
Abstract
Background
Given the high heterogeneity among breast tumors, associations between common germline genetic variants and survival that may exist within specific subgroups could go undetected in an unstratified set of breast cancer patients.
Methods
We performed genome-wide association analyses within 15 subgroups of breast cancer patients based on prognostic factors, including hormone receptors, tumor grade, age, and type of systemic treatment. Analyses were based on 91,686 female patients of European ancestry from the Breast Cancer Association Consortium, including 7531 breast cancer-specific deaths over a median follow-up of 8.1 years. Cox regression was used to assess associations of common germline variants with 15-year and 5-year breast cancer-specific survival. We assessed the probability of these associations being true positives via the Bayesian false discovery probability (BFDP < 0.15).
Results
Evidence of associations with breast cancer-specific survival was observed in three patient subgroups, with variant rs5934618 in patients with grade 3 tumors (15-year-hazard ratio (HR) [95% confidence interval (CI)] 1.32 [1.20, 1.45], P = 1.4E−08, BFDP = 0.01, per G allele); variant rs4679741 in patients with ER-positive tumors treated with endocrine therapy (15-year-HR [95% CI] 1.18 [1.11, 1.26], P = 1.6E−07, BFDP = 0.09, per G allele); variants rs1106333 (15-year-HR [95% CI] 1.68 [1.39,2.03], P = 5.6E−08, BFDP = 0.12, per A allele) and rs78754389 (5-year-HR [95% CI] 1.79 [1.46,2.20], P = 1.7E−08, BFDP = 0.07, per A allele), in patients with ER-negative tumors treated with chemotherapy.
Conclusions
We found evidence of four loci associated with breast cancer-specific survival within three patient subgroups. There was limited evidence for the existence of associations in other patient subgroups. However, the power for many subgroups is limited due to the low number of events. Even so, our results suggest that the impact of common germline genetic variants on breast cancer-specific survival might be limited.
Similar content being viewed by others

Introduction
Inherited common genetic variation is likely to influence survival in breast cancer patients [1]. Results from pre-clinical experiments have shown different metastatic behaviors in mice with different genetic backgrounds [2,3,4,5,6,7]. In addition, familial studies of breast cancer patients have shown that women with a first-degree relative with a poor prognosis breast cancer have a worse prognosis compared to women with a first-degree relative with a good prognosis cancer [8]. Moreover, genome-wide and candidate gene association studies have discovered common genetic variants associated with specific subtypes of breast cancer based on the expression of the estrogen receptor (ER) [9,10,11], progesterone receptor (PR), and the amplification of the human epidermal growth factor receptor 2 (HER2) [12, 13], which are known breast cancer prognostic factors [14, 15]. Finally, a number of studies have suggested that specific common germline genetic variants affect breast cancer prognosis both overall and within subgroups of patients [16,17,18,19,20,21,22,23,24].
Despite the supporting evidence, it remains challenging to identify common germline variants associated with breast cancer-specific survival. This may partially be explained by the good prognosis of breast cancer patients, which leads to underpowered analyses. Even large studies based on worldwide consortia cannot reach the number of breast cancer deaths necessary to detect small to moderate associations at a genome-wide significant level [19, 20, 25]. However, breast cancer is a heterogeneous disease, and it is possible that stronger associations between common germline variants and breast cancer-specific survival are present in certain patient subgroups, but cannot be detected in breast cancer overall. Previous studies provide modest evidence supporting this hypothesis [16, 19, 20].
The aim of our study was to evaluate the evidence for associations of inherited common genetic variants with breast cancer-specific survival within more homogeneous subgroups of breast cancer patients, defined by prognostic factors representative of tumor biology and/or by the type of systemic treatment. To this end, we performed genome-wide association analyses within clinically relevant, defined subgroups of patients based on hormone receptors, tumor grade, age at diagnosis, and type of systemic treatment [26, 27]. We also explored the subgroup-specific associations identified by previous studies [16, 19, 23, 28, 29], to confirm or refute those results.
Materials and methods
Study sample
We selected female breast cancer patients of European ancestry from studies participating in the Breast Cancer Association Consortium (BCAC). We included patients with available information about vital status and number of years from diagnosis to last follow-up who were diagnosed with a primary invasive breast cancer of any stage and were at least 18 years old at diagnosis. The final study sample consisted of 91,686 breast cancer patients from 70 BCAC studies. A description of the included studies is given in Additional file 1: Supplementary Table S1.
Information about histopathology, survival, and treatment was collected by individual studies and pooled and harmonized at the Netherlands Cancer Institute before incorporation into the BCAC database at the University of Cambridge (version 12, July 2019). All studies were approved by the relevant ethics committees and informed consent was obtained from all patients.
Patient subgroups
The subgroups of interest were defined based on age at diagnosis, estrogen receptor (ER) status, progesterone receptor (PR) status, human epidermal growth factor receptor 2 (HER2) status, tumor grade, and the use and type of systemic treatment, as available in the BCAC database. For age at diagnosis and tumor grade, we focused on subgroups characterized by worse prognosis. We thus defined 15 subgroups: (a) patients younger than age 40 years at diagnosis; (b) patients with grade 3 tumors; (c) patients with ER-positive (ER+) tumors, who received endocrine therapy (any kind); (d) patients diagnosed with ER-negative (ER−) tumors, who received chemotherapy (any kind); (e) patients with tumors that were hormone receptor (HR) positive (ER+ or PR+) and HER2-negative (HER2−); (f) patients with HR-positive (HR+), HER2− tumors, who received chemotherapy (any kind); (g) patients with HR+, HER2− tumors, who did not receive chemotherapy; (h) patients with HR+, HER2-positive (HER2+) tumors; (i) patients with HR-negative (HR−), HER2+ tumors, (j) patients with HR−, HER2− tumors; (k) patients who received Tamoxifen; (l) patients who received an aromatase inhibitor; (m) patients who received a Cyclophosphamide Methotrexate Fluorouracil (CMF)-like chemotherapy regimen; (n) patients who received taxanes; (o) patients who received anthracyclines.
The rationale and references to the literature supporting the choice of each subgroup for inclusion in a genome-wide association study on survival are given in Additional file 1: Supplementary Table S2. We did not include the subgroup of HER2+ tumors treated with Trastuzumab because of a relatively small number of patients and low event rate, leading to analyses that are more underpowered than those presented.
Patients with metastatic breast tumors at diagnosis (1.1 % of all included patients) were excluded from the subgroup analyses whose definition was based on the use and type of systemic therapy as generally they are treated with palliative intent [15, 30, 31].
In addition to the subgroup analyses, we also performed a genome-wide analysis of 15-year breast cancer-specific survival in all breast cancer patients. We performed this analysis to evaluate whether associations between common germline variants and breast cancer-specific survival in subgroups could be detected in the full dataset of patients. Genome-wide analyses for survival (unstratified by subtype) were previously performed [20] based on 12 GWAS datasets, but these included fewer patients from iCOGS and OncoArray (n = 84,757), with shorter follow-up, than were available in the current dataset. We focused our analyses on the iCOGS and OncoArray datasets, because the remaining 10 GWAS datasets used in the previous study did not include information about tumor characteristics, beyond ER status, or treatment, which were crucial for the subgroup analyses.
Due to the presence of missing values in the variables used to define the subgroups, not all patients could be classified by each subgroup. The number of patients included in each subgroup, together with the number of breast cancer-specific deaths, patient/tumor characteristics, treatment, and follow-up information, are shown in Additional file 1: Supplementary Tables S3-S4, and Additional file 2: Supplementary Table S5.
Imputation of missing values in clinical and pathological variables
For secondary adjusted analyses, we imputed missing values in the clinical and pathological variables using the Multiple imputation by Chained Equations (MICE) R package (v. 3.2.0), as described in Additional file 2: Supplementary Methods. A list of imputed variables and corresponding percentages of missing values and imputation methods is provided in Additional file 2: Supplementary Table S6.
Genotyping and imputation of genetic variants, ancestry analysis, and quality controls
Methods related to genotyping and genotype imputation have been described previously [17, 18, 20]. In brief, patients were genotyped with two different arrays: iCOGS and OncoArray [17, 32]. Only samples that were inferred to have European ancestry, based on genotype data, were included in the analyses. Non-genotyped variants were initially imputed based on the 1000 Genomes Project Phase 3 (October 2014) release as reference panel. More recently, non-genotyped single-nucleotide variants (SNVs) were re-imputed using a reference panel from the Haplotype Reference Consortium (HRC) [33] in order to improve imputation quality, especially for rarer variants. Analyses were performed on genotyped variants or imputed variants with a minor allele frequency (MAF) > 0.01. Imputed variants were included in the analyses if they had imputation r2 > 0.7. Approximately 10 million variants were analyzed.
Statistical analyses
The outcome in the analyses was breast cancer-specific survival (time to death due to breast cancer). Hazard ratios (HR) and 95% confidence intervals (CI) were estimated using delayed entry Cox regression models, where the time at risk was considered as starting from the time of study entry if the study entry was after diagnosis (22.9% within 1 year after diagnosis, and 27.3% more than 1 year after diagnosis) and from diagnosis if the time of study entry was missing (24.5%), at diagnosis (16.9%) or before diagnosis (8.4%). The time-to-event was right censored at the time of last follow-up, or at 15 years after diagnosis, whichever came first. Patients who died of unknown cause or causes other than breast cancer were censored at the time of death if death occurred before 15 years from diagnosis or at 15 years otherwise.
