
Abstract
Background
Molecular mechanisms leading to the adaptation of breast cancer (BC) cells to hypoxia are largely unknown. The anti-apoptotic Bcl-2 family member myeloid cell leukemia-1 (Mcl-1) is frequently amplified in BC; and elevated Mcl-1 levels have been correlated with poor prognosis. Here we investigated the pathophysiologic role of Mcl-1 in Her2-positive BC cells under hypoxic conditions.
Methods
RNA interference and a novel small molecule inhibitor, EU-5346, were used to examine the role of Mcl-1 in Her2-positive BC cell lines and primary BC cells (sensitive or intrinsically resistant to Her2 inhibitors) under hypoxic conditions (using a hypoxic incubation chamber). Mechanisms-of-action were investigated by RT-PCR, mitochondrial isolation, as well as immunoprecipitation/blotting analysis, and microscopy. The specificity against Mcl-1 of the novel small molecule inhibitor EU5346 was verified in Mcl-1Δ/null versus Mcl-1wt/wt Murine Embryonic Fibroblasts (MEFs). Proliferation, survival, and spheroid formation were assessed in response to Mcl-1 and Her2 inhibition.
Results
We demonstrate for a strong correlation between high Mcl-1 protein levels and hypoxia, predominantly in Her2-positive BC cells. Surprisingly, genetic depletion of Mcl-1 decreased Her2 and Hif-1α levels followed by inhibition of BC cell survival. In contrast, Mcl-1 protein levels were not downregulated after genetic depletion of Her2 indicating a regulatory role of Mcl-1 upstream of Her2. Indeed, Mcl-1 and Her2 co-localize within the mitochondrial fraction and form a Mcl-1/Her2- protein complex. Similar to genetically targeting Mcl-1 the novel small molecule Mcl-1 inhibitor EU-5346 induced cell death and decreased spheroid formation in Her2-positive BC cells. Of interest, EU-5346 induced ubiquitination of Mcl-1- bound Her2 demonstrating a previously unknown role for Mcl-1 to stabilize Her2 protein levels. Importantly, targeting Mcl-1 was also active in Her2-positive BC cells resistant to Her2 inhibitors, including a brain-primed Her2-positive cell line.
Conclusion
Our data demonstrate a critical role of Mcl-1 in Her2-positive BC cell survival under hypoxic conditions and provide the preclinical framework for the therapeutic use of novel Mcl-1- targeting agents to improve patient outcome in BC.
Similar content being viewed by others


Background
Breast cancer (BC) is the most common malignancy and the second most common cause of cancer-related mortality in women. The clinically relevant classification of BC is based on its histopathological appearance, tumor grade, lymph node involvement, and immunohistochemical properties including the presence of the estrogen receptor (ER), the progesterone receptor (PR), human epidermal growth factor receptor 2 (Her2; ErbB2, c-erbB2), and Ki-67 [1, 2]. Importantly, BC is known to be highly heterogenic with different intrinsic molecular subtypes. An additional BC classification based on molecular signatures has been developed recently with a clinical impact not only for patients’ prognosis but also the choice of treatment. Gene expression profiling has been utilized to classify BC into four different groups [3–6]. Specifically, BC is subgrouped into Luminal A, Luminal B, Her2-enriched, and basal-like BC [4]. Luminal-A BC, the most common subtype, is ER-positive and is characterized by the absence of Her2 expression, a low rate of proliferation (Ki67), and histologically low-grade tumors. Moreover, Luminal-A BC presents with high expression in mRNA and protein levels of the Luminal expression signature (e.g., ESR1, GATA3, FOXA1, XBP1, and MYB) [5, 7]. Luminal-B BC includes ER-positive but also ER-negative histologically high-grade tumors. In contrast to Luminal A, Luminal-B BC has an increased expression not only of proliferation genes such as KI67 and cyclin B1 but also frequently overexpresses epidermal growth factor receptor (EGFR) and HER2 [7]. Basal-like BC or triple-negative breast cancer (TNBC) is characterized by ER-negative, PR-negative, and Her2-negative tumors with high frequency of TP53 (80 %) and PIK3CA (9 %) mutation [5]. Her2-positive (Her2-enriched) BC is characterized by the amplification of the HER2 gene and other genes of the HER2 amplicon including STARD3 and GRB7 [6, 8]. Her2 in particular acts as a coreceptor and enhances signaling pathways of other Her family members. It is activated by homodimerization with Her2 or heterodimerization with other Her family members. Importantly, the heterodimer between Her2 and Her3 has the highest mitogenic potential [9]. Overexpression of Her2 occurs in 15–20 % of BC patients and is associated with worse biologic behavior; that is, increased rate of metastasis, and poor clinical outcome without Her2-targeted treatment [10]. Unprecedented therapeutic advances have been achieved during the last years by combining Her2 inhibitors trastuzumab (Herceptin®, Genentech, South San Francisco, CA, USA), lapatinib (Tykerb®, GlaxoSmithKline, Philadelphia, PA, USA; Tyverb®, GlaxoSmithKline, London, UK), and pertuzumab (Perjeta®, Genentech, South San Francisco, CA, USA) with chemotherapeutic regimens, and by the introduction of ado-trastuzumab emtansine (T-DM1, Kadcyla®, Genentech, South San Francisco, CA, USA) monotherapy. However, inherent and acquired resistance to these agents remains a significant barrier to further reduce mortality in this BC patient subtype, highlighting the urgent need for novel therapies [11]. In addition, these drugs do not penetrate the blood–brain barrier as easily as they reach the rest of the body, with lapatinib in combination with capecitabine and T-DM1 monotherapy being a possible exception [12].
