
Abstract
Human epidermal growth factor receptor 2 (HER2)-positive breast cancer (BC) is a highly aggressive subtype associated with poor prognosis. The advent of HER2-targeted drugs, including monoclonal antibodies, tyrosine-kinase inhibitors (TKIs) and antibody–drug conjugates, has yielded improved prognosis for patients. Compared with widely used monoclonal antibodies, small-molecule TKIs have unique advantages including oral administration and favorable penetration of blood–brain barrier for brain metastatic BC, and reduced cardiotoxicity. Pyrotinib is an irreversible TKI of the pan-ErbB receptor, and has recently been shown to be clinically effective for the treatment of HER2-positive BC in metastatic and neoadjuvant settings. This review highlights the development on the application of pyrotinib-based therapeutic approaches in the clinical settings of HER2-positive BC.
Similar content being viewed by others
Background
Breast cancer (BC) carries a high incidence and mortality in women worldwide [1]. Knowledge of BC pathogenesis and drug development has advanced and treatment strategies have improved, which has yielded increased long-term survival for patients.
BC is classified into four types based on molecular typing: luminal A, luminal B, human epidermal growth factor receptor 2 (HER2)-positive, and triple-negative [2, 3]. Among all types, HER2-positive BC accounts for approximately 15–20% of cases, and has highly aggressive biological properties [4].
HER2-targeted drugs have dominated treatment of HER2-positive BC [5]. Anti-HER2 drugs can be divided into three major categories: monoclonal antibodies (e.g., trastuzumab, pertuzumab, margetuximab, inetetamab), small-molecule tyrosine-kinase inhibitors (TKIs; e.g., pyrotinib, lapatinib, neratinib, tucatinib), and antibody–drug conjugates (ADCs: e.g., ado-trastuzumab emtansine [T-DM1], trastuzumab deruxtecan [T-DXd], disitamab vedotin [RC-48]). The advent of such drugs has provided more choices and chances for patients with HER2-positive BC [6].
Overview of HER2 signaling pathways and anti-HER2 drugs
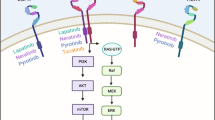
The HER (also known as ErbB) family consists of types 1–4, with a structure comprising extracellular, transmembrane, and intracellular domains. HER1 and HER4 have a receptor-dependent tyrosine-kinase domain. HER2 contains a receptor-independent tyrosine-kinase domain. HER3 lacks a tyrosine-kinase domain [7]. HER-1, -3, and -4 bind to ligands via the extracellular domain to elicit conformational changes that expose their dimerization domain. HER2, independent of ligands, can form homodimers and also heterodimers with HER-1, -3, and -4 in an open active conformation to regulate downstream signaling pathways (e.g., phosphoinositide 3-kinase/protein kinase B [PI3K/Akt], Ras/mitogen-activated protein kinase [MAPK]), thereby affecting the proliferation and apoptosis of cells (Fig. 1) [8,9,10].

Role of HER2-targeted drugs in HER signaling. The four HER family receptors share structural homology with a structure comprising extracellular, transmembrane, and intracellular domains. The extracellular region comprised of four subdomains (I–IV) involved in ligand binding and receptor dimerization; the intracellular region was linked to the single-pass, hydrophobic transmembrane domain, comprising of tyrosine-kinase domain and a tail region that contains several sites of tyrosine phosphorylation. Of note, HER2 harbors no ligand-binding cleft and HER3 has defective intracellular kinase domain. HER2 can form homodimers and also heterodimers with HER-1, -3, and -4 in an open active conformation to regulate downstream signaling pathways, notably phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) and Ras/mitogen-activated protein kinase (MAPK) pathways
Trastuzumab is a humanized immunoglobulin G1 antibody. It can target the extracellular domain IV of HER2, blocking its ligand-independent activation and downstream signaling pathways. These actions have regulatory effects on the proliferation and apoptosis of tumor cells, as well as antibody-dependent toxicity to HER2-overexpressed cells [11, 12]. Pertuzumab targets the extracellular domain II of HER2, can inhibit the heterodimerization of HER2 with HER-1, -3, and -4, block downstream signaling pathways, and regulate the proliferation and apoptosis of tumor cells (Fig. 1) [13, 14]. Trastuzumab and pertuzumab have demonstrated good efficacy for treatment of HER2-positive BC in clinical trials (CLEOPATRA, PUFFIN, NeoSphere, PEONY) [15,16,17,18]. However, 10–20% patients achieve no benefits due to resistance to the effects of trastuzumab (approximately one-third have primary resistance and two-thirds have secondary resistance) and different types of drug resistance are driven by different mechanisms [19]. The main mechanism of primary resistance is that the extracellular target receptors of HER2 are inactivated and thereby lack binding sites for trastuzumab, so downstream PI3K/Akt/mammalian target of rapamycin (mTOR) signal transduction is blocked [19, 20]. The mechanisms of secondary resistance mainly involve: cluster of differentiation (CD)44+/CD24− BC stem cells inhibiting the binding of trastuzumab to the extracellular domain of HER2 [21, 22]; signal masking by mucin-1 and mucin-4 [23, 24]; increased insulin-like growth factor I receptor signaling [25, 26]; altered beta-2 adrenergic receptor signaling [27]; blockade of phosphatase and tensin homolog (PETN)/PI3K/Akt signaling [28]; caveolae-mediated endocytosis [29]; cell-cycle changes that influence HER2 signaling [30].
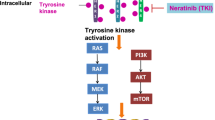
Clinical data suggest that ADC and small-molecule TKIs could be solutions to the resistance of HER2-targeted antibody drugs. TKIs can compete for the intracellular adenosine triphosphate (ATP)-binding region of the HER family to form an ATP-like structure. In this way, TKIs can inhibit the phosphorylation of tyrosine kinases, block the transduction of downstream signaling pathways, and thereby suppress the growth of tumor cells. Clinical evidence has demonstrated the significant efficacy of TKIs such as lapatinib, neratinib, and tucatinib in patients with HER2-positive BC [31,32,33,34]. Moreover, for patients with brain metastases, if monoclonal antibody drugs cannot cross the blood–brain barrier (BBB), then small-molecule TKIs can cross the BBB to achieve better therapeutic effects [35,36,37,38]. In addition, TKIs have oral dosage forms, multiple targets, and low toxicity.