With reference to the results of Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) [34], we additionally performed analyses within the subgroups whose definition was based on the use and type of systemic therapy, where we restricted the maximum follow-up time to 5 years after diagnosis (Additional file 2: Supplementary Table S5). The goal of those analyses was to investigate the potential short-term effects of germline variants on patients who received specific types of systemic treatment, since the effect might not be constant over time and treatment plans tend to focus on the first 5 years after diagnosis [15, 31].
Cox regression analysis was performed within each subgroup of interest, separately, and was stratified by country. All the analyses were performed separately by genotyping platform (iCOGS vs OncoArray), and the results were combined via a fixed-effects meta-analysis. The standard errors of the HR estimates were re-computed based on the likelihood ratio test statistic, as done previously [20] (Figs. 1 and 2). For variants that satisfied the inclusion criteria (MAF>0.01 and r2 > 0.7) on only one genotyping platform, we included the result for that specific platform (Tables 1 and 2). However, for variants with an association P < 5E−08, we also computed HR and 95% confidence interval in the other genotyping platform to verify that the direction of the association was the same (Additional file 2: Supplementary Table S7).

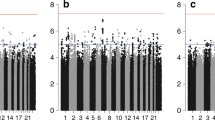
Genome-wide analyses of 15-year breast cancer-specific survival. The Manhattan plots show the results of the unadjusted analyses (stratified by country). Results are represented as −log10 of the p value from Cox regression models (y-axis). The x-axis shows the chromosome number, where chromosome 23 represents the X chromosome. The red line represents the genome-wide significant threshold 5E−08, the blue line corresponds to the threshold 1E−05. Breast cancer patients included in the analysis in panel: a younger than age 40 years at diagnosis; b diagnosed with a grade 3 tumor; c diagnosed with an ER+ tumor and treated with (any) endocrine therapy; d diagnosed with a ER− tumor and treated with (any) chemotherapy; e diagnosed with an ER+ or PR+, and HER2− tumor; f diagnosed with an ER+ or PR+, and HER2− tumor treated with (any) chemotherapy; g diagnosed with an ER+ or PR+, and HER2− tumor not treated with chemotherapy; h diagnosed with an ER+ or PR+, and HER2+ tumor; i diagnosed with an ER− and PR− and HER2+ tumor; j diagnosed with an ER− and PR− and HER2− tumor; k treated with tamoxifen; l treated with aromatase inhibitor; m treated with CMF-like chemotherapy; n treated with taxanes; o treated with anthracyclines; p all

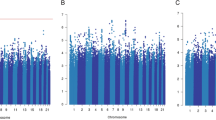
Genome-wide analyses of 5-year breast cancer-specific survival. The Manhattan plots show the results of the unadjusted analyses (stratified by country). Results are represented as −log10 of the p value from Cox regression models (y-axis). The x-axis shows the chromosome number, where chromosome 23 represents the X chromosome. The red line represents the genome-wide significant threshold 5E−08, the blue line corresponds to the threshold 1E−05. Breast cancer patients included in the analysis in panel: a diagnosed with a ER+ tumor and treated with (any) endocrine therapy; b diagnosed with a ER− tumor and treated with (any) chemotherapy; c diagnosed with a ER+ or PR+, and HER2− tumor treated with (any) chemotherapy; d diagnosed with a ER+ or PR+, and HER2− tumor not treated with chemotherapy; e treated with tamoxifen; f treated with aromatase inhibitor; g treated with CMF-like chemotherapy; h treated with taxanes; i treated with anthracyclines
Inflation of the likelihood ratio test statistics was estimated, within each subgroup, by dividing the median of the observed test statistics values by the median of a \( {\chi}_1^2 \) distribution (Additional file 2: Supplementary Figures S1 and S2). To assess the noteworthiness of the observed associations, we made use of the Bayesian false discovery probability (BFDP) measure [35]. To compute BFDPs, we set the prior probability of true association to 10−4 [36, 37], as done previously [20], and chose the prior distribution of the log hazard ratio of interest (effect size of a variant) to be a Normal distribution with mean 0 and standard error equal to 0.2 [36]. We describe associations with BFDP < 0.15 as “noteworthy” [20]. For each noteworthy result at a prior of 10−4, we also provided BFDPs under two, more restrictive, prior probabilities of true association (10−5 and 10−6; Additional file 3: Supplementary Tables S8-S9) [36, 37]. In addition, we estimated the power to detect genetic variant associated with 15-year and 5-year breast cancer-specific survival by subgroup (Additional file 3: Supplementary Table S10 and S11) as described in Additional file 2: Supplementary Methods.
For each genome-wide significant (P < 5E−08) [38] and/or noteworthy (BFDP < 0.15) association observed in the primary unadjusted subgroup analyses, we performed secondary analyses adjusted for age at diagnosis, tumor characteristics, and type of systemic treatment not used in the definition of the specific subgroup in which the association was detected (Tables 1 and 2). Secondary adjusted analyses were performed to account for residual heterogeneity; we used imputed covariates in order to keep the same sample size.
For each genome-wide significant or noteworthy association in the primary unadjusted analyses, we looked at the functional annotation of the surrounding genomic area, using the Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA GWAS) tool [39] (Additional file 2: Supplementary Figures S5 and S6). We also tested whether the expression of the nearest genes correlated with distant metastasis-free survival in breast cancer patients using KMplotter [40, 41].
PancanQTL [42] was used to identify cis-expression quantitative trait locus (eQTLs), trans-eQTLs, and survival eQTLs in breast cancer to see whether the genome-wide significant or noteworthy genetic variants from the primary analyses could be linked with the expression levels of genes affecting survival. In addition, for all the genome-wide significant and/or noteworthy associations detected in the primary analyses, we searched the GWAS catalog [43] to see whether there was already evidence of those being associated with breast cancer or other traits.
Results
After a median follow-up of 8.1 years, there were a total of 7531 breast cancer deaths among 91686 breast cancer patients (Additional file 1: Supplementary Tables S3-S4 and Additional file 2: Supplementary Table S5).
In the 15-year breast cancer-specific survival analyses, power for detecting genome-wide significant associations was < 0.45 for effect sizes (HRs) < 1.20 in all subgroups investigated and for all minor allele frequencies. The power was highest in the subgroups of grade 3 tumors, ER+ tumors treated with endocrine therapy, HR+ and HER2− tumors, and patients who received tamoxifen (Additional file 3: Supplementary Table S10). In the 5-year breast cancer-specific survival analyses, power was highest in the subgroups of patients with an ER− tumor who received chemotherapy, patients who received tamoxifen, and patients who received anthracyclines (Additional file 3: Supplementary Table S11).
Genome-wide significant and/or noteworthy associations with 15-year or 5-year breast cancer-specific survival were observed in the unadjusted analyses based on all patients (Additional file 2: Supplementary Table S7) and analyses of eight out of the 15 subgroups investigated (Tables 1 and 2; Figs. 1 and 2). The genomic inflation factor of the unadjusted genome-wide analyses varied from 0.981 to 1.028 (Additional file 2: Supplementary Figures S1- S4); it is therefore unlikely that the association results were affected by cryptic population substructure.
Two genome-wide significant associations were observed in the unstratified analysis based on patients genotyped using OncoArray (Additional file 2: Supplementary Table S7), namely variants rs57714252 (P = 4.7E−08) and rs4129285 (P = 4.9E−08), both situated in an intergenic region of chromosome 4 (Additional file 2: Supplementary Figure S5). These results were only based on the OncoArray data, since on iCOGS the variants did not satisfy the inclusion criteria for genotypes (iCOGS imputation r2 = 0.62). The corresponding estimates in the iCOGS data were in the opposite direction compared to the OncoArray estimates, and the results from the meta-analysis were not genome-wide significant and showed a large BFDP (Additional file 2: Supplementary Table S7).
Genome-wide significant associations were observed in the analysis restricted to patients diagnosed with a grade 3 tumor, with five correlated variants (Tables 1 and 2) located on chromosome X (Additional file 2: Supplementary Figure S5) in intron 1 of TBL1X. For the most significant variant, rs5934618, the alternative G allele was associated with increased risk of breast cancer death in unadjusted analyses (meta-analysis hazard ratio (HR) [95% confidence interval (CI)] 1.32 [1.20, 1.45], P = 1.4E−08, BFDP = 0.01; Table 2). The meta-analysis result remained substantially unchanged after adjusting for age at diagnosis, additional tumor characteristics, and treatment with (neo)adjuvant chemotherapy (HR [95% CI] 1.31 [1.18, 1.44], P = 7.9E−08, BFDP = 0.05; Table 2). The variant was not associated with the outcome in lower-grade tumors or in all patients combined (heterogeneity by grade P = 1.5E−03; Additional file 2: Supplementary Figure S7). All the five variants overlap chromatin features H3K4me3 and H3K27ac (associated with active transcription start sites) in multiple mammary cell types from normal breast tissue (Additional file 2: Supplementary Figure S8). Furthermore, there was evidence of TBL1X expression being associated with distant metastasis-free survival (HR [95%CI] for high vs low expression 1.71 [1.20,2.44], P = 2.7E−03) specifically in grade 3 patients, but not in patients with lower-grade disease (Additional file 2: Supplementary Figure S9). However, there was no evidence of association with TBL1X expression in normal breast tissue with any of the variants identified in our genome-wide analyses (Additional file 2: Supplementary Figure S10).