Hypoxic conditions develop during cancer progression because of rapidly proliferating tumor cells that reduce oxygen diffusion and impair perfusion of abnormal blood vessels in the tumor microenvironment. Cellular adaptation to hypoxia is predominantly mediated through protein stabilization of hypoxia-inducible factor (Hif) subunits. In BC, hypoxic regions have the potential to confer chemotherapy and radiation therapy resistance [13]. Molecular mechanisms which lead to the adaptation of BC cells to hypoxia are largely unknown.
Myeloid cell leukemia-1 (Mcl-1) is an anti-apoptotic protein of the Bcl-2 family characterized by its ability to oppose several apoptotic stimuli, a short half-life, its wide intracellular localization, and multiple pathways which tightly regulate Mcl-1 transcription, translation, and degradation [14]. Structurally, the N-terminus differs from the other anti-apoptotic Bcl-2 proteins in that it contains two polypeptide sequences enriched in proline, glutamic acid, serine, and threonine (PEST) [15]. PEST regions are made responsible for Mcl-1 degradation via the proteasome pathway, localization, and phosphorylation, thus providing the mechanistic base for the fine-tuned Mcl-1 protein functions in response to environmental stimuli and the cellular origin [14, 16, 17]. The pro-survival function of Mcl-1 is predominantly mediated by its binding to Bak and Bim. Conversely, release of Bak or Bim from their interaction with Mcl-1 induces apoptosis [18]. In addition, binding to the Bcl-2 homology 3 (BH3)-only protein NOXA enhances Mcl-1 degradation followed by caspase activation. In contrast, blockade of NOXA induction results in the suppression of apoptosis [19, 20]. Several past studies indicated that Mcl-1 is an important cancer target. Specifically, Mcl-1 is amplified in many cancers including BC and elevated Mcl-1 levels in BC correlate with increased tumor cell survival, growth, and poor prognosis [21–23]. Both epidermal growth factor and estrogen induce Mcl-1 expression via binding to their respective receptors and activation of downstream signaling pathways [24, 25]. Importantly, Mcl-1 mediates resistance against widely used anticancer therapies including paclitaxel [26] and gemcitabine [27] as well as early clinical BH3 mimetic drugs that block Bcl-2 and Bcl-xL [21, 28]. Moreover, trastuzumab sensitizes Her2-overexpressing cells to apoptosis by reducing anti-apoptotic Mcl-1 expression [29]. Based on these data, Mcl-1 holds great promise as a high-priority therapeutic target. Indeed, Mcl-1 is the current focus of widespread cancer drug development efforts, and a number of Mcl-1 inhibitors are in the cancer drug development pipeline worldwide [30].
In this study we determined a critical role of Mcl-1 in Her2-positive BC cell survival under hypoxic conditions also including brain-primed BC cells, and we present EU-5346 as a promising novel anti-Mcl-1 targeting agent. We thereby provide the preclinical framework for the therapeutic use of novel Mcl-1-targeting agents to improve patient outcome in BC.
