
Abstract
Background
Reports of dual carriers of pathogenic BRCA1 variants in trans are extremely rare, and so far, most individuals have been associated with a Fanconi Anemia-like phenotype.
Methods
We identified two families with a BRCA1 in-frame exon 20 duplication (Ex20dup). In one male individual, the variant was in trans with the BRCA1 frameshift variant c.2475delC p.(Asp825Glufs*21). We performed splicing analysis and used a transcription activation domain (TAD) assay to assess the functional impact of Ex20dup. We collected pedigrees and mapped the breakpoints of the duplication by long- and short-read genome sequencing. In addition, we performed a mitomycin C (MMC) assay from the dual carrier using cultured lymphoblastoid cells.
Results
Genome sequencing and RNA analysis revealed the BRCA1 exon 20 duplication to be in tandem. The duplication was expressed without skipping any one of the two exon 20 copies, resulting in a lack of wild-type transcripts from this allele. TAD assay indicated that the Ex20dup variant has a functional level similar to the well-known moderate penetrant pathogenic BRCA1 variant c.5096G > A p.(Arg1699Gln). MMC assay of the dual carrier indicated a slightly impaired chromosomal repair ability.
Conclusions
This is the first reported case where two BRCA1 variants with demonstrated functional impact are identified in trans in a male patient with an apparently normal clinical phenotype and no BRCA1-associated cancer. The results pinpoint a minimum necessary BRCA1 protein activity to avoid a Fanconi Anemia-like phenotype in compound heterozygous status and yet still predispose carriers to hormone-related cancers. These findings urge caution when counseling families regarding potential Fanconi Anemia risk. Furthermore, prudence should be taken when classifying individual variants as benign based on co-occurrence in trans with well-established pathogenic variants.
Similar content being viewed by others


Background
Inherited BRCA1 and BRCA2 variants are the main known cause of hereditary breast and ovarian cancer cases. While mono-allelic pathogenic BRCA1 variants are relatively common in the general population worldwide, biallelic pathogenic BRCA1 variants are rarely reported and, until recently, were assumed to be lethal during embryogenesis [1]. However, it has been suggested that dual carriers can survive due to several mechanisms, including some degree of retained wild-type activity from at least one allele or rescue mechanisms [1,2,3]. Nevertheless, these patients are still likely to develop early onset cancer and are often characterized by congenital anomalies and potentially chromosomal fragility. Previous reports of dual BRCA1 carriers are reviewed in Table 1.
Previously reported variants with residual function from dual in trans BRCA1 carriers include at least one variant with reduced penetrance or potential rescue mechanisms resulting in some level of BRCA1 function. In some studies, one of the variants was a missense variant that might retain some activity. For example, Domchek et al. reported a patient with early onset ovarian cancer who had a frameshift variant on one allele and a BRCA1 missense variant (c.5207T > C p.(Val1736Ala)) in trans [2]. They suggested that p.(Val1736Ala) was likely pathogenic but had sufficient residual BRCA1 activity allowing embryonal development and viability through adulthood. However, the authors assumed that the biallelic BRCA1 variants caused phenotypical differences and developmental delay in the patient, which they diagnosed as a “Fanconi Anemia (FA)-like phenotype” [2]. Similar results were reported by Sawyer et al. [4] and Keupp et al. [3]. In agreement with the idea that missense variants may retain some activity, a recent study showed reduced penetrance for patients above 50 years of age carrying pathogenic missense variants compared to those carrying protein truncating variants [5].
Protein truncating variants located in exon 11 (according to legacy exon numbering) of BRCA1 have been shown to retain minimal protein function since a naturally occurring alternative splice donor site results in the expression of an in-frame transcript lacking most of the exon [6]. Freire et al. [7], Chirita-Emandi et al. [8], and Seo et al. [1] also reported a total of six patients with biallelic protein truncating BRCA1 variants in exon 11. They all presented with features of FA and two of the patients developed cancers at 2 and 5 years age.
Here, we report a male lung cancer patient with apparently no FA features who was tested for BRCA1 variants due to a family history of breast, ovarian, and prostate cancer. We identified a pathogenic frameshift variant (c.2475delC) in BRCA1 exon 11, likely responsible for the familial cancer history and a duplication of BRCA1 exon 20 (Ex20dup) in trans.
We also identified the latter variant in a female patient diagnosed with breast cancer at age 46 from an unrelated family. We therefore focused on determining the functional impact of the BRCA1 Ex20dup variant to guide patient counseling and clinical management.
Methods
Patient material
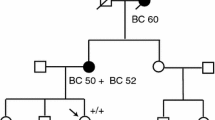
Blood samples were collected from probands from the two families. A sample from the proband in family 1 (F1) was used for immortalizing lymphocytes (LCL) by Epstein-Barr virus infection. Cells were treated with Puromycin before harvest to prevent nonsense-mediated mRNA decay. In addition, PAXgene® Blood tubes were collected from probands from both families (Fig. 1A, F1 III:1 and Fig. 1D, F2 IV:2).