In the same subgroup of grade 3 tumors, we observed a noteworthy, non-genome-wide significant association with variant rs66871326, located on chromosome 2 in an intron of C2orf80. For variant rs66871326, the alternative A allele was associated with decreased risk of breast cancer death (HR [95% CI] 0.85 [0.80,0.90], P = 2.1E−07, BFDP = 0.11). The corresponding BFDP increased to 0.49 after adjusting for age at diagnosis, additional tumor characteristics, and treatment with (neo)adjuvant chemotherapy (Table 1).
We identified six variants on chromosome 15 with genome-wide significant associations within the subgroup of patients diagnosed with an ER+ or PR+, HER2− tumor (Table 1). We identified two independent variants, namely rs8030394 and rs112813972, both situated in an intronic region of THSD4 (Additional file 2: Supplementary Figure S5). For the most significant variant, rs8030394, the T allele was associated with increased risk of breast cancer death (HR [95% CI]: 2.47 [1.81, 3.37], P = 1.1E−08, BFDP = 0.42). For the second variant, rs112813972, the C allele was associated with decreased risk of death (HR [95% CI] 0.40 [0.28,0.55], P = 4.0E−08, BFDP = 0.70). These associations were not genome-wide significant after adjusting for age at diagnosis, additional tumor characteristics, and treatment with (neo)adjuvant chemotherapy, and the corresponding BFDPs increased to 0.72 and 0.90, respectively (Table 1).
We observed genome-wide significant associations from the 15-year breast cancer-specific analyses in the subgroups of patients with an ER+ or PR+ and HER2− tumor who did and did not receive chemotherapy. Three correlated variants on chromosome 2 were identified within the subgroup of patients who received chemotherapy. The most significant variant, rs62192052, is located in an intronic region of DNER (Additional file 2: Supplementary Figure S5) and was associated with decreased risk of death (HR [95% CI] 0.15 [0.08, 0.28], P = 2.6E−09, per T allele; Table 1). Although the result remained genome-wide significant after adjusting for age at diagnosis and additional tumor characteristics, the BFDP from both the unadjusted and adjusted analysis was ≥ 0.99 (Table 1; Supplementary Table S8), indicating that this association is almost certainly a false positive. Variant rs56248395, located on chromosome 11 in an intron of NAV2 (Additional file 2: Supplementary Figure S5), was associated with breast cancer death in the subgroup of patients with an ER+ or PR+ and HER2− tumor who did not receive chemotherapy (HR [95% CI] 2.33 [1.72,3.15], P = 4.8E−08, per T allele; Table 1). This result had BFDP ≥ 0.59 and was only based on the OncoArray data, since on iCOGS the variants did not satisfy the inclusion criteria for genotypes (iCOGS imputation r2 = 0.66; Additional file 2: Supplementary Table S7). The corresponding estimates in the iCOGS data (HR [95% CI] 1.07 [0.80,1.41], P = 6.6E−01; Additional file 2: Supplementary Table S7) and from the meta-analysis (HR [95% CI] 1.53 [1.25,1.89], P = 5.2E−05; Table 2) were not genome-wide significant and not noteworthy (meta-analysis BFDP≥0.97; Table 2; Supplementary Table S9).
We observed three additional single SNP noteworthy associations from the 15-year breast cancer-specific survival analyses. The intergenic variant rs4679741 on chromosome 3 was associated with breast cancer death in the subgroup of patients with an ER+ tumor treated with endocrine therapy (HR [95% CI] 1.18 [1.11, 1.26], P = 1.6E−07, BFDP = 0.09, per G allele). This result became genome-wide significant after adjusting for age at diagnosis, tumor characteristics, and treatment with chemotherapy (HR [95% CI] 1.20 [1.13, 1.28], P = 1.1E−08, BFDP = 0.01). The BFDP of this association remained < 0.15 when considering 10−5 as prior probability of true association (Additional file 3: Supplementary Table S8). PanCanQTL did not show any cis-eQTLs, trans-eQTLs nor survival eQTLs for this variant. Variant rs1106333 on chromosome 3, whose nearest gene is GRIP2, was associated with risk of dying of breast cancer in the subgroup of patients with an ER− tumor who received chemotherapy (HR [95% CI] 1.68 [1.39,2.03], P = 5.6E−08, BFDP = 0.12 per A allele). This result also became genome-wide significant after adjusting for additional prognostic factors (HR [95% CI] 1.70 [1.41,2.05], P = 4.4E−08, BFDP = 0.11). PanCanGTL revealed the presence of a cis-eQTL linking variant rs1106333 with GRIP2 expression in prostate adenocarcinoma but not in breast cancer. There was no evidence of association of GRIP2 expression levels with distant metastasis-free survival within ER− breast cancer patients treated with chemotherapy based on KMPlotter data (Additional file 2: Supplementary Figure S11). The last association of interest was observed in the subgroup of patients who received anthracyclines with intergenic variant rs34072391 on chromosome 7, but was not noteworthy after adjustment for additional prognostic factors (Table 1).
In the 5-year survival analyses focused on the treatment subgroups, we observed two genome-wide significant associations within the subgroup of patients diagnosed with an ER− tumor who received chemotherapy. The most significant variant was rs78754389, located on chromosome 4 in an intronic region of gene ARAP2 (Additional file 2: Supplementary Figure S5; HR [95% CI] 1.79 [1.46,2.20], P = 1.7E−08, BFDP = 0.07 per A allele). This result remained both genome-wide significant and noteworthy after adjusting for age at diagnosis and additional tumor characteristics (HR [95% CI] 1.67 [1.39,2.00], P = 4.1E−08, BFDP = 0.09; Table 1). However, PanCanQTL did not show any cis-eQTLs, trans-eQTLs nor survival eQTLs for rs78754389 and there was no evidence of association of ARAP2 expression levels with distant metastasis-free survival within ER− breast cancer patients treated with chemotherapy based on KMPlotter data (Additional file 2: Supplementary Figure S12). The second genome-wide significant variant was rs117685664, located on chromosome 8 (HR [95% CI] 0.26 [0.16,0.42], P = 4.6E−08, per T allele). This association was not genome-wide significant after accounting for the age at diagnosis and additional tumor characteristics (Table 1), and the corresponding BFDPs from unadjusted and adjusted analysis were 1.00 and 0.99, respectively, indicating a false positive finding.
We also observed one additional noteworthy but not genome-wide significant association in the 5-year breast cancer-specific survival analyses in the subgroup of patients who received Tamoxifen with variant rs72775397, situated in the 3′ untranslated region of MCTP1 (HR [95% CI] 1.36 [1.21,1.53], P = 1.8E−07, BFDP = 0.11, per C allele). The association was attenuated after adjustment for additional prognostic factors (HR [95% CI] 1.11 [1.04,1.20], P = 3.8E−03, BFDP = 1.00).
We did not identify any genome-wide significant or noteworthy association in any of the remaining seven subgroups investigated (Figs. 1 and 2). In addition, none of the above reported associations have a BFDP < 0.15 when considering 10−6 as prior probability of true association (Additional file 3: Supplementary Tables S8). Moreover, none of the subgroup-specific genome-wide significant associations detected by previous studies using a smaller version (both in terms of number of cases and of length of follow-up) of the iCOGS and/or OncoArray BCAC datasets were replicated at P < 0.001 (Additional file 3: Supplementary Table S12).
Discussion
We investigated the association of over 10 million common germline genetic variants with breast cancer-specific survival within 15 patient subgroups based on prognostic factors representative of tumor biology or related to the type of systemic treatment. Our hypothesis was that focusing on more homogeneous subgroups of breast cancer patients might reveal otherwise undetected associations. Besides type of systemic treatment, the definition of the subgroups was based on current clinically used biological characteristics for tumor subtyping and treatment decisions: patient’s age, tumor histological grade, ER, PR, and HER2 status ( [31]; Supplementary table S2). We did not have gene expression or copy number aberration data available to classify tumors on the basis of specific biological processes [44, 45]; however, relevant survival differences have been reported among the four subtypes based on ER, PR, and HER2 status [27, 46, 47].
A concern about performing GWAS on several subgroups of patients is the increased proportion of false discoveries, also known as type I errors. For this reason and to overcome additional limitations of the association p values [35,36,37], we made use of the BFDP approach and only considered as robust candidates those associations with BFDP < 0.15 at a prior probability of 10−4.
We found evidence of four loci potentially associated with breast cancer survival: one in the subgroup of patients diagnosed with a grade 3 tumor, one in the subgroup of patients with an ER+ tumor and treated with endocrine therapy, and two in the subgroup of patients with an ER− tumor and treated with chemotherapy.
The most significant variant identified in the subgroup of grade 3 tumors, rs5934618, is situated in intron 1 of TBL1X, a gene which encodes the Transducin (beta)-like 1X-linked protein. Both TBL1X and the closely related gene TBLR1 have been implicated in the activation of the Wnt/beta-catenin signaling pathway, which has been reported to be overactivated in the progression and proliferation of several tumors, including breast tumors, where it has been linked with reduced overall survival [48,49,50]. Rs5934618 and the other four correlated variants identified in our genome-wide analysis overlap with chromatin features H3K4me3 and H3K27ac in normal breast; these histone marks are generally characteristics of gene promoters and/or enhancers and might indicate that one or more of these variants act through modulating expression of TBL1X. There was no direct evidence that any of these variants are expression single-nucleotide polymorphisms (eSNPs) for TBL1X, but this might reflect the tissues examined or that the variants only regulate the gene in a specific context.