Pyrotinib is an irreversible TKI of the pan-ErbB receptor. By binding covalently to the ATP-binding site of the intracellular kinase domain of HER, pyrotinib can inhibit the autophosphorylation of the homodimers/heterodimers of HER, thereby blocking the Ras/Raf/MEK/MAPK and PI3K/Akt signaling pathways. The binding model of pyrotinib with the kinase domain of HER2 suggests that they are connected by a hydrogen bond between the N1 atom of 3-cyanoquinoline and hinge region Met-801, and that an irreversible covalent double bond is present between the inhibitor and Cys-805 through the Michael addition reaction. This scenario affects downstream signaling and prevents the development and progression of tumors [39, 40]. A phase-Ib clinical trial of pyrotinib monotherapy for advanced breast cancer (ABC) revealed that the maximum tolerated dose was 400 mg/day; pyrotinib (p.o.) could be absorbed completely within 1 h, reach a maximum plasma concentration after 4 h, and achieve a stable plasma concentration after 8 days of administration [41, 42]. In a phase II trial of pyrotinib or lapatinib combined with capecitabine for HER2-positive ABC, the independent radiologic committee-assessed objective response rate (ORR) was 71.4% in the pyrotinib group and 49.2% in the control group, and overall progression-free survival (PFS) was 18.1 months in the pyrotinib group and 7.0 months in the control group (a reduction in the risk of disease progression: 64%) [43]. Studies on use of pyrotinib for treatment of HER2-positive BC are discussed further below.
Clinical evidence of pyrotinib in ABC
First-line therapy for ABC
CLEOPATRA and PUFFIN trials established trastuzumab plus pertuzumab to be first-line treatment for ABC [15, 16]. However, only ~ 11% of patients had been treated previously with trastuzumab in either trial, which differs from current clinical practice. Considering that trastuzumab and/or pertuzumab has been used frequently in the neoadjuvant/adjuvant setting, whether the TKI pyrotinib (with its unique molecular structure and mechanism of action) can provide more benefits for such patients merits investigation.
Recently, the European Society for Medical Oncology published the results of the PHILA study on the efficacy and safety of pyrotinib or placebo combined with trastuzumab and docetaxel as first-line therapy for 590 patients with HER2-positive recurrent/metastatic BC. Investigator-assessed median PFS was 24.3 months and 10.4 months for the two groups, respectively; the proportions of patients treated previously with trastuzumab were 15.5% and 14.3%, respectively; subgroup analysis revealed that median PFS was not reached and was 9.3 months for patients with previous trastuzumab therapy, respectively, and 21.9 months and 10.4 months for those without previous trastuzumab therapy, respectively [44]. In a pooled analysis of three randomized controlled trials on pyrotinib (NCT02422199, NCT03080805, NCT02973737) involving a total of 145 female patients who received pyrotinib as first-line treatment for ABC, blinded independent central review-assessed median PFS was 12.4 months, and ORR was 72.4%; 89.0% patients had used trastuzumab previously, with a median PFS of 12.5 months, which was similar to the whole cohort [45]. The PANDORA trial (NCT03876587) revealed favorable efficacy of pyrotinib plus docetaxel as first-line therapy for HER2-positive metastatic BC. Seventy-nine patients were enrolled, of whom 65 could be evaluated. ORR was 78.5% for 65 patients, 83.3% for patients with previous trastuzumab treatment (accounting for 30.4%), 74.5% for those without previous trastuzumab treatment (accounting for 68.6%), 89.5% for those with visceral metastases, and 73.3% for those without visceral metastases [46]. Those studies demonstrated the promising efficacy of pyrotinib as first-line therapy for HER2-positive ABC regardless of previous use of trastuzumab.
Second-line therapy for ABC
The PHOEBE trial assigned 267 patients to receive pyrotinib plus capecitabine or lapatinib plus capecitabine. Median PFS was 12.5 months and 6.8 months, respectively (hazard ratio [HR] = 0.39, 95% confidence interval [CI] 0.27–0.56, P < 0.0001). Median overall survival (OS) was not reached and was 26.9 months, respectively (HR = 0.69, 95% CI 0.48–0.98, P = 0.02). Subgroup analysis revealed significant benefits regardless of previous use of trastuzumab: median PFS was 12.5 months and 6.9 months for patients with previous trastuzumab treatment, respectively; median PFS was 12.5 months and 5.6 months for patients who had used trastuzumab before, respectively; OS was not reached [47, 48].
The PHENIX trial investigated the efficacy of pyrotinib plus capecitabine versus placebo plus capecitabine for patients who had had disease progression during or after trastuzumab treatment or who could not receive trastuzumab or lapatinib. Independent review committee-assessed median PFS was 11.1 months and 4.1 months, respectively (HR = 0.18, 95% CI 0.13–0.26, P < 0.001). In terms of secondary endpoints: ORR was 68.6% and 16.0%, respectively; clinical benefit was achieved in 76.8% and 22.3% of cases, respectively; median OS was 34.9 months and 23.6 months, respectively (HR = 0.74, 95% CI 0.54–1.02, P = 0.068). Subgroup analysis demonstrated that pyrotinib plus capecitabine was significantly superior to placebo plus capecitabine regardless of metastatic sites or the status of the hormone receptor and trastuzumab resistance [49, 50].