A Pedigree of a lung cancer patient F1 III:1 presenting with a BRCA1 c.2475delC frameshift variant detected by Sanger sequencing (B) and an exon 20 duplication (Ex20dup) in trans identified by two different MLPA test kits (C). Pedigree of a breast cancer patient F2 IV:2 (D) presenting with BRCA1 Ex20dup as detected by two MLPA kits (E). Probands in pedigrees (F1 III:1 and F2 IV:2) are marked by arrows. Variants detected in family members are indicated below the symbols of the tested individuals. If available, the patient age at disease onset in years is indicated
Variant detection
Genetic testing was performed using different technologies including Protein Truncation Test, Multiplexed Ligation-dependent Probe Amplification (MLPA) and Sanger sequencing [9].
Sequencing primers were designed to span the breakpoint region of the variant call from Whole Genome Sequencing (WGS) data (chr17: 41,206,829–41,211,992). The following primers were used: 5′-ATGTGATCTGGCCCTCATCT´-3′ intron 19 (61.79 °C), 5′-TAACTGGGCGTGGTGGTAG-3 intron 20 (61.46 °C). These primers were used for both touchdown PCR and Sanger sequencing reaction.
MLPA analysis was performed according to the manufacturer's instructions (MRC Holland, Amsterdam, the Netherlands) using kits P002 and P087 specific for the BRCA1 gene.
We used exon numbering in BRCA1 according to the traditional numbering, i.e., with no exon 4. Variants are described in accordance with the HGVS guidelines using the RefSeq transcript identifier NM_007294.4.
RNA extraction and analysis
Total RNA was extracted from a PAXgene sample using the PAXgene® Blood RNA Kit from PreAnalytiX by Qiagen according to the manufacturer’s protocol. RNA was extracted from LCL of the proband in family 1 (F1 III:1) using the RNeasy Mini Kit (Qiagen) following the instructions of the manufacturer including on-column DNA digest using DNaseI. RNA integrity and yield were subsequently assessed using an Agilent Bioanalyzer and NanoDrop ND-8000 instrument. For cDNA synthesis, the SuperScript III First-Strand Synthesis System from the RT-PCR Kit (Invitrogen, cat. No. 18080-051) was applied. An area spanning from exon 15 to exon 21 was amplified by PCR using the primers: 5′-CAACAGCTGGAAGAGTCTG-3′ and 5′-CCATAGCAACAGATTTCTAGC-3′. PCR products of approximately 700 bp and 800 bp in length were extracted from agarose gel. PCR products were re-amplified using the above noted primers before Sanger sequencing.
Whole genome sequencing
Long-read WGS was performed using Oxford Nanopore sequencing. High molecular weight DNA was extracted using Nanobind CBB Big DNA Kit (Circulomics). The sequencing library was prepared using a Ligation Sequencing Kit (Oxford Nanopore Technologies) and sequenced on a PromethION (Oxford Nanopore Technology) using a R9.4.1 flow cell. Data were mapped to the GRCh37 (hg19) reference genome using Minimap2. Genome Ribbon was used for visualization of long-read data (https://genomeribbon.com/).
Short-read WGS was performed using Illumina NovaSeq 6000 platform at 2 × 150bp read length. TruSeq™ DNA PCR-Free kit (Illumina, cat.no. 20015963) was used for sequencing library preparation. All processes were done according to the manufacturers’ instructions. Mean sequencing coverage of 30 × was achieved.
WGS data were de-multiplexed and mapped to GRCh37 (hg19) reference genome using BWA for paired-end alignments. SAM files were sorted and converted into BAM files by Picard. BAM files were sorted, indexed and duplicates were marked. Delly2 software was used for variant calling. IGV (Integrative Genomics Viewer) was used for further manual inspection of the region of interest.
Plasmids
The 5 × GAL4-luciferase reporter plasmid (Sun et al. [10]), pHKG3 (Bannister et al. [11]), and pYFP_BRCA1 (Fabbro et al. [12]) were kind gifts from Richard A. Maurer (Oregon Health Sciences University, Portland, Oregon), Tony Kouzarides (University of Cambridge), and Beric R. Henderson (Westmead Institute for Cancer Research, University of Sydney), respectively, while pRL-0 was obtained from Promega. pBluntII-BRCA11396-1863 was constructed by PCR using pYFP-BRCA1 as a template and the oligonucleotides 5′-TTTTGAATTCTCAACAGAAAGGGTC-3′ and 5′-TACTTATCTAGAGTTAGTAGTGGCTGTGG-3’. pHKG3-GAL4-BRCA1-BRCT1-2 was constructed by fusing a gene fragment coding for amino acids 1396 to 1863 of BRCA1 in-frame to the 3´end of the coding sequence of the DNA binding domain of GAL4 (amino acids 1–147). Finally, pcDNA3.1-GAL4-BRCA1-BRCT1-2 was constructed by cloning of GAL4-BRCA1-BRCT1-2 into pcDNA3.1 (Invitrogen). The pcDNA3.1 GAL4-BRCT1-2 wild-type plasmid was mutated using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent) and the following primers: 5′-ATTTCAGTGTCCATTCACACACAAACTCAGCATC-3′ (p.(Arg1699Trp)); 5′-AGTTTGTGTGTGAACAGACACTGAAATATTT-3’ (p.(Arg1699Gln)); 5’-CCTTCACCAACAGGCCCACAGATCAAC-3’ (p.(Met1775Arg)); 5’-CCACCAAGGTCCAAAGTGAGCAAGAGAATC-3’ (p.(Arg1751*)). Successful mutagenesis was verified via Sanger sequencing.