The remaining three variants potentially associated with breast cancer survival were as follows: rs4679741, identified in the subgroup of patients diagnosed with an ER+ tumor and treated with endocrine therapy; rs1106333 and rs78754389, identified in the subgroup of patients diagnosed with an ER− tumor and treated with chemotherapy. For variant rs4679741, it is unclear which the potential target genes might be, while there was no evidence linking the other two variants to the expression of the closest genes in breast cancer nor evidence of association between those genes and survival within the specific subgroups of breast cancer patients. Nevertheless, there are several mechanisms through which the four identified variants could affect survival. For example, they could act through regulation of an unannotated long noncoding RNA [51, 52] or microRNA [53, 54]. Further functional studies including epigenetic mechanisms are needed in order to gain more insights about the detected associations and to ascertain the potential underlying biological mechanisms.
We did not find strong evidence of germline variants associated with breast cancer-specific survival in any of the other subgroups of patients investigated. In addition, we did not replicate any of the subgroup-specific associations identified by previous studies. One of these associations, with variant rs4458204, was previously detected in the subgroup of patients with an ER− tumor who received chemotherapy [16]. The estimated HR (95% CI) was 1.81 (1.49–2.19) with association P = 1.9E−09. In our analysis of the same subgroup, we obtained a much lower HR estimate and the association was no longer statistically significant (HR (95% CI) 1.14 (0.99, 1.32), P = 6.0E−02), suggesting that the previous result was a false positive. Even though there is some overlap in terms of patients between the previous study and our current study, the latter is based on a substantially larger number of breast cancer patients and it includes more complete follow-up data.
A major strength of our study is the sample size, which was the largest to date and provided reasonable power to detect associations with breast cancer-specific survival within specific subgroups of patients. On the other hand, our study is subject to several limitations that are intrinsic to large consortium studies: these include variation in study design, time periods of diagnosis, and duration of follow-up, all of which can contribute to within subgroup heterogeneity. Some broad treatment-related subgroups, namely ER+ treated with any endocrine therapy and ER− treated with any chemotherapy, may include different treatments due to the wide period of diagnosis included in our study. On the other hand, the majority of patients were diagnosed between 2000 and 2009 (69.9% and 64.5% for ER+ treated with any endocrine therapy and ER− treated with any chemotherapy, respectively). If any impact on the results, the variation in treatment over time might have hampered the detection of associations between variants and survival in these subgroups. Several studies did not report the cause of death for all patients. Out of 14,606 deaths observed within the first 15 years after diagnosis, 7531 (51.6%) were due to breast cancer. Of the remaining 7075 deaths, 4905 (33.6%) were due to causes other than breast cancer, and for 2170 deaths (14.8%) it was unknown whether they were due to breast cancer or to other causes. This will have led to a loss of power, given that most of the deaths of unknown cause are likely to have been due to breast cancer. A related weakness of the study is its dependence on accuracy of cause of death certification and on coding practices of underlying cause of death in different countries. However, despite potential inaccuracies in cause of death, we considered it more valid to focus on deaths reported as due to breast cancer than by considering all deaths together, which would include those due to other causes. An additional limitation of the study is that in most subgroups we had very limited power to detect highly significant associations, particularly for small to moderate effect sizes (HRs 1.05–1.30), even for variants of relatively high minor allele frequency (MAF = 0.20). Therefore, we may have missed variants with low to moderate associations with survival.
Conclusions
In conclusion, we found evidence of four loci associated with breast cancer-specific survival within specific patient subgroups. The variants identified appear to be independent of known additional prognostic factors, as shown in the results of the adjusted analyses based on imputed clinic-pathological variables, and could, after proper validation, improve prognostic estimates and potentially help in better stratifying patients in treatment subgroups. However, the power for many subgroups is limited due to the low number of events. Even so, given the lack of evidence of strong associations in many of the patient subgroups investigated, and the fact that previously reported variants were not confirmed, our results suggest that the impact of common germline genetic variant on breast cancer-specific survival might be limited.
Availability of data and materials
All estimates from the genome-wide survival analyses are available through the BCAC website: http://bcac.ccge.medschl.cam.ac.uk. The datasets analyzed during the current study are not publicly available due to protection of participant privacy and confidentiality, and ownership of the contributing institutions, but may be made available in an anonymized form via the corresponding author on reasonable request and after approval of the involved institutions. To receive access to the data, a concept form must be submitted, which will then be reviewed by the BCAC Data Access Coordination Committee (DACC); see http://bcac.ccge.medschl.cam.ac.uk/bcacdata/.
Abbreviations
- BCAC:
-
Breast Cancer Association Consortium
- BFDP:
-
Bayesian false discovery probability
- CI:
-
Confidence interval
- CMF:
-
Cyclophosphamide Methotrexate Fluorouracil
- EBCTCG:
-
Early Breast Cancer Trialists’ Collaborative Group
- ER:
-
Estrogen receptor
- eQTL:
-
Expression quantitative trait locus
- eSNP:
-
Expression single-nucleotide polymorphism
- FUMA:
-
Functional mapping and annotation
- GWAS:
-
Genome-wide association study
- HER2:
-
Human epidermal growth factor receptor 2
- HR:
-
Hazard ratio or Hormone receptor
- HRC:
-
Haplotype Reference Consortium
- MAF:
-
Minor allele frequency
- MICE:
-
Multiple imputation by chained equations
- PR:
-
Progesterone receptor
- SNV:
-
Single-nucleotide variant
References
Ribelles N, Santonja A, Pajares B, Llácer C, Alba E. The seed and soil hypothesis revisited: current state of knowledge of inherited genes on prognosis in breast cancer. Cancer Treat Rev. 2014;40(2):293–9. https://doi.org/10.1016/j.ctrv.2013.09.010.
Hunter K. Host genetics influence tumour metastasis. Nat Rev Cancer. 2006;6(2):141–6. https://doi.org/10.1038/nrc1803.
Crawford NPS, Alsarraj J, Lukes L, Walker RC, Officewala JS, Yang HH, Lee MP, Ozato K, Hunter KW <em>Bromodomain 4</em> activation predicts breast cancer survival. Proceedings of the National Academy of Sciences. 2008;105(17):6380-6385, doi: https://doi.org/10.1073/pnas.0710331105.
Crawford NPS, Qian X, Ziogas A, Papageorge AG, Boersma BJ, Walker RC, et al. Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. Plos Genet. 2007;3(11):e214. https://doi.org/10.1371/journal.pgen.0030214.
Lukes L, Crawford NPS, Walker R, Hunter KW. The origins of breast cancer prognostic gene expression profiles. Cancer Res. 2009;69(1):310–8. https://doi.org/10.1158/0008-5472.CAN-08-3520.
Patel SJ, Molinolo AA, Gutkind S, Crawford NPS. Germline genetic variation modulates tumor progression and metastasis in a mouse model of neuroendocrine prostate carcinoma. Plos One. 2013;8(4):e61848. https://doi.org/10.1371/journal.pone.0061848.
Winter JM, Gildea DE, Andreas JP, Gatti DM, Williams KA, Lee M, et al. Mapping complex traits in a diversity outbred f1 mouse population identifies germline modifiers of metastasis in human prostate cancer. Cell Syst. 2017;4(1):31–45.e6.
Hartman M, Lindström L, Dickman PW, Adami H-O, Hall P, Czene K. Is breast cancer prognosis inherited? Breast Cancer Res. 2007;9(3):R39. https://doi.org/10.1186/bcr1737.
Stacey SN, Manolescu A, Sulem P, Thorlacius S, Gudjonsson SA, Jonsson GF, et al. Common variants on chromosome 5p12 confer susceptibility to estrogen receptor–positive breast cancer. Nat Genet. 2008;40(6):703–6. https://doi.org/10.1038/ng.131.
Couch FJ, Kuchenbaecker KB, Michailidou K, Mendoza-Fandino GA, Nord S, Lilyquist J, et al. Identification of four novel susceptibility loci for oestrogen receptor negative breast cancer. Nat Commun. 2016;7(1):11375. https://doi.org/10.1038/ncomms11375.
Milne RL, Kuchenbaecker KB, Michailidou K, Beesley J, Kar S, Lindström S, et al. Identification of ten variants associated with risk of estrogen-receptor-negative breast cancer. Nat Genet. 2017;49(12):1767–78. https://doi.org/10.1038/ng.3785.
Stevens KN, Fredericksen Z, Vachon CM, Wang X, Margolin S, Lindblom A, et al. 19p13.1 is a triple-negative–specific breast cancer susceptibility locus. Cancer Res. 2012;72(7):1795–803. https://doi.org/10.1158/0008-5472.CAN-11-3364.
Zhang H, Ahearn TU, Lecarpentier J, Barnes D, Beesley J, Qi G, et al. Genome-wide association study identifies 32 novel breast cancer susceptibility loci from overall and subtype-specific analyses. Nat Genet. 2020;52(6):572–81. https://doi.org/10.1038/s41588-020-0609-2.
Phipps AI, Li CI. Breast Cancer Biology and Clinical Characteristics. In: Li C, editor. Breast Cancer Epidemiology. New York, NY: Springer New York; 2010. p. 21–46. https://doi.org/10.1007/978-1-4419-0685-4_2.