Pyrotinib exhibits superior effects in prolonging PFS to other types of second-line therapy for ABC. Median PFS has been reported to be 9.6 months using T-DM1 alone [51], 8.4 months using lapatinib plus capecitabine [52], 8.2 months using trastuzumab plus capecitabine [53], and 2.8 months using trastuzumab plus lapatinib [54]. Multiple drugs have been approved for second-line therapy, but availability between countries/regions differs. Based on efficacy and safety evidence, pyrotinib has been recommended as preferred second-line therapy in Chinese clinical guidelines [55, 56].
Third-/later-line therapy for ABC
Third-/later-line treatment of ABC is complicated. Most patients develop drug resistance after experiencing various types of therapy (e.g., targeted, endocrine, chemotherapy), accompanied by multiple metastases. Treatment strategies should be formulated based on comprehensive factors.
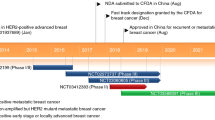
A real-world study evaluated the efficacy of pyrotinib plus capecitabine versus trastuzumab plus capecitabine as second-/later-line anti-HER2 therapy for patients with ABC: compared with the trastuzumab group (100 patients), the pyrotinib group (81 patients) showed significantly higher ORR (42.00% vs. 58.02%, P = 0.037) and significantly longer median PFS (7.11 months vs. 8.02 months, P = 0.035) [57]. In a real-world study investigating the efficacy of pyrotinib in the setting of lapatinib resistance (most patients had been treated with ≥ 2 lines of anti-HER2 regimens), 113 patients were assigned to receive a combination of pyrotinib plus capecitabine, vinorelbine, or trastuzumab; median PFS was 5.4 months for lapatinib-resistant patients and 9 months for lapatinib-naive patients [58]. Sun et al. [59] reported that, among 64 patients with ABC who had received multiple lines of treatment, 17.2% were resistant to lapatinib, with an ORR of 44.1% and a median PFS of ~ 10 months, after pyrotinib-based therapy. In a real-world study involving 94 patients (31.9% with resistance to lapatinib), for lapatinib-resistant and lapatinib-naive patients, pyrotinib-based treatment generated median PFS of 6.36 months and 9.02 months and median OS of 14.35 months and 20.73 months, respectively [60]. Another real-world study involving 218 patients (40.8% with previous use of lapatinib) showed that median PFS was 6.8 months with pyrotinib-based therapy as third-line treatment [61]. Those studies indicated that pyrotinib showed encouraging efficacy even after failure of multiple lines of therapy (Fig. 2).

Median PFS of lapatinib-resistant and -naive patients after later-line treatment with pyrotinib in advanced stage (mPFS, median progression-free survival; NA, not applicable)
Brain metastases
Patients with HER2-positive ABC are at high risk of developing brain metastases, which confers a poor prognosis [62]. In addition to local treatment, efficacious systemic treatment is vital for resolving brain metastases. The PERMEATE trial involving 78 patients with HER2-positive BC with brain metastases revealed that, for radiotherapy-naive and radiotherapy-resistant cohorts receiving pyrotinib plus capecitabine, the intracranial ORRs were 74.6% (95% CI 61.6–85.0) and 42.1% (95% CI 20.3–66.5), respectively, and median PFS was 11.3 months (95% CI 7.7–14.6) and 5.6 months (95% CI 3.4–10.0), respectively. Also, the most common adverse events of grade ≥ 3 were diarrhea (24%), reduced white blood cell count (14%), and reduced neutrophil count (14%), which were (in general) manageable [63].
Real-world studies have also demonstrated the stable and reliable efficacy of pyrotinib in patients with brain metastases [64,65,66]. In a real-world study of 113 patients, 31 patients with brain metastases receiving pyrotinib-containing treatment showed a median PFS of 6.7 months and an intracranial ORR of 28% [58]. Another real-world study reported various efficacy indicators of pyrotinib-based therapy in 42 patients with ABC suffering from brain metastases. ORR was 40.4%, disease control was obtained in 92.8% of cases, improvement in intracranial symptoms was noted in all patients, the median duration of intracranial improvement was 15 months, the median time to relieve brain metastases was 43 days, the median time to relieve other metastases was 50 days, the median time to progression of brain metastases was 16.6 months, and the median time to disease progression was 11.1 months [67]. In a retrospective study involving 61 HER2-positive patients with brain metastases treated by pyrotinib-based regimens, median PFS was 8.6 months, median OS was 18.0 months, and the combination of pyrotinib with nab-paclitaxel was superior to the combination with capecitabine and vinorelbine with respect to PFS and OS. Those studies suggested that the unique structure and low molecular weight of pyrotinib enabled BBB crossing, thereby generating favorable therapeutic effects upon brain metastases. Ongoing research may provide more evidence for the therapeutic value of pyrotinib in patients with ABC with brain metastases, and further optimize the use of pyrotinib.
Clinical evidence of pyrotinib in early BC
Neoadjuvant therapy
According to guidelines set by the National Comprehensive Cancer Network in 2022 and American Society of Clinical Oncology in 2021 [68, 69], neoadjuvant therapy is recommended for patients with HER2-positive BC with tumor size > 2 cm and/or a positive lymph node status (LN+). Neoadjuvant therapy for HER2-positive BC has evolved from single trastuzumab targeting to trastuzumab-based dual targeting. The NOAH study established the role of single-target drugs in neoadjuvant therapy for HER2-positive BC [70]. In NeoSphere and PEONY studies, total pathologic complete response (tpCR) with trastuzumab plus pertuzumab was achieved in 39.3% of cases, which was significantly superior to that of single-target therapy and chemotherapy [17, 18]. Small-molecule TKIs and macromolecule monoclonal antibodies act on intracellular and extracellular target sites simultaneously to exhibit synergistic anti-HER2 effects. The NeoALTTO trial assigned 455 patients to receive trastuzumab plus lapatinib, lapatinib alone, or trastuzumab alone, and pathologic complete response (pCR) was achieved in 51.3%, 24.7%, and 29.5% of cases, respectively, which demonstrated the superior efficacy of trastuzumab plus TKI in the neoadjuvant setting [33]. A meta-analysis of 1410 patients (from CALGB 40601, CHER-LOB, NSABP-B41, and NeoALTTO trials) revealed that, compared with trastuzumab monotherapy, lapatinib plus trastuzumab improved recurrence-free survival significantly (HR = 0.62, 95% CI 0.46–0.85) and OS (HR = 0.65, 95% CI 0.43–0.98) upon combination with neoadjuvant chemotherapy [71]. Those results indicated that a combination of trastuzumab with TKIs could be a promising neoadjuvant strategy.