Cloning of BRCA1 exon 20 dup variant
cDNA was generated from RNA purified from the F1 proband’s LCL using the RevertAid TM H Minus First-Strand cDNA Synthesis Kit. Using the primers 5’-CACCGAATTCCAGAGGGATACCATGCAACATAAC-3’ and 5’-TCTAGATCAGTAGTGGCTGTGGGGG-3’ a 1491 bp fragment spanning the BRCT1-2 region including the Ex20dup was amplified and cloned into the pENTR™/D-Topo vector using the pENTR Directional TOPO® Cloning kit (Invitrogen). After sequence confirmation via Sanger sequencing (see Additional file 1: Table S1 for sequencing primer sequences), the vector was digested using EcoRI and BsgI (NEB) and the Ex20dup spanning region ligated into the pcDNA3.1 reporter plasmid replacing GAL4-BRCT1-2 wild type.
Cell lines HEK293 and T47D cells were purchased from ATCC (American Type Culture Collection) and cultured in high glucose (4.5 g/L) Dulbecco’s Modified Eagle Medium (DMEM GlutaMAX™) supplemented with 1 mM sodium pyruvate (Thermo Fischer), 10% Fetal Bovine Serum (Biowest) and 1% penicillin/streptomycin (Thermo Fischer). Cells were grown at 37°C and 5% CO2 in a humidified incubator.
Transcriptional activation assay
300.000 HEK293 cells or 150.000 T47D cells were seeded in triplicates in 6-well plates. After 24 h, cells were co-transfected using Fugene 6 (6 μl/well) with 2 μg of pcDNA3.1 GAL4-BRCT1-2 wild type or mutated fusion protein variant, 1 μg of a GAL4 Luciferase reporter vector and 0.1 μg of PrLO plasmid for normalization. Cells were incubated for 48 h at 37°C and 5% CO2 and Firefly luciferase and Renilla luciferase were measured in a GloMax® 96 Luminometer using the Dual-Luciferase® Reporter Assay Kit (Promega) following the manufacturer’s protocol.
Two-tailed unpaired Student`s t-test was used to compare relative luciferase activities between the different variants. p < 0.05 was considered significant.
Results
The proband, a male patient (Fig. 1A, F1 III:1) was diagnosed with lung cancer (planocelluar carcinoma) at an age of 64 years. He had smoked 20 cigarettes/day since he was 14 years old. Genetic testing revealed two BRCA1 variants: a frameshift variant c.2475delC (p.(Asp825Glufs*21), Fig. 1B) in exon 11 and a duplication of exon 20 (Fig. 1C). The patient had an apparently normal phenotype with respect to clinical features of FA. As the patient died from the lung cancer, we were unable to do a clinical assessment for Fanconi Anemia, however, going through the patient journal we did not identify even subtle features of FA. MMC assay for chromosomal breakage was performed on an immortalized lymphoblast culture. Zero out of 20 metaphases examined had more than 10 chromosomal breaks in agreement with a normal phenotype. Nevertheless, an average of 2.9 breaks was observed compared to 0.6 in a control LCL cell line (data not shown). The patient underwent surgery for lung cancer but did not receive adjuvant therapy, since no lymph node metastases were detected. However, one year later a metastasis was detected in the lung. He received three rounds of carboplatin and vinorelbine. During the treatment, he experienced low grade neuropathy in the hands and fatigue but otherwise few adverse effects.
The maternal branch of the family was strongly affected by cancer; the patient’s mother (II:6) died of bilateral ovarian cancer by the age of 56, three maternal aunts died of breast cancer (II:12) or ovarian cancer (II:9; II:13) at the age of 41, 49 and 63, respectively, and an uncle had prostate cancer (II:11). Clinical information from the paternal family was sparse; however, the father and his four siblings all lived until 70–93 years of age (Fig. 1A, II:1–II:5). The death certificate from the father indicated no cancer. The proband’s daughter (IV:2) tested positive for the c.2475delC variant and was tested negative for the Ex20dup variant (MLPA data not shown). She was diagnosed with breast cancer at the age of 28. Her grandaunt (II:12) also carried c.2475delC but not Ex20dup as confirmed by MLPA (data not shown). The proband’s son (IV:1) inherited the Ex20dup variant (Fig. 1A) and is to date disease-free at the age of 49. As the variants were separately passed on to the daughter and the son of the patient (Fig. 1A), the variants were confirmed to be in trans.
In another family (F2, Fig. 1D), BRCA1 Ex20dup was identified in a female who developed breast cancer (invasive ductal carcinoma, estrogen receptor positive (100%), HER2 normal expression) at 46 years (Fig. 1E, F2 IV:2). The family history of this patient did not provide obvious evidence for hereditary breast and ovarian cancer syndrome (HBOC).
