frequented by older adults: for example, bowling clubs and social clubs. To optimise involvement of Indigenous New Zealanders (Māori), recruitment will be undertaken in marae (Māori meeting place) and through the Māori health arm of Ngai Tahu, the principal iwi (tribe) in the South Island of New Zealand.
Potential participants identified through these avenues will be sent a letter outlining the study. The letter will include a postage-paid response card which those interested in taking part in the study can return to the research secretary. Individuals indicating willingness to participate will be phoned by the research nurse who will conduct a brief eligibility screen and will invite potential participants to attend a full screening assessment.
Screening assessment
A screening assessment will be conducted by the research nurse to ensure potential participants meet the inclusion and exclusion criteria. Potential participants’ GPs will be asked to confirm whether there any contraindications to the use of nortriptyline.
The study Clinical Review Team will decide each potential participant’s eligibility for the study. Once eligibility has been confirmed a baseline assessment will be undertaken. Participants will be asked to record NSAID and other analgesic use for 2 weeks before their baseline assessment.
Baseline assessment, informed consent and assignment of intervention
At the baseline assessment appointment the research nurse will further explain the study, answer any questions and seek informed consent. All participants will provide written informed consent to their participation in the study and will then be assigned a unique sequentially-numbered study identifier according to the order in which he or she is enrolled in the trial. The participant will then complete the baseline assessment and anthropometric measures before the research nurse dispenses the study medication.
Sequence generation
Participants will be randomly divided into two groups (A and B) of equal size using a computer-generated randomisation schedule with permuted blocks of random size prepared by the study statistician. The randomisation schedule will not be stratified as the risk of important imbalances in prognostic factors is small for a trial of this size [29].
Allocation concealment and implementation
The study medication (nortriptyline or identical placebo) will be packaged in identical containers. Each container will be pre-labelled (by the study pharmacist contracted to provide the study medication) with a study identifier according to randomisation schedule. The contracted pharmacist will determine which group of participants, A or B, will be allocated to receive nortriptyline. Neither the study statistician nor the contracted pharmacist will have any contact with the participants, nor will they be able to influence treatment allocation. Study numbers and study medication will be issued sequentially to participants.
Blinding
The study investigators, the research nurse dispensing the medication and assessing outcomes, and the participants will remain blind to the treatment allocation. After the final assessment, the participant’s study arm allocation will be un-blinded and this information will be communicated to the participant’s GP, thus allowing for continued prescription (if desired) of the active medication for those participants in the active treatment arm. The task of un-blinding will be performed independently by a member of the host department staff who is not involved with the study, and allocation will not be revealed to the research nurse or any of the investigators. We will assess the effectiveness of the allocation blinding by asking participants and the research nurse at the final assessment which arm of the study they believe the participant had been allocated to. To prevent accidental un-blinding there will be no contact between the study team and the participant until after data cleaning, database lockdown and analysis are complete.
Study medication
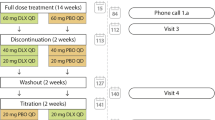
Nortriptyline dosing has high inter-individual variability: the effective and tolerated daily dose ranges from < 25 mg to > 100 mg [30, 31]. To allow for this, participants will pass through an 8-week blinded dose-adjustment period to allow titration of study medication according to analgesic effect and tolerability. This process closely resembles usual clinical practice when initiating a TCA.
Dose-adjustment period (weeks 0–8)
All participants will receive capsules containing nortriptyline 25 mg or identical placebo capsules and will be instructed to start study medication at a dose of 1 capsule at night. Every 2 weeks the research nurse will telephone participants and record their response to the study drug: participants will be asked whether they have experienced any change in their knee pain and whether they have experienced adverse effects. Participants who have achieved satisfactory pain relief will be instructed to continue their current dose. If pain relief is inadequate and side effects are tolerated then the participant will be instructed to increase their daily dose by one capsule. If side effects are intolerable then the daily dose will be reduced by one capsule. Dose adjustment will be carried out over 8 weeks to allow participants to reach the potential maximum dose of 4 capsules daily of study medication (a potential maximum dose of 100 mg nortriptyline daily) if required and tolerated. At each of these telephone reviews adverse events will be recorded and serious adverse events (SAEs) will be immediately notified to the Clinical Review Team.
Steady dose treatment period (weeks 8–14)
Following the initial dose-adjustment period, participants will continue at their maximum tolerated or effective dose for 6 weeks, a period consistent with published recommendations on OA research [32]. During this period participants will not be routinely contacted but they will be encouraged to contact the research nurse by phone if they have questions relating to their study medication.