We researched the use of pyrotinib in neoadjuvant therapy for HER2-positive BC: 19 patients received four cycles of ECP (epirubicin, cyclophosphamide, pyrotinib) and then four cycles of THP (docetaxel, trastuzumab, pyrotinib) before surgery, and tpCR was achieved in 73.7% (95% CI 48.8–90.9), and ORR was 100% (95% CI 82.4–100) of cases [72]. Subsequent clinical trials confirmed the favorable activity of pyrotinib in neoadjuvant therapy. In the PHEDRA trial (NCT03588091), 355 patients were assigned randomly to receive pyrotinib or placebo in combination with trastuzumab and docetaxel for four cycles before surgery; the pyrotinib group showed a significantly higher rates of tpCR (41.0% vs. 22.0%) and breast pCR (43.8% vs. 23.7%) (assessed by an independent review committee) than the placebo group [73]. The multicenter phase II Panphila trial reported a pCR rate of 55.1% in 69 patients with HER2-positive BC receiving six cycles of neoadjuvant therapy with TCbHPy (docetaxel, carboplatin, trastuzumab, pyrotinib) [74]. In the phase II NeoATP trial, the pCR rate reached 69.81% in 53 patients with HER2-positive local ABC (stage IIA–IIIC) receiving four cycles of pyrotinib plus trastuzumab and paclitaxel-cisplatin as neoadjuvant treatment [75]. A retrospective study of 545 patients revealed that in the neoadjuvant setting, the pCR rate with TCbHPy was superior to that with TCbH and comparable to that with TCbHP (docetaxel, carboplatin, trastuzumab, pertuzumab) in HER2-positive local ABC [76]. Those results demonstrated that pyrotinib could significantly improve the pCR and ORR of patients under neoadjuvant treatment (Fig. 3), thereby increasing the possibility of rapid tumor shrinkage and cure at an early stage. As shown in the studies stated above, chemotherapy regimens in combination with trastuzumab and pyrotinib vary. Optimizing chemotherapy combinations and balancing neoadjuvant efficacy and toxicity are key problems to be explored further. Clinical studies on neoadjuvant therapy using pyrotinib are summarized in Table 1.

Comparison of pathologic complete response rate between neoadjuvant chemotherapy regimens from different clinical studies (T, taxanes; H, trastuzumab; P, pertuzumab; L, lapatinib; Cb, carboplatin; F, fluorouracil; E, epirubicin; Py, pyrotinib; Chemo, chemotherapy)
Adjuvant therapy
Since failure of the ALLTO trial [77], few studies have investigated the efficacy of adjuvant TKIs for HER2-positive BC. The BCIRG006, NSABP B-31/NCCTG N9831, and HERA studies demonstrated that trastuzumab administered in the adjuvant setting can control disease progression effectively [78,79,80]. The KATHERINE trial revealed that adjuvant T-DM1 greatly increased the 3-year invasive disease-free survival (iDFS) rate of patients with HER2-positive BC who did not achieve pCR who had received neoadjuvant therapy. In the APHINITY trial, pertuzumab plus trastuzumab with chemotherapy significantly increased the 6-year iDFS rate compared with trastuzumab with chemotherapy, especially for LN+ patients [81]. The ExteNET trial is the only one with successful results with TKIs in the adjuvant setting. That study randomly assigned 2840 patients treated with adjuvant trastuzumab and chemotherapy to receive neratinib or placebo for 1 year; compared with placebo, neratinib increased the 5-year iDFS rate significantly by 2.5% (87.7% vs. 90.2%) and by 3.7% (86.6% vs. 91.2%) in the LN+ subgroup analysis [82]. Whether pyrotinib can be used in intensive adjuvant therapy, especially for high-risk patients (LN+, non-pCR), merits attention. An ongoing phase III trial (CTR20191261) is exploring extended adjuvant therapy (pyrotinib following trastuzumab) in LN+ patients who had been treated with trastuzumab and/or pertuzumab. That study could provide more data for adjuvant application of TKIs. Clinical studies on adjuvant pyrotinib therapy are summarized in Table 2.
Toxicity of pyrotinib and management
Owing to its unique structure and pharmacological mechanism of action, pyrotinib exhibits favorable efficacy and effective tumor control in HER2-positive BC but, simultaneously, its adverse reactions (e.g., diarrhea) trouble patients. In the PHOEBE, PHENIX, and PANDORA trials, the incidence rates of diarrhea of grade ≥ 3 were 31%, 33%, and 38.2%, respectively [46, 47, 49]. The PHADRA and PHILA trials also reported a high rate of diarrhea of grade ≥ 3 [44, 73]. Fortunately, this problem has some solution in intent-to-treat analysis. The PANDORA trial revealed that prophylaxis using loperamide reduced the incidence of diarrhea of grade ≥ 3 significantly from 38.2 to 8.9% [46]. ChiCTR2200060339 [83] and ChiCTR2100051163 [84] are also exploring active management of diarrhea to reduce diarrhea of grade ≥ 3. In clinical practice, to increase adherence and extend treatment cycles, the tolerability of pyrotinib can be improved by: establishing patients’ expectations of adverse reactions; reducing patients’ psychological burden such as fear; preventive treatment with loperamide; avoiding long-term diarrhea-induced negative conditions such as anorexia and fatigue.