A third, Italian, family was previously reported [13] in which the BRCA1 Ex20dup was identified in a patient diagnosed with early onset breast cancer (HER2 negative; hormone receptor status unknown), who succumbed to the disease at age 34. The updated family history included a total of four additional breast cancer cases: the proband’s mother diagnosed at age 55, one maternal aunt diagnosed at 50, and another maternal aunt and her daughter (a first cousin of the proband’s), diagnosed at 74 and 54, respectively. Cascade testing could not be extended to any of the family members with breast cancer, but it was offered to two unaffected first-degree blood relatives, the proband’s daughter and sister, yielding negative results. Family history also included a lung cancer diagnosis in the proband’s maternal grandfather. No further clinical or pathological information could be retrieved.
To identify the genomic location of the exon 20 duplication, we performed long-read WGS on DNA of the proband (F1 III:1). Alignment of 50 kb reads supports a tandem duplication with forward orientation (Fig. 2A). To further fine-map the breakpoints, Illumina WGS was performed. As schematically summarized in Fig. 2B, multiple inverted reads mapped to the genomic BRCA1 location chr17:41,203,000-chr17:41,218,000. Moreover, data analysis using the IGV software suggested a tandem duplication of 5.163 kb region spanning from chr17:41,206,829-chr17:41,211,992 (Additional file 1: Figure S1). Subsequently, we validated the breakpoint by Sanger sequencing (Fig. 2C) indicating location of a breakpoint between chr17:41,206,830-chr17:41,206,840 and chr17:41,211,993-chr17:41,212,003. Since the sequence of 11 bp (GCTCACTGCAA) is identical in these regions, a more precise breakpoint definition was not possible. The formal HGVS nomenclature for the variant is based on this c.5194-2841_5277 + 2229dup. Sanger sequencing of family 2 (F2 IV:2, Fig. 2C) confirmed the same breakpoint; hence, the variants are identical. However, extended pedigree analysis did not link the two families although they stem from the same geographic region in contrast to the family from Italy. In this Italian family, a duplication of an 8706 bp region was reported with breakpoints located in position chr17:41,213,666 and chr17:41,204,961, respectively [12]. Thus, different breakpoints were identified in the Italian family in comparison with the Danish families. To confirm that the duplication of exon 20 (84 bp) results in an in-frame transcript, RNA originating from immortalized lymphocytes of F1 III:1 was reverse transcribed and analyzed. Sanger sequencing of cloned cDNA fragments spanning from exon 16 to 24 confirmed the presence of a BRCA1 Ex20dup transcript (Fig. 3A, Additional file 1: Figure S2). Moreover, RNA of F2 IV:2 was extracted from blood, reversely transcribed and analyzed by Sanger Sequencing to confirm the presence of a BRCA1 Ex20dup transcript and to exclude exon 20-related alternative splicing (Additional file 1: Figure S2). The PCR-based amplification of an area spanning from exon 15 to exon 21 generated two products of different lengths (Fig. 3B, uncropped gel image in Additional file 1: Figure S3).

Schematic illustration of the breakpoint region identified via Oxford Nanopore and Illumina whole-genome sequencing and Sanger sequencing. A Oxford Nanopore long-read sequencing confirmed in-frame exon 20 duplication. B Multiple inverted Illumina reads fine-mapped the duplication to chr17:41,203,000-chr17:41,218,000. C Sanger sequencing of the PCR-amplified amplicon junction (primer locations are indicated by the black half arrows) aligned to the BRCA1 reference sequence. Eleven consistent bp between chr17:41,206,830-chr17:41,206,840 and chr17:41,211,993-chr17:41,212,003 were identified flanked by corresponding sequences of intron 19 and intron 20. The ref. seq of intron 19 is shown in red and the ref. seq of intron 20 is shown in black. Non-matching bases are displayed in light gray

Analysis of the BRCA1 Ex20dup transcript. A Sanger sequencing results of cloned cDNA fragments of Family 1 patient III:1 confirm the expression of an in-frame BRCA1 Ex20dup transcript. Black arrows in the upper panel indicate exon transitions. B PCR-amplification of cDNA retrieved from Family 2 patient IV:2 provides two products spanning from exon 15 to exon 21. A heterozygous single nucleotide polymorphism (rs1799966, underlined) located on exon 16 allows discrimination between the transcripts originating from the wild-type allele (approx. 700 bp) and the BRCA1 Ex20dup carrying allele (approx. 800 bp). The corresponding uncropped original gel image is shown in Additional file 1: Figure S3
Furthermore, using the same RT-PCR product from F2 IV:2, we also tested the possibility of splicing-rescue i.e. skipping of one of the exon 20 copies by splicing from the Ex20dup allele. We did this by sequencing a heterozygous G/A single nucleotide polymorphism (rs1799966) located in exon 16. This showed that the G nucleotide is present on the allele with the duplication of exon 20 and not detectable on the wild-type allele only displaying the A nucleotide (Fig. 3B).