Participants will be free to use their usual pain relieving medication as prescribed by their GP during the study period. Participants’ GPs will be informed of their patients’ participation in the study and will be asked not to prescribe nortriptyline or any other antidepressants to participants during the study period.
Outcome measures
In line with recommended practice in OA trials, we will assess outcomes in the domains of pain, physical function and participant global assessment [33].
Primary outcome
Difference between treatment arms in mean pain score at 14 weeks, measured using the WOMAC pain subscale and adjusted for pain score at baseline.
Secondary outcomes
The following outcomes will be assessed at 14 weeks in a structured interview:
-
1.
Physical function using the WOMAC function subscale
-
2.
Participant-rated global assessment using a visual analogue scale (VAS)
-
3.
Difference in the proportion of participants reporting a treatment effect, defined according to the Osteoarthritis Research Society International (OARSI) set of responder criteria [34]
-
4.
Quality of life using the 36-Item Short-Form Health Survey 36 (SF36) survey
-
5.
Participant-recorded NSAID and other analgesic use in the final 2 weeks of the study period
-
6.
Adverse events will be coded using the Common Terminology Criteria for Adverse Events (CTCAE) [35]. Tricyclic adverse effects will be recorded using the Antidepressant Side-Effect Checklist, [36] measured at 14 weeks.
The WOMAC knee OA index measures the three domains of pain, disability and joint stiffness using a set of 24 questions. It is amongst the most widely used measures of lower limb symptoms and has been well-established as a valid, reliable and responsive measure of pain and disability in OA [37, 38]. The WOMAC index used in this study will be version 3.1 in the 11-point numerical rating scale format standardised in English for a New Zealand population. The pain component of the WOMAC index covers five situations: walking on a flat surface, going up or down stairs, night pain, sitting or lying, and standing. The level of pain experienced in each of the 5 situations is scored from 0 to 10 (no pain to extreme pain) to give a combined maximum score of 50. Minimal training is required to administer the WOMAC, and completion time is approximately 12 minutes per participant.
Baseline assessment measurements
In addition to the primary and secondary outcome measures, the research nurse will also record participants’:
-
1.
Age
-
2.
Sex
-
3.
Ethnicity (using New Zealand Census 2006 categories)
-
4.
Height and weight for calculation of body mass index (BMI)
-
5.
Study knee disease duration
-
6.
Location and number of other osteoarthritic joints
-
7.
Use of assistive devices
-
8.
Current medication use:
-
a.
Analgesics (including NSAIDs)
-
b.
All other medications
-
a.
-
9.
Other chronic conditions.
Final assessment
The research nurse will assess each participant at the study clinic at week 14. The following data will be recorded:
-
1.
Pain using the WOMAC pain scale
-
2.
Physical function using the WOMAC function scale
-
3.
Participant-rated global assessment using a VAS
-
4.
Quality of life using the SF36 survey
-
5.
NSAID and other analgesic use in the previous 2 weeks
-
6.
Tricyclic adverse effects using the Antidepressant Side-Effect Checklist
-
7.
Other adverse events
-
8.
Participant’s belief about treatment arm allocation
Study medication withdrawal
Withdrawal of TCAs (including nortriptyline) may be associated with antidepressant discontinuation symptoms. It is, therefore, recommended that these medications are withdrawn in a gradual fashion rather than stopping abruptly. We will recommend a tapered withdrawal to participants in the active arm of the study at the completion of the study. The daily dose will be reduced by 1 capsule (25 mg nortriptyline) every week. To ensure that the investigators and the research nurse remain blinded, this information will be communicated to the participant in a pre-prepared letter which will be posted to the participant by a non-blinded member of the host department. Participants in the placebo arm will also receive a pre-prepared letter informing them of their treatment allocation and that they can simply stop taking their study medication.
Sample size
The minimum important clinical difference for a reduction in pain measured using the WOMAC osteoarthritis index has been determined to be about 10 % of the scale maximum, or a total difference of 5 points on the WOMAC pain numerical rating scale (range 0 to 50) [39]. A sample size of 85 per group will give at least 90 % power at a 2-sided significance level of 0.05 to detect a difference in treatment effect of 5 points between the nortriptyline and placebo groups. The sample size was calculated using a pooled standard deviation 10 points estimated from previous studies, [40] and conservatively assuming no correlation between baseline and follow-up scores. This sample size also allows for detection of the minimum important clinical difference in the proportion of participants responding to treatment according to the Outcome Measures in Rheumatology-OARSI (OMERACT-OARSI) criteria [34]. The sample size will be inflated to 100 participants per group (200 in total) to account for a possible 15 % loss-to-follow-up.