Biomarkers of pyrotinib efficacy
A phase-I clinical study reported that the efficacy of pyrotinib could be predicted by the levels of phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) and TP53 mutations in circulating tumor DNA rather than in tumor cells [41]. The NeoATP study [75] revealed that pCR was more likely to be achieved in patients who were estrogen receptor-negative progesterone receptor-negative, HER2 3+ by immunohistochemistry (IHC), with a HER2/CEP17 ratio ≥ 4, and HER2 copy number ≥ 14. pCR was not related to PIK3CA status, Ki67 index, or stromal tumor-infiltrating lymphocytes (TILs). The Panphila study [74] confirmed that patients with hormone receptor-negative disease and HER2 IHC 3+ were more likely to achieve pCR. In addition, the pCR rate was independent of the TIL level regardless of whether the threshold of the TIL level was defined as 5% or 50%, and the TIL level was similar in pCR and non-pCR cohorts. Multiplex IHC results revealed associations of pCR with stromal levels of CD20, CD8, CD4, and forkhead box P3 (FOXP3) and epithelial levels of CD20, CD8, and CD4 before treatment. Among them, stromal levels of CD20, CD8, and CD4 and the epithelial level of CD8 were determined to be independent predictors of pCR according to multivariable logistic regression analysis. Based on stromal immune markers, unsupervised hierarchical clustering analysis revealed that patients with high levels of CD20, CD8, CD4, and FOXP3 simultaneously had a higher possibility of pCR. We assessed 425 genes in tumor samples from patients receiving neoadjuvant therapy with pyrotinib, trastuzumab, and chemotherapy. We concluded that the PIK3CA mutation was an independent predictor of therapeutic effects; patients with a PIK3CA mutation were less likely to achieve pCR, whereas the TIL level was not associated with pCR [85]. Those biomarker studies could preliminarily guide the selection of patients more likely to benefit from pyrotinib-based regimens. Ongoing biomarker studies may provide more information on the use of pyrotinib for BC.
Conclusions
At present, among four approved anti-HER2 TKI drugs in China, pyrotinib has more robust clinical evidence and covers more people in clinical practice. Compared with lapatinib, PHOEBE study demonstrated that pyrotinib can significantly prolong PFS in metastatic setting [47]. In terms of neratinib, NEfERT-T trial failed to prove that neratinib–paclitaxel was superior to trastuzumab–paclitaxel in first-line HER2-positive ABC [37]. Compared with tucatinib, whose benefit is limited to metastatic setting, evidence supports clinical benefit of pyrotinib in both early and advanced stage.
Pyrotinib shows encouraging efficacy in neoadjuvant, advanced-stage, first-/second-/later-line, and brain-metastases settings, as well as in triple-positive patients. With excellent therapeutic effects, pyrotinib is changing the landscape of BC treatment. Future research should focus on how to select and identify patients who are more likely to benefit from pyrotinib-containing combinations. For example, does combination with pyrotinib have greater efficacy for patients who progress rapidly after (neo)adjuvant treatment with macromolecular antibodies such as trastuzumab? Can pyrotinib prevent and reduce the risk of metastases to the central nervous system? Why are patients sensitive or resistant to pyrotinib, and could the related molecular markers be identified? In which populations can combination with pyrotinib better compensate for the deficiency of macromolecular antibody drugs? Another focus is how to identify (at an early stage) patients prone to pyrotinib-related diarrhea and formulate strategies for optimal management of diarrhea, which can help deepen understanding of the toxicity of pyrotinib and improve its safety and patient adherence. Such explorations will help maximize the benefits of patients taking pyrotinib.
Availability of data and materials
Not applicable.
Abbreviations
- BC:
-
Breast cancer
- HER2:
-
Human epidermal growth factor receptor 2
- TKI:
-
Tyrosine-kinase inhibitor
- ADC:
-
Antibody–drug conjugate
- T-DM1:
-
Ado-trastuzumab emtansine
- PI3K:
-
Phosphoinositide 3-kinase
- CD:
-
Cluster of differentiation
- ATP:
-
Adenosine triphosphate
- BBB:
-
Blood–brain barrier
- MAPK:
-
Mitogen-activated protein kinase
- ABC:
-
Advanced breast cancer
- ORR:
-
Objective response rate
- PFS:
-
Progression-free survival
- HR:
-
Hazard ratio
- CI:
-
Confidence interval
- LN:
-
Lymph node
- tpCR:
-
Total pathologic complete response
- pCR:
-
Pathologic complete response
- iDFS:
-
Invasive disease-free survival
- PIK3CA:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- IHC:
-
Immunohistochemistry
- FOXP3:
-
Orkhead box P3
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30.
Feng Y, Spezia M, Huang S, et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018;5(2):77–106.
Yeo SK, Guan JL. Breast cancer: multiple subtypes within a tumor? Trends Cancer. 2017;3(11):753–60.
Loibl S, Gianni L. HER2-positive breast cancer. Lancet. 2017;389(10087):2415–29.
Bradley R, Braybrooke J, Gray R, et al. Trastuzumab for early-stage, HER2-positive breast cancer: a meta-analysis of 13 864 women in seven randomised trials. Lancet Oncol. 2021;22(8):1139–50.
Schlam I, Swain SM. HER2-positive breast cancer and tyrosine kinase inhibitors: the time is now. NPJ Breast Cancer. 2021;7(1):56.
Citri A, Skaria KB, Yarden Y. The deaf and the dumb: the biology of ErbB-2 and ErbB-3. Exp Cell Res. 2003;284(1):54–65.
Kreutzfeldt J, Rozeboom B, Dey N, et al. The trastuzumab era: current and upcoming targeted HER2+ breast cancer therapies. Am J Cancer Res. 2020;10(4):1045–67.
Zhao J, Mohan N, Nussinov R, et al. Trastuzumab blocks the receiver function of HER2 leading to the population shifts of HER2-containing homodimers and heterodimers. Antibodies (Basel). 2021;10(1):7.
Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11(2):263–75.
Varchetta S, Gibelli N, Oliviero B, et al. Elements related to heterogeneity of antibody-dependent cell cytotoxicity in patients under trastuzumab therapy for primary operable breast cancer overexpressing Her2. Cancer Res. 2007;67(24):11991–9.