Kurian AW, Carlson RW. Principles of Breast Cancer Therapy. In: Li C, editor. Breast Cancer Epidemiology. New York: Springer New York; 2010. p. 371–88. https://doi.org/10.1007/978-1-4419-0685-4_17.
Li J, Lindström LS, Foo JN, Rafiq S, Schmidt MK, Pharoah PDP, et al. 2q36.3 is associated with prognosis for oestrogen receptor-negative breast cancer patients treated with chemotherapy. Nat Commun. 2014;5(1):4051.
Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, et al. The OncoArray Consortium: A Network for Understanding the Genetic Architecture of Common Cancers. Cancer Epidemiol Biomarkers Prev. 2017;26(1):126–35. https://doi.org/10.1158/1055-9965.EPI-16-0106.
Guo Q, Schmidt MK, Kraft P, Canisius S, Chen C, Khan S, et al. Identification of Novel Genetic Markers of Breast Cancer Survival. JNCI. 2015;107(5): djv081. https://doi.org/10.1093/jnci/djv081.
Kadalayil L, Khan S, Nevanlinna H, Fasching PA, Couch FJ, Hopper JL, et al. Germline variation in ADAMTSL1 is associated with prognosis following breast cancer treatment in young women. Nat Commun. 2017;8(1):1632. https://doi.org/10.1038/s41467-017-01775-y.
Escala-Garcia M, Guo Q, Dörk T, Canisius S, Keeman R, Dennis J, et al. Genome-wide association study of germline variants and breast cancer-specific mortality. Br J Cancer. 2019;120(6):647–57. https://doi.org/10.1038/s41416-019-0393-x.
Escala-Garcia M, Abraham J, Andrulis IL, Anton-Culver H, Arndt V, Ashworth A, et al. A network analysis to identify mediators of germline-driven differences in breast cancer prognosis. Nat Commun. 2020;11(1):312. https://doi.org/10.1038/s41467-019-14100-6.
Rafiq S, Khan S, Tapper W, Collins A, Upstill-Goddard R, Gerty S, et al. A genome wide meta-analysis study for identification of common variation associated with breast cancer prognosis. Plos One. 2014;9(12):e101488. https://doi.org/10.1371/journal.pone.0101488.
Kiyotani K, Mushiroda T, Tsunoda T, Morizono T, Hosono N, Kubo M, et al. A genome-wide association study identifies locus at 10q22 associated with clinical outcomes of adjuvant tamoxifen therapy for breast cancer patients in Japanese. Hum Mol Genet. 2011;21(7):1665–72. https://doi.org/10.1093/hmg/ddr597.
Stevens KN, Vachon CM, Lee AM, Slager S, Lesnick T, Olswold C, et al. Common breast cancer susceptibility loci are associated with triple-negative breast cancer. Cancer Res. 2011;71(19):6240–9. https://doi.org/10.1158/0008-5472.CAN-11-1266.
Pirie A, Guo Q, Kraft P, Canisius S, Eccles DM, Rahman N, et al. Common germline polymorphisms associated with breast cancer-specific survival. Breast Cancer Res. 2015;17(1):58. https://doi.org/10.1186/s13058-015-0570-7.
Dackus GM, ter Hoeve ND, Opdam M, Vreuls W, Varga Z, Koop E, et al. Long-term prognosis of young breast cancer patients (≤ 40 years) who did not receive adjuvant systemic treatment: protocol for the PARADIGM initiative cohort study. BMJ Open. 2017;7(11):e017842. https://doi.org/10.1136/bmjopen-2017-017842.
Blows FM, Driver KE, Schmidt MK, Broeks A, van Leeuwen FE, Wesseling J, et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: a collaborative analysis of data for 10,159 cases from 12 studies. Plos Med. 2010;7(5):e1000279. https://doi.org/10.1371/journal.pmed.1000279.
Fagerholm R, Schmidt MK, Khan S, Rafiq S, Tapper W, Aittomäki K, et al. The SNP rs6500843 in 16p13.3 is associated with survival specifically among chemotherapy-treated breast cancer patients. Oncotarget. 2015;6(10):7390–407.
Lei J, Rudolph A, Moysich KB, Rafiq S, Behrens S, Goode EL, et al. Assessment of variation in immunosuppressive pathway genes reveals TGFBR2 to be associated with prognosis of estrogen receptor-negative breast cancer after chemotherapy. Breast Cancer Res. 2015;17(1):18. https://doi.org/10.1186/s13058-015-0522-2.
Cardoso F, Senkus E, Costa A, Papadopoulos E, Aapro M, André F, et al. 4th ESO–ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)††These guidelines were developed by the European School of Oncology (ESO) and the European Society for Medical Oncology (ESMO). Ann Oncol. 2018;29(8):1634–57. https://doi.org/10.1093/annonc/mdy192.
Cardoso F, Kyriakides S, Ohno S, Penault-Llorca F, Poortmans P, Rubio IT, et al. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol. 2019;30(8):1194–220. https://doi.org/10.1093/annonc/mdz173.
Michailidou K, Hall P, Gonzalez-Neira A, Ghoussaini M, Dennis J, Milne RL, et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nature Genet. 2013;45(4):353–61. https://doi.org/10.1038/ng.2563.
the Haplotype Reference Consortium, McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nature Genet. 2016;48:1279.
EBCTCG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–717.
Wakefield J. A Bayesian Measure of the Probability of False Discovery in Genetic Epidemiology Studies. Am J Hum Genet. 2007;81(2):208–27. https://doi.org/10.1086/519024.
Stephens M, Balding DJ. Bayesian statistical methods for genetic association studies. Nat Rev Genet. 2009;10(10):681–90. https://doi.org/10.1038/nrg2615.
Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls.
Goeman JJ, Solari A. Multiple hypothesis testing in genomics. Stat Med. 2014;33(11):1946–78. https://doi.org/10.1002/sim.6082.
Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. https://doi.org/10.1038/s41467-017-01261-5.
Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123(3):725–31. https://doi.org/10.1007/s10549-009-0674-9.
Lánczky A, Nagy Á, Bottai G, Munkácsy G, Szabó A, Santarpia L, et al. miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat. 2016;160(3):439–46. https://doi.org/10.1007/s10549-016-4013-7.
Gong J, Mei S, Liu C, Xiang Y, Ye Y, Zhang Z, et al. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res. 2017;46(D1):D971–D6.
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2018;47(D1):D1005–D12.
Yersal O, Barutca S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J Clin Oncol. 2014;5(3):412–24. https://doi.org/10.5306/wjco.v5.i3.412.
Russnes HG, Lingjærde OC, Børresen-Dale A-L, Caldas C. Breast cancer molecular stratification: from intrinsic subtypes to integrative clusters. Am J Pathol. 2017;187(10):2152–62. https://doi.org/10.1016/j.ajpath.2017.04.022.
Onitilo AA, Engel JM, Greenlee RT, Mukesh BN. Breast cancer subtypes based on ER/PR and Her2 expression: comparison of clinicopathologic features and survival. Clin Med Res. 2009;7(1-2):4–13. https://doi.org/10.3121/cmr.2008.825.
Howlader N, Cronin KA, Kurian AW, Andridge R. Differences in breast cancer survival by molecular subtypes in the United States. Cancer Epidemiology Biomarkers &. Prevention. 2018;27(6):619–26.
Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017;36(11):1461–73. https://doi.org/10.1038/onc.2016.304.
Lin S-Y, Xia W, Wang JC, Kwong KY, Spohn B, Wen Y, et al. β-Catenin, a novel prognostic marker for breast cancer: Its roles in cyclin D1 expression and cancer progression. Proc Natl Acad Sci. 2000;97(8):4262–6. https://doi.org/10.1073/pnas.060025397.
Inagawa S, Itabashi M, Adachi S, Kawamoto T, Hori M, Shimazaki J, et al. Expression and prognostic roles of beta-catenin in hepatocellular carcinoma: correlation with tumor progression and postoperative survival. Clin Cancer Res. 2002;8(2):450–6.
Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29(4):452–63. https://doi.org/10.1016/j.ccell.2016.03.010.
Sanchez Calle A, Kawamura Y, Yamamoto Y, Takeshita F, Ochiya T. Emerging roles of long non-coding RNA in cancer. Cancer Sci. 2018;109(7):2093–100. https://doi.org/10.1111/cas.13642.
Peng Y, Croce CM. The role of microRNAs in human cancer. Signal Transduction Targeted Ther. 2016;1(1):15004. https://doi.org/10.1038/sigtrans.2015.4.
Volinia S, Galasso M, Sana ME, Wise TF, Palatini J, Huebner K, et al. Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc Natl Acad Sci. 2012;109(8):3024–9. https://doi.org/10.1073/pnas.1200010109.