Finally, we analyzed if the identified BRCA1 Ex20dup variant would result in a protein with retained C-terminal functionality. Thus, we performed a classical Transcriptional Activation (TA) assay using the human embryonic kidney cell line HEK293 and the epithelial breast cancer cell line T47D [14]. BRCT (BRCA1 Carboxy Terminal) 1–2 domains of BRCA1 were cloned and expressed as a fusion protein with GAL4, and cells were co-transfected with a luciferase reporter vector under a GAL4 promoter to assess transcriptional activation capacity of the BRCT1-2 functional domain. Apart from the wild type and Ex20dup BRCA1 sequences, four pathogenic variants that also impaired BRCA1 DNA binding ability were analyzed. A schematic overview of the reporter construct and analyzed BRCA1 variants is displayed in Fig. 4A. The known pathogenic control variants p.(Arg1751*), p.(Met1775Arg) [15] and p.(Arg1699Trp) [5] were proven to cause almost complete loss of function, with residual activities ranging from 2.4% to 16.9% in comparison with the wild type BRCA1 sequence. The p.(Arg1699Gln), known to be associated with a moderate risk relative to an average truncating variant and p.(Arg1699Trp) [16, 17], displayed partial but significant loss of function with 22.6% residual activity in HEK293 cells and 58.7% activity in T47D cells. Comparable values were determined for the Ex20dup variant with 27.3% and 66.2% relative activity in HEK293 and T47D cells, respectively. Thus, the Ex20dup variant exerted a significantly lower transcriptional activity in comparison with the wild-type (Fig. 4B,C), retained a significantly higher activity than the pathogenic control variants p.(Arg1751*), p.(Met1775Arg) and p.(Arg1699Trp), but had similar activity as the moderate penetrance p.(Arg1699Gln) variant. It should be noted that reduction in activity for these two variants was less marked in the hormone sensitive T47D cell line, compared to the HEK293 line.

Analysis of the transcriptional activity of the BRCT domains carrying Ex20dup in comparison with wild-type BRCA1 (wt) regions and BRCA1 variants conferring a high (p.(Arg1699Trp), p.(Arg1751*) and p.(Met1775Arg)) or intermediate (p.(Arg1699Gln)) risk for breast and ovarian cancer. A Overview of cloned variants (BRCA1 exon 13 to exon 24) fused to the GAL4 DNA binding domain for subsequent Transcriptional Activation assay. The assays were performed in B human embryonic kidney cells HEK293 (n = 2) and C epithelial breast cancer cells T47D (n = 3). The relative luciferase activity normalized to the BRCT wt domain is shown. Error bars represent standard deviations and significance was determined via an unpaired, two-tailed Student’s t-test. The black stars indicate significance of reduction compared to wt, and the red stars indicate differences between Ex20dup and individual variants with *p < 0.05, **p < 0.01 and, ***p < 0.001, ns: not significant
Discussion
In this study, we identified a dual BRCA1 variant male carrier with a tandem duplication of exon 20 and a well-known pathogenic variant c.2475delC in trans. At the time of study initiation, the clinical consequence of this in-frame duplication was considered uncertain; it had been reported once in an Italian family where it was suggested to be pathogenic with no further evidence provided for class assignment. However, exon 20 contains part of the BRCT domains, which mediate complex functions of BRCA1 in DNA damage response via, e.g., phospho-protein interactions [18] and in transcriptional activation [19, 20]. Several known pathogenic missense variants have been reported in this domain including c.5213G > A p.(Gly1738Glu) in exon 20, illustrating the importance of this region [21]. We performed allele-specific analysis to eliminate the possibility of rescue of function by exon skipping. Furthermore, we investigated the functional consequences of the duplication by assaying the transcriptional activation using a well-established assay for the BRCA1 BRCT domains [14]. Analysis in both HEK293 and the breast cancer cell line T47D showed that Ex20dup has comparable TAD activity with the missense variant p.(Arg1699Gln), acknowledging increased variability between assay repeats for the T47D assays in particular. The latter variant is well-known to have reduced penetrance compared to average truncating variant, causing risk of breast and ovarian cancer by age 70 years of 20% and 6%, respectively [16, 17, 22].
Evidence from mouse models suggested that one copy of BRCA1 is necessary for embryonic development [23]. However, recent data have shown that if a minimal level of functional protein is produced from the variant allele, for example due to an alternative isoform rescuing minimal function, the fetus is able to survive. Variants located in exon 11 after nucleotide c.787 are a prominent example because of a naturally occurring in-frame isoform, which lacks the majority of exon 11 (c.788–4096), hence more than half of the gene, and yet retains minimal function in PARP inhibitor and cisplatin resistance assays [6]. All reported dual BRCA1 carriers present combinations of variants that may retain some protein function. Furthermore, most patients carrying two known presumed “high risk” pathogenic or likely pathogenic variants had some degree of physical features of FA (Table 1).