Derakhshani A, Rezaei Z, Safarpour H, et al. Overcoming trastuzumab resistance in HER2-positive breast cancer using combination therapy. J Cell Physiol. 2020;235(4):3142–56.
Badache A, Hynes NE. A new therapeutic antibody masks ErbB2 to its partners. Cancer Cell. 2004;5(4):299–301.
Adams CW, Allison DE, Flagella K, et al. Humanization of a recombinant monoclonal antibody to produce a therapeutic HER dimerization inhibitor, pertuzumab. Cancer Immunol Immunother. 2006;55(6):717–27.
Swain SM, Miles D, Kim S-B, et al. Pertuzumab, trastuzumab, and docetaxel for HER2-positive metastatic breast cancer (CLEOPATRA): end-of-study results from a double-blind, randomised, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21(4):519–30.
Xu B, Li W, Zhang Q, et al. Pertuzumab, trastuzumab, and docetaxel for Chinese patients with previously untreated HER2-positive locally recurrent or metastatic breast cancer (PUFFIN): a phase III, randomized, double-blind, placebo-controlled study. Breast Cancer Res Treat. 2020;182(3):689–97.
Gianni L, Pienkowski T, Im YH, et al. 5-year analysis of neoadjuvant pertuzumab and trastuzumab in patients with locally advanced, inflammatory, or early-stage HER2-positive breast cancer (NeoSphere): a multicentre, open-label, phase 2 randomised trial. Lancet Oncol. 2016;17(6):791–800.
Shao Z, Pang D, Yang H, et al. Efficacy, safety, and tolerability of pertuzumab, trastuzumab, and docetaxel for patients with early or locally advanced ERBB2-positive breast cancer in Asia: the PEONY phase 3 randomized clinical trial. JAMA Oncol. 2020;6(3):e193692.
Luque-Cabal M, Garcia-Teijido P, Fernandez-Perez Y, et al. Mechanisms behind the resistance to trastuzumab in HER2-amplified breast cancer and strategies to overcome it. Clin Med Insights Oncol. 2016;10(Suppl 1):21–30.
Asic K. Dominant mechanisms of primary resistance differ from dominant mechanisms of secondary resistance to targeted therapies. Crit Rev Oncol Hematol. 2016;97:178–96.
Seo AN, Lee HJ, Kim EJ, et al. Expression of breast cancer stem cell markers as predictors of prognosis and response to trastuzumab in HER2-positive breast cancer. Br J Cancer. 2016;114(10):1109–16.
Martin-Castillo B, Oliveras-Ferraros C, Vazquez-Martin A, et al. Basal/HER2 breast carcinomas: integrating molecular taxonomy with cancer stem cell dynamics to predict primary resistance to trastuzumab (Herceptin). Cell Cycle. 2013;12(2):225–45.
Raina D, Uchida Y, Kharbanda A, et al. Targeting the MUC1-C oncoprotein downregulates HER2 activation and abrogates trastuzumab resistance in breast cancer cells. Oncogene. 2014;33(26):3422–31.
Pai P, Rachagani S, Lakshmanan I, et al. The canonical Wnt pathway regulates the metastasis-promoting mucin MUC4 in pancreatic ductal adenocarcinoma. Mol Oncol. 2016;10(2):224–39.
Saisana M, Griffin SM, May FEB. Importance of the type I insulin-like growth factor receptor inHER2, FGFR2andMET-unamplified gastric cancer with and without Ras pathway activation. Oncotarget. 2016;7(34):54445–62.
Toth G, Szoor A, Simon L, et al. The combination of trastuzumab and pertuzumab administered at approved doses may delay development of trastuzumab resistance by additively enhancing antibody-dependent cell-mediated cytotoxicity. MAbs. 2016;8(7):1361–70.
Liu D, Yang Z, Wang T, et al. beta2-AR signaling controls trastuzumab resistance-dependent pathway. Oncogene. 2016;35(1):47–58.
Berns K, Horlings HM, Hennessy BT, et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007;12(4):395–402.
Chung YC, Chang CM, Wei WC, et al. Metformin-induced caveolin-1 expression promotes T-DM1 drug efficacy in breast cancer cells. Sci Rep. 2018;8(1):3930.
Scaltriti M, Eichhorn PJ, Cortes J, et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc Natl Acad Sci U S A. 2011;108(9):3761–6.
Murthy RK, Loi S, Okines A, et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N Engl J Med. 2020;382(7):597–609.
Chan A, Moy B, Mansi J, et al. Final efficacy results of neratinib in HER2-positive hormone receptor-positive Early-stage breast cancer from the phase III ExteNET trial. Clin Breast Cancer. 2021;21(1):80-91.e7.
Baselga J, Bradbury I, Eidtmann H, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379(9816):633–40.
Sirhan Z, Thyagarajan A, Sahu RP. The efficacy of tucatinib-based therapeutic approaches for HER2-positive breast cancer. Mil Med Res. 2022;9(1):39.
O’Sullivan CC, Davarpanah NN, Abraham J, et al. Current challenges in the management of breast cancer brain metastases. Semin Oncol. 2017;44(2):85–100.
Xuhong JC, Qi XW, Zhang Y, et al. Mechanism, safety and efficacy of three tyrosine kinase inhibitors lapatinib, neratinib and pyrotinib in HER2-positive breast cancer. Am J Cancer Res. 2019;9(10):2103–19.
Awada A, Colomer R, Inoue K, et al. Neratinib plus paclitaxel vs trastuzumab plus paclitaxel in previously untreated metastatic ERBB2-positive breast cancer: the NEfERT-T randomized clinical trial. JAMA Oncol. 2016;2(12):1557–64.
Gelmon KA, Boyle FM, Kaufman B, et al. Lapatinib or trastuzumab plus taxane therapy for human epidermal growth factor receptor 2-positive advanced breast cancer: final results of NCIC CTG MA.31. J Clin Oncol. 2015;33(14):1574–83.