Acknowledgements
We thank all the individuals who took part in these studies and all the researchers, clinicians, technicians, and administrative staff who have enabled this work to be carried out. ABCFS thank Maggie Angelakos, Judi Maskiell, Gillian Dite. ABCTB Investigators: Christine Clarke, Deborah Marsh, Rodney Scott, Robert Baxter, Desmond Yip, Jane Carpenter, Alison Davis, Nirmala Pathmanathan, Peter Simpson, J. Dinny Graham, Mythily Sachchithananthan. Samples are made available to researchers on a non-exclusive basis. BCEES thanks Allyson Thomson, Christobel Saunders, Terry Slevin, BreastScreen Western Australia, Elizabeth Wylie, Rachel Lloyd. The BCINIS study would not have been possible without the contributions of Dr. K. Landsman, Dr. N. Gronich, Dr. A. Flugelman, Dr. W. Saliba, Dr. F. Lejbkowicz, Dr. E. Liani, Dr. I. Cohen, Dr. S. Kalet, Dr. V. Friedman, Dr. O. Barnet of the NICCC in Haifa, and all the contributing family medicine, surgery, pathology and oncology teams in all medical institutes in Northern Israel. BIGGS thanks Niall McInerney, Gabrielle Colleran, Andrew Rowan, Angela Jones. The BREOGAN study would not have been possible without the contributions of the following: Manuela Gago-Dominguez, Jose Esteban Castelao, Angel Carracedo, Victor Muñoz Garzón, Alejandro Novo Domínguez, Maria Elena Martinez, Sara Miranda Ponte, Carmen Redondo Marey, Maite Peña Fernández, Manuel Enguix Castelo, Maria Torres, Manuel Calaza (BREOGAN), José Antúnez, Máximo Fraga and the staff of the Department of Pathology and Biobank of the University Hospital Complex of Santiago-CHUS, Instituto de Investigación Sanitaria de Santiago, IDIS, Xerencia de Xestion Integrada de Santiago-SERGAS; Joaquín González-Carreró and the staff of the Department of Pathology and Biobank of University Hospital Complex of Vigo, Instituto de Investigacion Biomedica Galicia Sur, SERGAS, Vigo, Spain. The BSUCH study acknowledges the Principal Investigator, Barbara Burwinkel, and, thanks Peter Bugert, Medical Faculty Mannheim. CCGP thanks Styliani Apostolaki, Anna Margiolaki, Georgios Nintos, Maria Perraki, Georgia Saloustrou, Georgia Sevastaki, Konstantinos Pompodakis. CGPS thanks staff and participants of the Copenhagen General Population Study. For the excellent technical assistance: Dorthe Uldall Andersen, Maria Birna Arnadottir, Anne Bank, Dorthe Kjeldgård Hansen. The Danish Cancer Biobank is acknowledged for providing infrastructure for the collection of blood samples for the cases. Investigators from the CPS-II cohort thank the participants and Study Management Group for their invaluable contributions to this research. They also acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, as well as cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program. The authors would like to thank the California Teachers Study Steering Committee that is responsible for the formation and maintenance of the Study within which this research was conducted. A full list of California Teachers Study team members is available at https://www.calteachersstudy.org/team. DIETCOMPLYF thanks the patients, nurses and clinical staff involved in the study. The DietCompLyf study was funded by the charity Against Breast Cancer (Registered Charity Number 1121258) and the NCRN. We thank the participants and the investigators of EPIC (European Prospective Investigation into Cancer and Nutrition). ESTHER thanks Hartwig Ziegler, Sonja Wolf, Volker Hermann, Christa Stegmaier, Katja Butterbach. FHRISK thanks NIHR for funding. GC-HBOC thanks Stefanie Engert, Heide Hellebrand, Sandra Kröber and LIFE - Leipzig Research Centre for Civilization Diseases (Markus Loeffler, Joachim Thiery, Matthias Nüchter, Ronny Baber). The GENICA Network: Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, and University of Tübingen, Germany [HB, RH, Wing-Yee Lo], German Cancer Consortium (DKTK) and German Cancer Research Center (DKFZ), Partner Site Tübingen, Tübingen, Germany [HB], gefördert durch die Deutsche Forschungsgemeinschaft (DFG) im Rahmen der Exzellenzstrategie des Bundes und der Länder - EXC 2180 - 390900677 [HB], Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany [Yon-Dschun Ko, Christian Baisch], Institute of Pathology, University of Bonn, Germany [Hans-Peter Fischer], Molecular Genetics of Breast Cancer, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Germany [Ute Hamann], Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, Germany [TB, Beate Pesch, Sylvia Rabstein, Anne Lotz]; and Institute of Occupational Medicine and Maritime Medicine, University Medical Center Hamburg-Eppendorf, Germany [Volker Harth]. HABCS would like to thank Peter Schürmann, Natalia Bogdanova, Nikki Adrian Krentel, Regina Meier, Frank Papendorf, Michael Bremer, Johann H. Karstens, Hans Christiansen and Peter Hillemanns for their contributions to this study. HEBCS thanks Johanna Kiiski, Carl Blomqvist, Sofia Khan, Kristiina Aittomäki, Kirsimari Aaltonen, Karl von Smitten, Irja Erkkilä. ICICLE thanks Kelly Kohut, Michele Caneppele, Maria Troy. KARMA and SASBAC thank the Swedish Medical Research Counsel. KBCP thanks Eija Myöhänen, Helena Kemiläinen. kConFab/AOCS wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (which has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia, and the National Institute of Health (USA)) for their contributions to this resource, and the many families who contribute to kConFab. LMBC thanks Gilian Peuteman, Thomas Van Brussel, EvyVanderheyden and Kathleen Corthouts. MABCS thanks Milena Jakimovska (RCGEB “Georgi D. Efremov”), Emilija Lazarova, Marina Iljovska (University Clinic of Radiotherapy and Oncology), Katerina Kubelka-Sabit, Dzengis Jashar and Mitko Karadjozov (Adzibadem-Sistina Hospital) for their contributions and commitment to this study. MARIE thanks Petra Seibold, Dieter Flesch-Janys, Judith Heinz, Nadia Obi, Alina Vrieling, Sabine Behrens, Ursula Eilber, Muhabbet Celik, Til Olchers and Stefan Nickels. MBCSG (Milan Breast Cancer Study Group): Paolo Radice, Siranoush Manoukian, Jacopo Azzollini, Erica Rosina, Daniela Zaffaroni, Bernardo Bonanni, Irene Feroce, Mariarosaria Calvello, Aliana Guerrieri Gonzaga, Monica Marabelli, Davide Bondavalli and the personnel of the Cogentech Cancer Genetic Test Laboratory. The MCCS was made possible by the contribution of many people, including the original investigators, the teams that recruited the participants and continue working on follow-up, and the many thousands of Melbourne residents who continue to participate in the study. We thank the coordinators, the research staff and especially the MMHS participants for their continued collaboration on research studies in breast cancer. The following are NBCS Collaborators: Anne-Lise Børresen-Dale (Prof. Em.), Kristine K. Sahlberg (PhD), Lars Ottestad (MD), Rolf Kåresen (Prof. Em.) Dr. Ellen Schlichting (MD), Marit Muri Holmen (MD), Toril Sauer (MD), Vilde Haakensen (MD), Olav Engebråten (MD), Bjørn Naume (MD), Alexander Fosså (MD), Cecile E. Kiserud (MD), Kristin V. Reinertsen (MD), Åslaug Helland (MD), Margit Riis (MD), Jürgen Geisler (MD), OSBREAC and Grethe I. Grenaker Alnæs (MSc). For NHS and NHS2 the study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. We would like to thank the participants and staff of the NHS and NHS2 for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The authors assume full responsibility for analyses and interpretation of these data. OBCS thanks Katri Pylkäs, Arja Jukkola, Saila Kauppila, Meeri Otsukka, Leena Keskitalo and Kari Mononen for their contributions to this study. The OFBCR thanks Teresa Selander, Nayana Weerasooriya and Steve Gallinger. ORIGO thanks E. Krol-Warmerdam, and J. Blom for patient accrual, administering questionnaires, and managing clinical information. The LUMC survival data were retrieved from the Leiden hospital-based cancer registry system (ONCDOC) with the help of Dr. J. Molenaar. PBCS thanks Louise Brinton, Mark Sherman, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao, Michael Stagner. The ethical approval for the POSH study is MREC /00/6/69, UKCRN ID: 1137. We thank staff in the Experimental Cancer Medicine Centre (ECMC) supported Faculty of Medicine Tissue Bank and the Faculty of Medicine DNA Banking resource. PREFACE thanks Sonja Oeser and Silke Landrith. PROCAS thanks NIHR for funding. The RBCS thanks Jannet Blom, Saskia Pelders, Wendy J.C. Prager – van der Smissen, and the Erasmus MC Family Cancer Clinic. SBCS thanks Sue Higham, Helen Cramp, Dan Connley, Ian Brock, Sabapathy Balasubramanian and Malcolm W.R. Reed. We thank the SEARCH and EPIC teams. SKKDKFZS thanks all study participants, clinicians, family doctors, researchers and technicians for their contributions and commitment to this study. We thank the SUCCESS Study teams in Munich, Duessldorf, Erlangen and Ulm. SZBCS thanks Ewa Putresza. UCIBCS thanks Irene Masunaka. UKBGS thanks Breast Cancer Now and the Institute of Cancer Research for support and funding of the Generations Study, and the study participants, study staff, and the doctors, nurses and other health care providers and health information sources who have contributed to the study. We acknowledge NHS funding to the Royal Marsden/ICR NIHR Biomedical Research Centre. The authors thank the WHI investigators and staff for their dedication and the study participants for making the program possible.