Our male patient with biallelic BRCA1 variants had an apparently normal phenotype despite carrying an pathogenic exon 11 frameshift variant and an exon 20 duplication with similar function to a well-known moderate penetrance variant p.(Arg1699Gln). He had few side effects during chemotherapy which also indicates some BRCA1 protein function. Interestingly, the patient reported by Keupp et al. carried p.(Arg1699Gln) in trans with the p.(Cys61Gly) missense variant, had a mild physical FA phenotype and no detectable chromosomal fragility, but the patient experienced severe chemotoxicity [3]. The p.(Cys61Gly) variant was recently shown to confer a similar risk for early onset of breast and ovarian cancer as protein truncating variants like c.2475delC, but lower risk of breast cancer for patients older than 50 years [5]. Further studies including functional assay analysis may help to identify the minimal level of BRCA1 activity required to avoid FA in dual carriers.
The similarity in transactivating function of the Ex20dup variant to the established reduced penetrance p.(Arg1699Gln) variant could be interpreted to mean that Ex20dup has a similar risk profile to p.(Arg1699Gln), but caution is advisable without insight into the interpretation of experimental and clinical data for reduced penetrance variants in general. The ACMG/AMP guidelines [24], and those specified for BRCA1/2 (https://cspec.genome.network/cspec/ui/svi/), are designed and calibrated for classifying variants for Mendelian disease. Applying these BRCA1-specified criteria to the Ex20dup variant, there is conflicting evidence toward and against pathogenicity. Criteria applicable include: population frequency data (absence in gnomADSV; PM2_supporting), variant type and location (proven in-frame duplication within domain; PVS1_Strong); lack of recessive Fanconi Anemia phenotype (no physical features, chromosome normal range, no chemotoxicity, cancer age 64y (> 50y); BS2); breast tumor features against pathogenicity (LR 0.32, for ER positive HER2 positive tumor; BP5_Supporting) according to [25]. Regarding segregation analysis, both families were uninformative in this regard: family 1—the variant was from the father’s side without disease and not genotyped, and the carrier son age 49 does not have a disease; family 2—the variant was from the mother’s side and the carrier mother had a cancer type inconsistent with BRCA1-related disease, and no other cancers were reported in the other ungenotyped relatives. Last, although this variant was not previously assayed as part of larger-scale studies accepted for ongoing VCEP use, assay strength of supporting is appropriate based on the Brnich recommendations PS3_Supporting, [26]. Overall, according to a point approach [27], 6 points would be in favor of pathogenicity and 5 points against pathogenicity, and the variant remains of unknown significance. More clinical data, such as that from larger-scale segregation, penetrance and case–control studies, will be required to determine what level of cancer risk may be associated with this specific Ex20dup variant.
We also note the challenges of comparing similar but not identical duplication (or deletion) events. Although an Ex20dup variant was identified in a third family with several cases of breast cancer and lung cancer [13], the breakpoint in the Italian proband was different from the one in our reported Danish families, indicating that a separate analysis of function and clinical studies would be required to investigate its clinical relevance.
Conclusion
Based on functional data, we propose that the duplication of exon 20 identified in Danish probands may represent a variant with reduced penetrance for breast and ovarian cancer. Further, our results may provide an indication of the level of BRCA1 function that might prevent development of FA physical and chromosomal features in dual carriers. This is an important consideration for classification of BRCA1 variants of unknown significance, since co-occurrence in trans with a known pathogenic variant is often used as an argument for benign classification of variants [28]. The cases reported so far (Table 1) indicate that caution should be taken in this approach depending on the location and type of pathogenic variant.
Our findings may also be informative for risk assessment in potential FA families as it shows that variants with some BRCA1 function may not predispose to classical FA features.
Availability of data and materials
Data and material analyzed during the study are available from the corresponding author on reasonable request.
Abbreviations
- Ex20dup:
-
Exon 20 duplication
- TAD:
-
Transcription activation domain assay
- MMC:
-
Mitomycin C assay
- FA:
-
Fanconi Anemia
- LCL:
-
Immortalizing lymphocytes
- MLPA:
-
Multiplexed Ligation-dependent Probe Amplification
- WGS:
-
Whole genome sequencing
References
Seo A, Steinberg-Shemer O, Unal S, Casadei S, Walsh T, Gumruk F, Shalev S, Shimamura A, Akarsu NA, Tamary H, et al. Mechanism for survival of homozygous nonsense mutations in the tumor suppressor gene BRCA1. Proc Natl Acad Sci USA. 2018;115(20):5241–6.
Domchek SM, Tang J, Stopfer J, Lilli DR, Hamel N, Tischkowitz M, Monteiro AN, Messick TE, Powers J, Yonker A, et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013;3(4):399–405.
Keupp K, Hampp S, Hubbel A, Maringa M, Kostezka S, Rhiem K, Waha A, Wappenschmidt B, Pujol R, Surralles J, et al. Biallelic germline BRCA1 mutations in a patient with early onset breast cancer, mild Fanconi anemia-like phenotype, and no chromosome fragility. Mol Genet Genomic Med. 2019;7(9):e863.