Ma F, Li Q, Chen S, Zhu W, Fan Y, Wang J, Luo Y, Xing P, Lan B, Li M, Yi Z. Phase I study and biomarker analysis of pyrotinib, a novel irreversible Pan-ERBB receptor tyrosine kinase inhibitor, in patients with human epidermal growth factor receptor 2–positive metastatic breast cancer. J Clin Oncol. 2017;35:3105–12.
Li X, Yang C, Wan H, et al. Discovery and development of pyrotinib: a novel irreversible EGFR/HER2 dual tyrosine kinase inhibitor with favorable safety profiles for the treatment of breast cancer. Eur J Pharm Sci. 2017;110:51–61.
Meng J, Liu XY, Ma S, et al. Metabolism and disposition of pyrotinib in healthy male volunteers: covalent binding with human plasma protein. Acta Pharmacol Sin. 2019;40(7):980–8.
Zhu Y, Li L, Zhang G, et al. Metabolic characterization of pyrotinib in humans by ultra-performance liquid chromatography/quadrupole time-of-flight mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1033–1034:117–27.
Ma F, Ouyang Q, Li W, et al. Pyrotinib or lapatinib combined with capecitabine in HER2-positive metastatic breast cancer with prior taxanes, anthracyclines, and/or trastuzumab: a randomized, phase II study. J Clin Oncol. 2019;37(29):2610–9.
Xu B, Yan M, Ma F, Li W, Ouyang Q, Tong Z, Teng Y, Wang S, Wang Y, Geng C, Luo T. Pyrotinib or placebo in combination with trastuzumab and docetaxel for HER2-positive metastatic breast cancer (PHILA): a randomized phase III trial. Ann Oncol. 2022;33(suppl_7):S808–69.
Guan X, Ma F, Xu B. Pooled analyses of randomized controlled trials on pyrotinib plus capecitabine and a rethink of the first-line options for HER2-positive relapsed or metastatic breast cancer. Cancer Innov. 2022;1(2):119–23.
Wang X, Huang J, Zheng Y, et al. Pyrotinib in combination with docetaxel as first-line treatment for HER2-positive metastatic breast cancer (PANDORA): a single-arm, multicenter phase 2 trial. Cancer Res. 2022;82(4_Supplement):S646.
Xu B, Yan M, Ma F, et al. Pyrotinib plus capecitabine versus lapatinib plus capecitabine for the treatment of HER2-positive metastatic breast cancer (PHOEBE): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021;22(3):351–60.
Xu B, Yan M, Ma F, et al. Updated overall survival (OS) results from the phase 3 PHOEBE trial of pyrotinib versus lapatinib in combination with capecitabine in patients with HER2-positive metastatic breast cancer. Cancer Res. 2022;82(4_Supplement):GS3-02.
Yan M, Bian L, Hu X, et al. Pyrotinib plus capecitabine for human epidermal growth factor receptor 2-positive metastatic breast cancer after trastuzumab and taxanes (PHENIX): a randomized, double-blind, placebo-controlled phase 3 study. Transl Breast Cancer Res. 2020;1:13.
Jiang Z, Yan M, Bian L, et al. Overall survival (OS) results from the phase III PHENIX trial of HER2+ metastatic breast cancer treated with pyrotinib plus capecitabine. Cancer Res. 2022;82(4_Supplement):PD8-05.
Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367(19):1783–91.
Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355(26):2733–43.
von Minckwitz G, du Bois A, Schmidt M, et al. Trastuzumab beyond progression in human epidermal growth factor receptor 2-positive advanced breast cancer: a German breast group 26/breast international group 03–05 study. J Clin Oncol. 2009;27(12):1999–2006.
Blackwell KL, Burstein HJ, Storniolo AM, et al. Randomized study of Lapatinib alone or in combination with trastuzumab in women with ErbB2-positive, trastuzumab-refractory metastatic breast cancer. J Clin Oncol. 2010;28(7):1124–30.
Jiang Z, Li J, Chen J, et al. Chinese society of clinical oncology (CSCO) Breast Cancer Guidelines 2022. Transl Breast Cancer Res. 2022;3.
Bian L, Li F, Jiang Z. Thoughts on therapy strategy in the era of “after anti-HER2 TKI” in CSCO BC Guidelines 2022. Transl Breast Cancer Res. 2022;3.
Miao Y, Chen J, Deng R, et al. Clinical efficacy of pyrotinib combined with capecitabine in the second-line or above treatment for HER-2 positive advanced breast cancer and its association with cell-free DNA. J Oncol. 2022;2022:9449489.
Lin Y, Lin M, Zhang J, et al. Real-world data of pyrotinib-based therapy in metastatic HER2-positive breast cancer: promising efficacy in lapatinib-treated patients and in brain metastasis. Cancer Res Treat. 2020;52(4):1059–66.
Sun Y, Chen B, Li J, et al. Real-world analysis of the efficacy and safety of a novel irreversible HER2 tyrosine kinase inhibitor pyrotinib in patients with HER2-positive metastatic breast cancer. Cancer Manag Res. 2021;13:7165–74.
Ouyang DJ, Chen QT, Anwar M, et al. The efficacy of pyrotinib as a third- or higher-line treatment in HER2-positive metastatic breast cancer patients exposed to lapatinib compared to lapatinib-naive patients: a real-world study. Front Pharmacol. 2021;12:682568.
Li C, Bian X, Liu Z, et al. Effectiveness and safety of pyrotinib-based therapy in patients with HER2-positive metastatic breast cancer: a real-world retrospective study. Cancer Med. 2021;10(23):8352–64.
Pedrosa R, Mustafa DA, Soffietti R, et al. Breast cancer brain metastasis: molecular mechanisms and directions for treatment. Neuro Oncol. 2018;20(11):1439–49.