Data management and statistical analysis
Participants’ data will be recorded in individual participant record booklets. Data will be entered into an Access database (Microsoft, Redmond, WA, USA) and then exported to the latest available versions of R and Stata (StataCorp, College Station, TX, USA) [41].
Data analysis will be performed on an intention-to-treat basis with primary and secondary outcomes compared between participants randomised to nortriptyline versus those randomised to placebo regardless of adherence to entry criteria, study medication actually taken, treatment withdrawal, or protocol deviation. A per-protocol analysis will also be performed. Unit of analysis will be at the level of the patient, where patients with OA in both knees will choose the most symptomatic to be the one under study. No interim analyses will be undertaken. All statistical tests will be 2-sided and a level of significance (alpha) of ≤ 0.05 set for all confidence intervals and p values. Demographic characteristics and baseline data will be summarised using descriptive statistics, and presented by treatment group. The mean and standard deviation of participants’ WOMAC pain score at 14 weeks follow-up, and change between baseline and 14 weeks, will be calculated for each treatment group.
The primary outcome of the study will be the size of the treatment effect (mean difference in pain between treatment groups at 14 weeks adjusting for differences at baseline), which will be determined using linear regression modelling including treatment group as fixed effect and baseline pain scores as a covariate. A secondary analysis of this primary outcome will be conducted using multivariable linear regression, adjusting for pre-randomisation variables reasonably expected to predict a favourable outcome (duration of disease, medication use at baseline, and use of assistive devices), and participants’ use of other analgesics (paracetamol, NSAIDs, and opioids) in the 2 final weeks of the study period (weeks 12–14). Secondary continuous outcome measures (WOMAC function and stiffness, patient global assessment, and quality of life measures) will be analysed in a similar manner.
As secondary analysis, patients will be dichotomised according to the OMERACT-OARSI set of responder criteria, defined as a high improvement in pain or function, or a moderate improvement in at least two of pain, function, or patient’s global assessment [34]. Incident rates will be calculated for this and other binary outcomes (including use of other pain medications, adverse events, and tricyclic side effects) and compared using chi-square or Fisher exact tests. Generalised linear regression models will be used to calculate absolute and relative differences in risk between treatment groups with 95 % confidence intervals, adjusting for other variables as appropriate. Self-reported treatment dosage over the past 2 weeks will be summarised for each follow-up, as will the number of participants discontinuing treatment and reasons for doing so. The incidence of all suspect serious treatment reactions will be presented in line with the CONSORT 2010 recommendations [42].
Participants who are missing outcome data will be included in the analysis using modern multiple imputation methods. To determine the robustness of results per protocol analysis will be performed, excluding participants with major protocol violations such as cross-over treatments, withdrawals and loss-to-follow-up. Dose-response will be investigated by entering final dose achieved as a predictor in regression models.
Ethical approval
The study was approved by the Northern A Health and Disability Ethics Committee. Ethics ref 14/NTA/139.
Discussion
This protocol outlines the design of the first randomised controlled trial of a TCA in the management of OA. As there is currently a paucity of effective and safe analgesics for this common condition, the results of the study may have widespread clinical and public health implications.
Trial status
The trial was registered with the Australian New Zealand Clinical Trials Registry on 27 June 2014. Trial identifier: ACTRN12614000683639.
Status: recruiting.
Abbreviations
- ACR:
-
American College of Rheumatology
- BMI:
-
body mass index
- CDHB:
-
Canterbury District Health Board
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- GP:
-
general practitioner
- fMRI:
-
functional magnetic resonance imaging
- NSAID:
-
non-steroidal anti-inflammatory drug
- OA:
-
osteoarthritis
- OMERACT-OARSI:
-
Outcome Measures in Rheumatology-Osteoarthritis Research Society International
- PET:
-
positron emission tomography
- RCT:
-
randomised controlled trial
- SAEs:
-
serious adverse events
- SF36:
-
36-Item Short Form Health Survey
- SNRIs:
-
serotonin and noradrenalin reuptake inhibitors
- TCA:
-
tricyclic antidepressant
- VAS:
-
visual analogue scale
- WOMAC:
-
Western Ontario and McMaster Universities
References
Jinks C, Ong B, Richardson J. A mixed methods study to investigate needs assessment for knee pain and disability: population and individual perspectives. BMC Musculoskelet Disord. 2007;8(1):59.
Arthritis Care. OA Nation the most comprehensive UK report of people with osteoarthritis. 2004. https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&cad=rja&uact=8&ved=0CB0QFjAAahUKEwj8qJPg8rHIAhXFl5QKHdJ3A8s&url=http%3A%2F%2Fwww.arthritiscare.org.uk%2F%403235%2FForhealthpZTLTzlfpxbjMpdvQZaaUJ4g.