Sawyer SL, Tian L, Kahkonen M, Schwartzentruber J, Kircher M, University of Washington Centre for Mendelian G, Consortium FC, Majewski J, Dyment DA, Innes AM, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015;5(2):135–42.
Li H, Engel C, Hoya M, Peterlongo P, Yannoukakos D, Livraghi L, Radice P, Thomassen M, Hansen TVO, Gerdes AM, et al. Risks of breast and ovarian cancer for women harboring pathogenic missense variants in BRCA1 and BRCA2 compared with those harboring protein truncating variants. Genet Med. 2022;24(10):2208.
Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O’Brien SW, et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016;76(9):2778–90.
Freire BL, Homma TK, Funari MFA, Lerario AM, Leal AM, Velloso E, Malaquias AC, Jorge AAL. Homozygous loss of function BRCA1 variant causing a Fanconi-anemia-like phenotype, a clinical report and review of previous patients. Eur J Med Genet. 2018;61(3):130–3.
Chirita-Emandi A, Andreescu N, Popa C, Mihailescu A, Riza AL, Plesea R, Ioana M, Arghirescu S, Puiu M. Biallelic variants in BRCA1 gene cause a recognisable phenotype within chromosomal instability syndromes reframed as BRCA1 deficiency. J Med Genet. 2021;58(9):648–52.
Thomassen M, Hansen TV, Borg A, Lianee HT, Wikman F, Pedersen IS, Bisgaard ML, Nielsen FC, Kruse TA, Gerdes AM. BRCA1 and BRCA2 mutations in Danish families with hereditary breast and/or ovarian cancer. Acta Oncol. 2008;47(4):772–7.
Sun P, Enslen H, Myung PS, Maurer RA. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994;8(21):2527–39.
Bannister AJ, Brown HJ, Sutherland JA, Kouzarides T. Phosphorylation of the c-Fos and c-Jun HOB1 motif stimulates its activation capacity. Nucleic Acids Res. 1994;22(24):5173–6.
Fabbro M, Rodriguez JA, Baer R, Henderson BR. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J Biol Chem. 2002;277(24):21315–24.
Agata S, Viel A, Della Puppa L, Cortesi L, Fersini G, Callegaro M, Dalla Palma M, Dolcetti R, Federico M, Venuta S, et al. Prevalence of BRCA1 genomic rearrangements in a large cohort of Italian breast and breast/ovarian cancer families without detectable BRCA1 and BRCA2 point mutations. Genes Chromosom Cancer. 2006;45(9):791–7.
Millot GA, Carvalho MA, Caputo SM, Vreeswijk MP, Brown MA, Webb M, Rouleau E, Neuhausen SL, Hansen T, Galli A, et al. A guide for functional analysis of BRCA1 variants of uncertain significance. Hum Mutat. 2012;33(11):1526–37.
Quiles F, Fernandez-Rodriguez J, Mosca R, Feliubadalo L, Tornero E, Brunet J, Blanco I, Capella G, Pujana MA, Aloy P, et al. Functional and structural analysis of C-terminal BRCA1 missense variants. PLoS ONE. 2013;8(4):e61302.
Spurdle AB, Whiley PJ, Thompson B, Feng B, Healey S, Brown MA, Pettigrew C, kConFab, Van Asperen CJ, Ausems MG, et al. BRCA1 R1699Q variant displaying ambiguous functional abrogation confers intermediate breast and ovarian cancer risk. J Med Genet. 2012;49(8):525–32.
Moghadasi S, Meeks HD, Vreeswijk MP, Janssen LA, Borg A, Ehrencrona H, Paulsson-Karlsson Y, Wappenschmidt B, Engel C, Gehrig A, et al. The BRCA1 c.5096G>A p.Arg1699Gln (R1699Q) intermediate risk variant: breast and ovarian cancer risk estimation and recommendations for clinical management from the ENIGMA consortium. J Med Genet. 2018;55(1):15–20.
Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302(5645):636–9.
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71.
Monteiro AN, August A, Hanafusa H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc Natl Acad Sci USA. 1996;93(24):13595–9.
Parsons MT, Tudini E, Li H, Hahnen E, Wappenschmidt B, Feliubadalo L, Aalfs CM, Agata S, Aittomaki K, Alducci E, et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: an ENIGMA resource to support clinical variant classification. Hum Mutat. 2019;40(9):1557–78.
Lovelock PK, Spurdle AB, Mok MT, Farrugia DJ, Lakhani SR, Healey S, Arnold S, Buchanan D, kConFab I, Couch FJ, et al. Identification of BRCA1 missense substitutions that confer partial functional activity: potential moderate risk variants. Breast Cancer Res. 2007;9(6):R82.
Hakem R, de la Pompa JL, Sirard C, Mo R, Woo M, Hakem A, Wakeham A, Potter J, Reitmair A, Billia F, et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996;85(7):1009–23.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Spurdle AB, Couch FJ, Parsons MT, McGuffog L, Barrowdale D, Bolla MK, Wang Q, Healey S, Schmutzler R, Wappenschmidt B, et al. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: a large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res. 2014;16(6):3419.
Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, Kanavy DM, Luo X, McNulty SM, Starita LM, et al. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 2019;12(1):3.