Yan M, Ouyang Q, Sun T, et al. Pyrotinib plus capecitabine for patients with human epidermal growth factor receptor 2-positive breast cancer and brain metastases (PERMEATE): a multicentre, single-arm, two-cohort, phase 2 trial. Lancet Oncol. 2022;23(3):353–61.
Vaklavas C, Roberts BS, Varley KE, et al. TBCRC 002: a phase II, randomized, open-label trial of preoperative letrozole with or without bevacizumab in postmenopausal women with newly diagnosed stage 2/3 hormone receptor-positive and HER2-negative breast cancer. Breast Cancer Res. 2020;22(1):22.
Fares J, Kanojia D, Rashidi A, et al. Landscape of combination therapy trials in breast cancer brain metastasis. Int J Cancer. 2020;147(7):1939–52.
Montemurro F, Delaloge S, Barrios CH, et al. Trastuzumab emtansine (T-DM1) in patients with HER2-positive metastatic breast cancer and brain metastases: exploratory final analysis of cohort 1 from KAMILLA, a single-arm phase IIIb clinical trial. Ann Oncol. 2020;31(10):1350–8.
Gao M, Fu C, Li S, et al. The efficacy and safety of pyrotinib in treating HER2-positive breast cancer patients with brain metastasis: a multicenter study. Cancer Med. 2022;11(3):735–42.
National Comprehensive Cancer Network (NCCN). Clinical practice guidelines in oncology: breast cancer. Version 2.2022—December 20, 2021.
Giordano SH, Franzoi MAB, Temin S, et al. Systemic therapy for advanced human epidermal growth factor receptor 2-positive breast cancer: ASCO guideline update. J Clin Oncol. 2022;40(23):2612–35.
Gianni L, Eiermann W, Semiglazov V, et al. Neoadjuvant and adjuvant trastuzumab in patients with HER2-positive locally advanced breast cancer (NOAH): follow-up of a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet Oncol. 2014;15(6):640–7.
Guarneri V, Griguolo G, Miglietta F, et al. Survival after neoadjuvant therapy with trastuzumab-lapatinib and chemotherapy in patients with HER2-positive early breast cancer: a meta-analysis of randomized trials. ESMO Open. 2022;7(2):100433.
Xuhong J, Qi X, Tang P, et al. Neoadjuvant pyrotinib plus trastuzumab and chemotherapy for stage I-III HER2-positive breast cancer: a phase II clinical trial. Oncologist. 2020;25(12):e1909–20.
Wu J, Liu Z, Yang H, et al. Pyrotinib in combination with trastuzumab and docetaxel as neoadjuvant treatment for HER2-positive early or locally advanced breast cancer (PHEDRA): a randomized, double-blind, multicenter, phase 3 study. Cancer Res. 2022;82(4_Supplement):PD8-08.
Liu Z, Wang C, Chen X, et al. Pathological response and predictive role of tumour-infiltrating lymphocytes in HER2-positive early breast cancer treated with neoadjuvant pyrotinib plus trastuzumab and chemotherapy (Panphila): a multicentre phase 2 trial. Eur J Cancer. 2022;165:157–68.
Yin W, Wang Y, Wu Z, et al. Neoadjuvant trastuzumab and pyrotinib for locally advanced HER2-positive breast cancer (NeoATP): primary analysis of a phase II study. Clin Cancer Res. 2022.
Zhu J, Jiao D, Wang C, et al. Neoadjuvant efficacy of three targeted therapy strategies for HER2-positive breast cancer based on the same chemotherapy regimen. Cancers (Basel). 2022;14(18).
Sonnenblick A, de Azambuja E, Agbor-Tarh D, et al. Lapatinib-related rash and breast cancer outcome in the ALTTO phase III randomized trial. J Natl Cancer Inst. 2016;108(8).
Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365(14):1273–83.
Perez EA, Romond EH, Suman VJ, et al. Trastuzumab plus adjuvant chemotherapy for human epidermal growth factor receptor 2-positive breast cancer: planned joint analysis of overall survival from NSABP B-31 and NCCTG N9831. J Clin Oncol. 2014;32(33):3744–52.
Cameron D, Piccart-Gebhart MJ, Gelber RD, et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet. 2017;389(10075):1195–205.
Piccart M, Procter M, Fumagalli D, et al. Adjuvant pertuzumab and trastuzumab in early HER2-positive breast cancer in the APHINITY trial: 6 years’ Follow-Up. J Clin Oncol. 2021;39(13):1448–57.
Martin M, Holmes FA, Ejlertsen B, et al. Neratinib after trastuzumab-based adjuvant therapy in HER2-positive breast cancer (ExteNET): 5-year analysis of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(12):1688–700.
Hongyuan Li QC. A multicenter, prospective cohort study to improve tolerability of pyrotinib in HER2-positive early stage breast cancer. https://www.chictr.org.cn/showproj.html?proj=169650.
Wang S. Phase II exploratory, randomized, open-label, multicenter clinical study of probiotics combined with or without loperamide in the prevention of pyrotinib-induced diarrhea. https://www.chictr.org.cn/showproj.html?proj=133840.
Shi Q, Xuhong J, Luo T, et al. PIK3CA mutations are associated with pathologic complete response rate to neoadjuvant pyrotinib and trastuzumab plus chemotherapy for HER2-positive breast cancer. Br J Cancer. 2023;128(1):121–9.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from Chongqing Science and Health Project (2023GDRC011), Chaoyang Cancer Research Foundation (Y-Young2022-0205), Shanghai Cancer Prevention and Anti-cancer Development Foundation (CYBER-2022-004) and Health Industry Research Project (201302016).
Author information
Authors and Affiliations
Contributions
JJ, XQ, JX, QS, and YZ conceived the study. XQ and QS drafted the manuscript. JJ and YZ revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qi, X., Shi, Q., Xuhong, J. et al. Pyrotinib-based therapeutic approaches for HER2-positive breast cancer: the time is now. Breast Cancer Res 25, 113 (2023). https://doi.org/10.1186/s13058-023-01694-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-023-01694-5