The global burden of disease: 2004 update October 2014. http://www.who.int/healthinfo/global_burden_disease/2004_report_update/en/.
Ministry of Health. A Portrait of Health: Key results of the 2006/07 New Zealand Health Survey. 2008. http://www.health.govt.nz/publication/portrait-health-key-results-2006-07-new-zealand-health-survey
Arthritis New Zealand. The economic cost of arthritis in New Zealand in 2010. Report by Access Economics Pty Limited for: Arthritis New Zealand. 2010
Hunter DJ, Felson DT. Osteoarthritis. BMJ. 2006;332(7542):639–42.
National Institute for Health and Care Excellence (NICE). Osteoarthritis Care and management in adults. Clinical guideline CG177 2014. http://www.nice.org.uk/guidance/cg177/evidence/cg177-osteoarthritis-full-guideline3.
Miceli-Richard C, Le Bars M, Schmidely N, Dougados M. Paracetamol in osteoarthritis of the knee. Ann Rheum Dis. 2004;63(8):923–30.
Towheed T, Maxwell L, Judd M, Catton M, Hochberg Marc C, Wells GA. Acetaminophen for osteoarthritis. Cochrane Database Syst Rev. 2006;1
Coxib and Traditional NSAID Trialists’ (CNT) Collaboration. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–79.
Nüesch E, Rutjes Anne WS, Husni E, Welch V, Jüni P. Oral or transdermal opioids for osteoarthritis of the knee or hip. Cochrane Database Syst Rev. 2009;4
Gwynne-Jones D. Quantifying the demand for hip and knee replacement in Otago, New Zealand. N Z Med J. 2013;126:7–17.
Baliki M, Geha P, Jabakhanji R, Harden N, Schnitzer T, Apkarian AV. A preliminary fMRI study of analgesic treatment in chronic back pain and knee osteoarthritis. Mol Pain. 2008;4(1):47.
Gwilym SE, Keltner JR, Warnaby CE, Carr AJ, Chizh B, Chessell I, et al. Psychophysical and functional imaging evidence supporting the presence of central sensitization in a cohort of osteoarthritis patients. Arthritis Care Res. 2009;61(9):1226–34.
Kulkarni B, Bentley DE, Elliott R, Julyan PJ, Boger E, Watson A, et al. Arthritic pain is processed in brain areas concerned with emotions and fear. Arthritis Rheum. 2007;56(4):1345–54.
Sofat N, Ejindu V, Kiely P. What makes osteoarthritis painful? The evidence for local and central pain processing. Rheumatology. 2011;50(12):2157–65.
Sullivan M, Bentley S, Fan M-Y, Gardner G. A single-blind placebo run-in study of venlafaxine XR for activity-limiting osteoarthritis pain. Pain Med. 2009;10(5):806–12.
Chappell AS, Desaiah D, Liu-Seifert H, Zhang S, Skljarevski V, Belenkov Y, et al. A double-blind, randomized, placebo-controlled study of the efficacy and safety of duloxetine for the treatment of chronic pain due to osteoarthritis of the knee. Pain Pract. 2011;11(1):33–41.
Abou-Raya S, Abou-Raya A, Helmii M. Duloxetine for the management of pain in older adults with knee osteoarthritis: randomised placebo-controlled trial. Age Aging. 2012;41(5):646–52.
Frakes EP, Risser RC, Ball TD, Hochberg MC, Wohlreich MM. Duloxetine added to oral nonsteroidal anti-inflammatory drugs for treatment of knee pain due to osteoarthritis: results of a randomized, double-blind, placebo-controlled trial. Curr Med Res Opin. 2011;27(12):2361–72. PubMed.
Savigny P, Kuntze S, Watson P, Underwood M, Ritchie G, Cotterell M, et al. Low back pain: early management of persistent non-specific low back pain. 2009. https://www.nice.org.uk/guidance/CG88.
Üçeyler N, Häuser W, Sommer C. A systematic review on the effectiveness of treatment with antidepressants in fibromyalgia syndrome. Arthritis Care Res. 2008;59(9):1279–98.
Häuser W, Bernardy K, Üçeyler N, Sommer C. Treatment of fibromyalgia syndrome with antidepressants: a meta-analysis. JAMA. 2009;301(2):198–209.
Scott WA. The relief of pain with an antidepressant in arthritis. Practitioner. 1969;202(212):802–7.
Drug information handbook. 11th ed: American Pharmaceutical Association. 2003.