Funding
BCAC is funded by Cancer Research UK [C1287/A16563], the European Union’s Horizon 2020 Research and Innovation Programme (grant numbers 634935 and 633784 for BRIDGES and B-CAST respectively), and by the European Community’s Seventh Framework Programme under grant agreement number 223175 (grant number HEALTH-F2-2009-223175) (COGS). The EU Horizon 2020 Research and Innovation Programme funding source had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Genotyping of the OncoArray was funded by the NIH Grant U19 CA148065, and Cancer Research UK Grant C1287/A16563 and the PERSPECTIVE project supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research (grant GPH-129344), and the Ministère de l’Économie, Science et Innovation du Québec through Genome Québec and the PSRSIIRI-701 grant, and the Quebec Breast Cancer Foundation. Funding for the iCOGS infrastructure came from: the European Community's Seventh Framework Programme under grant agreement n° 223175 (HEALTH-F2-2009-223175) (COGS), Cancer Research UK (C1287/A10118, C1287/A10710, C12292/A11174, C1281/A12014, C5047/A8384, C5047/A15007, C5047/A10692, C8197/A16565), the National Institutes of Health (CA128978) and Post-Cancer GWAS initiative (1 U19 CA148537, 1 U19 CA148065 and 1 U19 CA148112 - the GAME-ON initiative), the Department of Defence (W81XWH-10-1-0341), the Canadian Institutes of Health Research (CIHR) for the CIHR Team in Familial Risks of Breast Cancer, and Komen Foundation for the Cure, the Breast Cancer Research Foundation, and the Ovarian Cancer Research Fund. The DRIVE Consortium was funded by U19 CA148065.
The Australian Breast Cancer Family Study (ABCFS) was supported by grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The ABCFS was also supported by the National Health and Medical Research Council of Australia, the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and the Victorian Breast Cancer Research Consortium. J.L.H. is a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellow. M.C.S. is a NHMRC Senior Research Fellow. The ABCS study was supported by the Dutch Cancer Society [grants NKI 2007-3839; 2009 4363]. The Australian Breast Cancer Tissue Bank (ABCTB) was supported by the National Health and Medical Research Council of Australia, The Cancer Institute NSW, and the National Breast Cancer Foundation. The ACP study is funded by the Breast Cancer Research Trust, UK. KM and AL are supported by the NIHR Manchester Biomedical Research Centre, the Allan Turing Institute, and, by the ICEP (Cancer Research UK (C18281/A19169). The AHS study is supported by the intramural research program of the National Institutes of Health, the National Cancer Institute (grant number Z01-CP010119), and the National Institute of Environmental Health Sciences (grant number Z01-ES049030). The work of the BBCC was partly funded by ELAN-Fond of the University Hospital of Erlangen. The BCEES was funded by the National Health and Medical Research Council, Australia and the Cancer Council Western Australia. For the BCFR-NY, BCFR-PA, and BCFR-UT, this work was supported by grant UM1 CA164920 from the National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the BCFR. The BCINIS study is supported in part by the Breast Cancer Research Foundation (BCRF). For BIGGS, ES is supported by NIHR Comprehensive Biomedical Research Centre, Guy’s & St. Thomas’ NHS Foundation Trust in partnership with King’s College London, UK. IT is supported by the Oxford Biomedical Research Centre. The BREast Oncology GAlician Network (BREOGAN) is funded by Acción Estratégica de Salud del Instituto de Salud Carlos III FIS PI12/02125/Cofinanciado FEDER, PI17/00918/Cofinanciado FEDER; Acción Estratégica de Salud del Instituto de Salud Carlos III FIS Intrasalud (PI13/01136); Programa Grupos Emergentes, Cancer Genetics Unit, Instituto de Investigacion Biomedica Galicia Sur. Xerencia de Xestion Integrada de Vigo-SERGAS, Instituto de Salud Carlos III, Spain; Grant 10CSA012E, Consellería de Industria Programa Sectorial de Investigación Aplicada, PEME I + D e I + D Suma del Plan Gallego de Investigación, Desarrollo e Innovación Tecnológica de la Consellería de Industria de la Xunta de Galicia, Spain; Grant EC11-192. Fomento de la Investigación Clínica Independiente, Ministerio de Sanidad, Servicios Sociales e Igualdad, Spain; and Grant FEDER-Innterconecta. Ministerio de Economia y Competitividad, Xunta de Galicia, Spain. The BSUCH study was supported by the Dietmar-Hopp Foundation, the Helmholtz Society and the German Cancer Research Center (DKFZ). CCGP is supported by funding from the University of Crete. The CECILE study was supported by Fondation de France, Institut National du Cancer (INCa), Ligue Nationale contre le Cancer, Agence Nationale de Sécurité Sanitaire, de l'Alimentation, de l'Environnement et du Travail (ANSES), Agence Nationale de la Recherche (ANR). The CGPS was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council, and Herlev and Gentofte Hospital. The American Cancer Society funds the creation, maintenance, and updating of the CPS-II cohort. The California Teachers Study and the research reported in this publication were supported by the National Cancer Institute of the National Institutes of Health under award number U01-CA199277; P30-CA033572; P30-CA023100; UM1-CA164917; and R01-CA077398. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The collection of cancer incidence data used in the California Teachers Study was supported by the California Department of Public Health pursuant to California Health and Safety Code Section 103885; Centers for Disease Control and Prevention’s National Program of Cancer Registries, under cooperative agreement 5NU58DP006344; the National Cancer Institute’s Surveillance, Epidemiology and End Results Program under contract HHSN261201800032I awarded to the University of California, San Francisco, contract HHSN261201800015I awarded to the University of Southern California, and contract HHSN261201800009I awarded to the Public Health Institute. The opinions, findings, and conclusions expressed herein are those of the author(s) and do not necessarily reflect the official views of the State of California, Department of Public Health, the National Cancer Institute, the National Institutes of Health, the Centers for Disease Control and Prevention or their Contractors and Subcontractors, or the Regents of the University of California, or any of its programs. The University of Westminster curates the DietCompLyf database funded by Against Breast Cancer Registered Charity No. 1121258 and the NCRN. The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by: Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF) (Germany); the Hellenic Health Foundation, the Stavros Niarchos Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (UK). The ESTHER study was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe). FHRISK is funded from NIHR grant PGfAR 0707-10031. D.G.E. is supported by the all Manchester NIHR Biomedical Research Centre (IS-BRC-1215-20007). The GC-HBOC (German Consortium of Hereditary Breast and Ovarian Cancer) is supported by the German Cancer Aid (grant no 110837, coordinator: Rita K. Schmutzler, Cologne). This work was also funded by the European Regional Development Fund and Free State of Saxony, Germany (LIFE - Leipzig Research Centre for Civilization Diseases, project numbers 713-241202, 713-241202, 14505/2470, 14575/2470). The GENICA was funded by the Federal Ministry of Education and Research (BMBF) Germany grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114, the Robert Bosch Foundation, Stuttgart, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, the Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, as well as the Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany. The GESBC was supported by the Deutsche Krebshilfe e. V. [70492] and the German Cancer Research Center (DKFZ). The HABCS study was supported by the Claudia von Schilling Foundation for Breast Cancer Research. The HEBCS was financially supported by the Helsinki University Hospital Research Fund, the Finnish Cancer Society, and the Sigrid Juselius Foundation. ICICLE was supported by Breast Cancer Now, CRUK and Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. Financial support for KARBAC was provided through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet, the Swedish Cancer Society, The Gustav V Jubilee foundation and Bert von Kantzows foundation. The KARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer. The KBCP was financially supported by the special Government Funding (EVO) of Kuopio University Hospital grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, and by the strategic funding of the University of Eastern Finland. kConFab is supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. Financial support for the AOCS was provided by the United States Army Medical Research and Materiel Command [DAMD17-01-1-0729], Cancer Council Victoria, Queensland Cancer Fund, Cancer Council New South Wales, Cancer Council South Australia, The Cancer Foundation of Western Australia, Cancer Council Tasmania and the National Health and Medical Research Council of Australia (NHMRC; 400413, 400281, 199600). G.C.T. and P.W. are supported by the NHMRC. RB was a Cancer Institute NSW Clinical Research Fellow. LMBC is supported by the ‘Stichting tegen Kanker’. DL is supported by the FWO. The MABCS study is funded by the Research Centre for Genetic Engineering and Biotechnology “Georgi D. Efremov,” MASA. The MARIE study was supported by the Deutsche Krebshilfe e.V. [70-2892-BR I, 106332, 108253, 108419, 110826, 110828], the Hamburg Cancer Society, the German Cancer Research Center (DKFZ) and the Federal Ministry of Education and Research (BMBF) Germany [01KH0402 and 01ER1306]. MBCSG is supported by grants from the Italian Association for Cancer Research (AIRC). The MCBCS was supported by the NIH grants CA192393, CA116167, CA176785 an NIH Specialized Program of Research Excellence (SPORE) in Breast Cancer [CA116201], and the Breast Cancer Research Foundation and a generous gift from the David F. and Margaret T. Grohne Family Foundation. The Melbourne Collaborative Cohort Study (MCCS) cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further augmented by Australian National Health and Medical Research Council grants 209057, 396414 and 1074383 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry and the Australian Institute of Health and Welfare, including the National Death Index and the Australian Cancer Database. The MEC was supported by NIH grants CA63464, CA54281, CA098758, CA132839 and CA164973. The MISS study is supported by funding from ERC-2011-294576 Advanced grant, Swedish Cancer Society, Swedish Research Council, Local hospital funds, Berta Kamprad Foundation, Gunnar Nilsson. The MMHS study was supported by NIH grants CA97396, CA128931, CA116201, CA140286 and CA177150. The NBCS has received funding from the K.G. Jebsen Centre for Breast Cancer Research; the Research Council of Norway grant 193387/V50 (to A-L Børresen-Dale and V.N. Kristensen) and grant 193387/H10 (to A-L Børresen-Dale and V.N. Kristensen), South Eastern Norway Health Authority (grant 39346 to A-L Børresen-Dale) and the Norwegian Cancer Society (to A-L Børresen-Dale and V.N. Kristensen). The Northern California Breast Cancer Family Registry (NC-BCFR) and Ontario Familial Breast Cancer Registry (OFBCR) were supported by grant U01CA164920 from the USA National Cancer Institute of the National Institutes of Health. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The Carolina Breast Cancer Study (NCBCS) was funded by Komen Foundation, the National Cancer Institute (P50 CA058223, U54 CA156733, U01 CA179715), and the North Carolina University Cancer Research Fund. The NHS was supported by NIH grants P01 CA87969, UM1 CA186107, and U19 CA148065. The NHS2 was supported by NIH grants U01 CA176726 and U19 CA148065. The OBCS was supported by research grants from the Finnish Cancer Foundation, the Academy of Finland (grant number 250083, 122715 and Center of Excellence grant number 251314), the Finnish Cancer Foundation, the Sigrid Juselius Foundation, the University of Oulu, the University of Oulu Support Foundation and the special Governmental EVO funds for Oulu University Hospital-based research activities. The ORIGO study was supported by the Dutch Cancer Society (RUL 1997-1505) and the Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL CP16). The PBCS was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. Genotyping for PLCO was supported by the Intramural Research Program of the National Institutes of Health, NCI, Division of Cancer Epidemiology and Genetics. The PLCO is supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, National Institutes of Health. The POSH study is funded by Cancer Research UK (grants C1275/A11699, C1275/C22524, C1275/A19187, C1275/A15956 and Breast Cancer Campaign 2010PR62, 2013PR044. PROCAS is funded from NIHR grant PGfAR 0707-10031. The RBCS was funded by the Dutch Cancer Society (DDHK 2004-3124, DDHK 2009-4318). The SASBAC study was supported by funding from the Agency for Science, Technology and Research of Singapore (A*STAR), the US National Institute of Health (NIH) and the Susan G. Komen Breast Cancer Foundation. The SBCS was supported by Sheffield Experimental Cancer Medicine Centre and Breast Cancer Now Tissue Bank. SEARCH is funded by Cancer Research UK [C490/A10124, C490/A16561] and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. The University of Cambridge has received salary support for PDPP from the NHS in the East of England through the Clinical Academic Reserve. SKKDKFZS is supported by the DKFZ. The SMC is funded by the Swedish Cancer Foundation and the Swedish Research Council (VR 2017-00644) grant for the Swedish Infrastructure for Medical Population-based Life-course Environmental Research (SIMPLER). The SZBCS was supported by Grant PBZ_KBN_122/P05/2004 and the program of the Minister of Science and Higher Education under the name “Regional Initiative of Excellence” in 2019–2022 project number 002/RID/2018/19 amount of financing 12 000 000 PLN. The UCIBCS component of this research was supported by the NIH [CA58860, CA92044] and the Lon V Smith Foundation [LVS39420]. The UKBGS is funded by Breast Cancer Now and the Institute of Cancer Research (ICR), London. ICR acknowledges NHS funding to the NIHR Biomedical Research Centre. The USRT Study was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. The WHI program is funded by the National Heart, Lung, and Blood Institute, the US National Institutes of Health and the US Department of Health and Human Services (HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C and HHSN271201100004C). This work was also funded by NCI U19 CA148065-01.
Author information
Authors and Affiliations
Consortia
Contributions
M.K.S. conceived the study. A.M. performed the main data analyses and drafted the initial manuscript. M.K.S. and A.M. interpreted the data. M.E.-G., J.B., and S.C. provided computational support for the data analyses. R.K., Q.W., M.K.B., and J.D. provided database support. All authors contributed to the critical revision and editing of the final version of the manuscript for publication. All authors were involved in the data generation or provision and read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki. All individual studies included in the analyses were approved by the appropriate institutional ethical review boards. All study participants provided informed consent.
Consent for publication
Not applicable.
Competing interests
Matthias W. Beckmann and Peter A. Fasching conduct research funded by Amgen, Novartis, and Pfizer (not related to this study). Peter A. Fasching received Honoraria from Roche, Novartis, and Pfizer (not related to this study). Allison W. Kurian’s institution received a research funding from Myriad genetics for an unrelated project (not related to this study). The other authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table S1
, Supplementary Table S2, Supplementary Table S3, and Supplementary Table S4. Supplementary Table S1. Shows an overview of the Breast Cancer Association Consortium studies included in the analyses. Supplementary Table S2. Includes the rationale and references to the literature supporting the choice of each subgroup for inclusion in a genome-wide association study on breast cancer-specific survival. Supplementary Table S3. Shows the characteristics of the breast cancer patients included in the 15-year breast cancer-specific survival analyses, overall and by subgroup. Supplementary Table S4. Shows the characteristics of the breast cancer patients included in the 5-year breast cancer-specific survival analyses for subgroups defined based on type of systemic treatment received.
Additional file 2: Supplementary Methods
, Supplementary Table S5, Supplementary Table S6, Supplementary Table S7, Supplementary Figure S1, Supplementary Figure S2, Supplementary Figure S3, Supplementary Figure S4, Supplementary Figure S5, Supplementary Figure S6, Supplementary Figure S7, Supplementary Figure S8, Supplementary Figure S9, Supplementary Figure S10, Supplementary Figure S11, Supplementary Figure S12. The Supplementary methods section includes details about: multiple imputation of missing data; power calculations. Supplementary Table S5. Shows an overview of number of breast cancer patients, breast cancer deaths and follow-up information by subgroup and endpoint. Supplementary Table S6. Shows a list of imputed variables with corresponding percentage of missing values, imputation method and processing. Supplementary Table S7. Shows an overview of the GWAS significant associations (P < 5 E-08) and noteworthy (BFDP< 0.15) associations from the unadjusted 15-year and 5-year breast cancer-specific survival analyses by subgroup. Supplementary Figure S1. Shows the Q-Q plots of the meta-analysis results of all variants for 15-year breast cancer-specific survival. Supplementary Figure S2. Shows the Q-Q plots of the meta-analysis results of all variants for 5-year breast cancer-specific survival. Supplementary Figure S3. Shows the Q-Q plots of the meta-analysis results for 15-year breast cancer-specific survival and the corresponding genome inflation factors by minor allele frequency. Supplementary Figure S4. Shows the Q-Q plots of the meta-analysis results for 5-year breast cancer-specific survival and the corresponding genome inflation factors by minor allele frequency. Supplementary Figure S5. Shows the regional plots of genome-wide significant (P < 5E-08) independent associated variants from the 15-year and 5-year genome-wide breast cancer-specific survival analyses. Supplementary Figure S6. Shows the regional plots of noteworthy (BFDP< 0.15), non-genome-wide significant (P > 5E-08) variants from the 15-year and 5-year genome-wide breast cancer-specific survival analyses. Supplementary Figure S7 shows the unadjusted association of variant rs5934618 with 15-year breast cancer-specific survival by tumor grade and in all breast cancer patients. Supplementary Figure S8. Shows the functional annotation and position of variants rs5934618, rs4830644, rs3810742, rs4830642, and rs72611496 relative to TBL1X. Supplementary Figure S9. Shows Kaplan-Meier distant metastasis-free survival plots (based on KMPlotter data) for high versus low expression level of gene TBL1X by tumor grade. Supplementary Figure S10. Shows the association of genetic variants with TBL1X expression, based on GTEx v8 data on samples of normal breast tissue from 396 individuals (male and female). Supplementary Figure S11. Shows a Kaplan-Meier distant metastasis-free survival plot for high versus low expression level of gene GRIP2, restricted to patients with an ER- tumor who received chemotherapy (based on KMPlotter data). Supplementary Figure S12. Shows a Kaplan-Meier distant metastasis-free survival plot for high versus low expression level of gene ARAP2, restricted to patients with an ER- tumor who received chemotherapy (based on KMPlotter data).
Additional file 3: Supplementary Table S8
, Supplementary Table S9, Supplementary Table S10, Supplementary Table S11, and Supplementary Table S12. Supplementary Table S8. Shows the BFDPs under two more restrictive prior probabilities of true association (10-5 and 10-6) for the results presented in Table 1. Supplementary Table S9. Shows the BFDPs under two more restrictive prior probabilities of true association (10-5 and 10-6) for the results presented in Table 2. Supplementary Table S10. Shows power calculation by subgroup, at the two-sided 5E-08 level for varying genotype hazard ratio (GHR) and minor allele frequency (MAF), based on number of cases and event rate from the 15-year breast cancer-specific analyses. Supplementary Table S11. Shows power calculation by subgroup, at the two-sided 5E-08 level for varying genotype hazard ratio (GHR) and minor allele frequency (MAF), based on number of cases and event rate from the 5-year breast cancer-specific analyses. Supplementary Table S12. Shows the subgroup-specific associations detected by previous studies and corresponding estimates from the current study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Morra, A., Escala-Garcia, M., Beesley, J. et al. Association of germline genetic variants with breast cancer-specific survival in patient subgroups defined by clinic-pathological variables related to tumor biology and type of systemic treatment. Breast Cancer Res 23, 86 (2021). https://doi.org/10.1186/s13058-021-01450-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-021-01450-7