Tavtigian SV, Harrison SM, Boucher KM, Biesecker LG. Fitting a naturally scaled point system to the ACMG/AMP variant classification guidelines. Hum Mutat. 2020;41(10):1734–7.
Byrjalsen A, Steffensen AY, Hansen TVO, Wadt K, Gerdes AM. Classification of the spliceogenic BRCA1 c.4096+3A>G variant as likely benign based on cosegregation data and identification of a healthy homozygous carrier. Clin Case Rep. 2017;5(6):876–9.
de la Hoya M, Soukarieh O, López-Perolio I, Vega A, Walker LC, van Ierland Y, Baralle D, Santamariña M, Lattimore V, Wijnen J, et al. Combined genetic and splicing analysis of BRCA1 c.[594–2A>C; 641A>G] highlights the relevance of naturally occurring in-frame transcripts for developing disease gene variant classification algorithms. Hum Mol Genet. 2016;25(11):2256–68.
Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FBL, Hoogerbrugge N, Spurdle AB, Tavtigian SV, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29(11):1282–91.
Lindor NM, Guidugli L, Wang X, Vallée MP, Monteiro ANA, Tavtigian S, Goldgar DE, Couch FJ. A review of a multifactorial probability based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat. 2012;33(5):900–3.
Bouwman P, van der Gulden H, van der Heijden I, Drost R, Klijn CN, Prasetyanti P, Pieterse M, Wientjens E, Seibler J, Hogervorst FBL, et al. A high-throughput functional complementation assay for classification of BRCA1 missense variants. Cancer Discov. 2013;3(10):1142–55.
Petitalot A, Dardillac E, Jacquet E, Nhiri N, Guirouilh-Barbat J, Julien P, Bouazzaoui I, Bonte D, Feunteun J, Schnell JA, et al. Combining homologous recombination and phosphopeptide-binding data to predict the impact of BRCA1 BRCT variants on cancer risk. Mol Cancer Res. 2019;17(1):54–69.
Fernandes VC, Golubeva VA, Di Pietro G, Shields C, Amankwah K, Nepomuceno TC, de Gregoriis G, Abreu RBV, Harro C, Gomes TT, et al. Impact of amino acid substitutions at secondary structures in the BRCT domains of the tumor suppressor BRCA1: implications for clinical annotation. J Biol Chem. 2019;294(15):5980–92.
Vallon-Christersson J, Cayanan C, Haraldsson K, Loman N, Bergthorsson JT, Brøndum-Nielsen K, Gerdes AM, Møller P, Kristoffersson U, Olsson H, et al. Functional analysis of BRCA1 C-terminal missense mutations identified in breast and ovarian cancer families. Hum Mol Genet. 2001;10(4):353–60.
Kwong A, Ho CYS, Shin VY, Au CH, Chan TL, Ma ESK. A Case Report of Germline Compound Heterozygous Mutations in the BRCA1 Gene of an Ovarian and Breast Cancer Patient. Int J Mol Sci. 2021;22(2):889.
Borlin PR, Brazzola P, Frontzek K, Zanoni P, Morscher RJ, Hench J, Frank S, Kottke R, Rushing EJ, Goeggel Simonetti B, et al. Cancer in children with biallelic BRCA1 variants and Fanconi anemia-like features: Report of a malignant brain tumor in a young child. Pediatr Blood Cancer. 2022;69:e29680.
Acknowledgements
We thank Richard A. Maurer (Oregon Health Sciences University, Portland, Oregon), Tony Kouzarides (University of Cambridge), and Beric R. Henderson (Westmead Institute for Cancer Research. University of Sydney) for the kind gifts of plasmids. Lars Jønson is thanked for the construction of the pcDNA3.1 GAL4 BRCA11396-1863 plasmid.
Funding
Open access funding provided by University of Southern Denmark. The research foundation at Odense University Hospital. ABS was supported by an Australian NHMRC Investigator Fellowship (APP177524).
Author information
Authors and Affiliations
Contributions
M.T. and T. A. K. designed and supervised the overall project. I.B., À.M.R., T.T.N.D., I.M. and D.S. performed and analyzed the in vitro experiments. I.M. and M.J.L. performed the WGS data analysis. H.R.N., S.E.B., A.-B.Sk., L.K.E.H., A.D.N., A.V. and C.M.R. provided critical clinical data and/or research material. I.B., T.T.N.D. and M.T. wrote the initial manuscript and Q.H, U.B.J, Tv.O.H, M.T.P., E.T. and A.B.Sp. critically reviewed and edited the manuscript. All authors reviewed the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed as part of the clinical analysis of the patients. Written or oral consent was given for the study. As a part of the clinical evaluation, this study did not demand ethical approval.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary Tables and Figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Block, I., Mateu-Regué, À., Do, T.T.N. et al. Male with an apparently normal phenotype carrying a BRCA1 exon 20 duplication in trans to a BRCA1 frameshift variant. Breast Cancer Res 26, 6 (2024). https://doi.org/10.1186/s13058-023-01755-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-023-01755-9