Watson CPN, Vernich L, Chipman M, Reed K. Nortriptyline versus amitriptyline in postherpetic neuralgia: a randomized trial. Neurology. 1998;51(4):1166–71.
Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, et al. Development of criteria for the classification and reporting of osteoarthritis: classification of osteoarthritis of the knee. Arthritis Rheum. 1986;29(8):1039–49.
Bellamy N, Buchanan WW, Goldsmith CH, Campbell J, Stitt LW. Validation study of WOMAC: a health status instrument for measuring clinically important patient relevant outcomes to antirheumatic drug therapy in patients with osteoarthritis of the hip or knee. J Rheumatol. 1988;15(12):1833–40.
Kernan WN, Viscoli CM, Makuch RW, Brass LM, Horwitz RI. Stratified randomization for clinical trials. J Clin Epidemiol. 1999;52(1):19–26.
NZF New Zealand Formulary. Nortriptyline 2013; 13 Available from: http://www.nzf.org.nz/nzf_2260.html?searchterm=nortriptyline.
Wolf CR, Smith G, Smith RL. Pharmacogenetics. BMJ. 2000;320(7240):987–90.
Altman R, Brandt K, Hochberg M, Moskowitz R, Bellamy N, Bloch DA, et al. Design and conduct of clinical trials in patients with osteoarthritis: recommendations from a task force of the Osteoarthritis Research Society: results from a workshop. Osteoarthritis Cartilage. 1996;4(4):217–43.
Bellamy N, Kirwan J, Boers M, Brooks P, Strand V, Tugwell P, et al. Recommendations for a core set of outcome measures for future phase III clinical trials in knee, hip, and hand osteoarthritis. Consensus development at OMERACT III. J Rheumatol. 1997;24(4):799–802.
Pham T, van der Heijde D, Altman RD, Anderson JJ, Bellamy N, Hochberg M, et al. OMERACT-OARSI Initiative: Osteoarthritis Research Society International set of responder criteria for osteoarthritis clinical trials revisited. Osteoarthritis Cartilage. 2004;12(5):389–99.
U.S. Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 4.0. Published: 28 May 2009 (v4.03: 14 June, 2010).
Uher R, Farmer A, Henigsberg N, Rietschel M, Mors O, Maier W, et al. Adverse reactions to antidepressants. Br J Psychiatry. 2009;195(3):202–10.
Rogers JC, Irrgang JJ. Measures of adult lower extremity function: The American Academy of Orthopedic Surgeons Lower Limb Questionnaire, The Activities of Daily Living Scale of the Knee Outcome Survey (ADLS), Foot Function Index (FFI), Functional Assessment System (FAS), Harris Hip Score (HHS), Index of Severity for Hip Osteoarthritis (ISH), Index of Severity for Knee Osteoarthritis (ISK), Knee Injury and Osteoarthritis Outcome Score (KOOS), and Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC™). Arthritis Care Res. 2003;49(S5):S67–84.
McConnell S, Kolopack P, Davis AM. The Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC): a review of its utility and measurement properties. Arthritis Care Res. 2001;45(5):453–61.
Ehrich EW, Davies GM, Watson DJ, Bolognese JA, Seidenberg BC, Bellamy N. Minimal perceptible clinical improvement with the Western Ontario and McMaster Universities osteoarthritis index questionnaire and global assessments in patients with osteoarthritis. J Rheumatol. 2000;27(11):2635–41.
Babul N, Noveck R, Chipman H, Roth SH, Gana T, Albert K. Efficacy and safety of extended-release, once-daily tramadol in chronic pain: a randomized 12-week clinical trial in osteoarthritis of the knee. J Pain Symptom Manage. 2004;28(1):59–71.
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL http://www.R-project.org/. 2014.
Ioannidis JPA, Evans SJW, Gøtzsche PC, O’Neill RT, Altman DG, Schulz K, et al. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med. 2004;141(10):781–8.
Acknowledgments
This study is funded with a project grant from the Health Research Council of New Zealand. This funding source had no role in the design of this study and will not have any role during its execution, analyses, interpretation of the data, or decision to submit results.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BH conceived of the study and led the study design and drafted the protocol. JW, LT, LS, DM, JA, BT and GH all contributed to the study design. JW led the statistical planning. All authors contributed to refinement of the study protocol and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hudson, B., Williman, J.A., Stamp, L.K. et al. Nortriptyline in knee osteoarthritis (NortIKA Study): study protocol for a randomised controlled trial. Trials 16, 448 (2015). https://doi.org/10.1186/s13063-015-0961-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13063-015-0961-1