Methods
Materials
Lapatinib ditosylate and ABT-199 were purchased from Santa Cruz Biotechnology (Heidelberg, Germany); cobalt (II) chloride hexahydrate from Sigma Aldrich (Schnelldorf, Germany); trastuzumab (Herceptin®) from Roche (San Francisco, CA, USA); and ZVAD pan-caspase inhibitor z-VAD-fmk from Bachem (Bubendorf, Switzerland). Antibodies against human Mcl-1 (S-19), extracellular signal-regulated kinase 2 (ERK2), and Ubiquitin (P4D1) were obtained from Santa Cruz Biotechnology; antibodies against Her2/ErbB2 (D8F12), Poly (ADP-ribose) polymerase (PARP), Bcl-xL, and Prohibitin-1 from Cell Signaling Technology (Boston, MA, USA); the antibody against α-tubulin from Sigma Aldrich; the antibody against Hif-1α, EpCAM, and ErbB2/Her2 from BD Biosciences (Heidelberg, Germany); the antibody against Bcl-2 (ab18210) from Abcam (Boston, MA, USA); and the antibody against murine Mcl-1 from Rockland Immunochemicals Inc., Limerick, PA 19468, USA.
Isolation of primary tumor cells
BC cells were collected by centrifugation of pleural fluid. The cell pellet was resuspended with RPMI medium and placed in culture flasks; >95 % purity of BC cells was confirmed using anti-EpCAM and anti-Her2 antibodies by fluorescence-activated cell sorting (FACS) analysis. Samples were isolated from patients with advanced disease and resistant against multiple agents including hormone therapy, Her2 inhibitors, capecitabine, paclitaxel, eribulin, and everolimus. The collection and use of patient BC cells has been approved by the ethics committee of the Medical Faculty, University of Heidelberg (approval number 022/2013). Informed consent was obtained in accordance with the Declaration of Helsinki.
Cell culture and conditions
MCF-7 cells were a kind gift from Dr Beckhove (German Cancer Research Center (DKFZ), Heidelberg, Germany), MDA MB-157 and MDA MB-468 cells were from Dr Oskarsson (DKFZ), HCC-1954 and MCF-10A cells were from Dr Wiemann (DKFZ), and SKBR3 cells were from Dr Trumpp (DKFZ). JIMT-1-BR3 cells (brain metastatic BC cell line) were a kind gift from Dr Steeg (Laboratory of Molecular Pharmacology, NCI’s Women's Malignancies Branch, National Cancer Institute/National Institutes of Health, Bethesda, MD, USA). JIMT-1-BR3 cells were obtained by a method detailed in previous publication [31]. All other cell lines were purchased from the Leibniz Institute DSMZ—German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). MCF-7, MDA MB 231, and HCC-1954 cells were cultured in RPMI 1640 medium (Gibco, Life Technologies, Grand Island, NY, USA) supplemented with 10 % heat-inactivated fetal bovine serum (FBS; PAA Laboratories, Cölbe, Germany), 1 % penicillin/streptomycin and 2 mM l-glutamine (all from Gibco, Life Technologies). MDA MB 453, MDA MB-157, MDA MB-468, JIMT-1, and JIMT-1 BR3 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Life Technologies) supplemented with 10 % heat-inactivated FBS, 1 % penicillin/streptomycin and 2 mM l-glutamine. BT-474 and SKBR3 cells were maintained similarly to MDA MB 453 cells, but with the addition of minimum essential medium (MEM) nonessential amino acids (Gibco, Life Technologies). MCF-10A cells were maintained in DMEM/F12 supplemented with 10 % heat-inactivated FBS, 1 % penicillin/streptomycin, 2.5 mg insulin, 5 mg hydrocortisone (Sigma), 8 μl cholera toxin, and 10 μg human epidermal growth factor (Sigma Aldrich). Murine embryonic fibroblast (MEF) cell lines Mcl-1wt/wt and Mcl-1Δ/nullwere generated by SV40 large T transformation followed by Tet-Cre-mediated deletion. Single cell clones were selected and then grown in DMEM supplemented with 10 % heat-inactivated FBS, 1 % penicillin/streptomycin, 2 mM l-glutamine, 2-mercaptoethanol (Sigma Aldrich), and MEM nonessential amino acids (Gibco, Life Technologies) from early passages. Both Mcl-1wt/wt and Mcl-1Δ/null MEFs were extensively characterized as being hypersensitive to various death stimuli with restorable resistance upon re-expression of Mcl-1 [32]. For hypoxia, cells were incubated in a hypoxia chamber (COY Laboratory Products, Ann Arbor, MI, USA) with a computerized oxygen controller to maintain an atmosphere of 0.5– 1 % oxygen, 5 % carbon dioxide, and 37 °C.
Small interfering RNAs and cell transfection
BC cells were transiently transfected with indicated amounts of small interfering RNA (siRNA) siGENOME SMART pools of MCL1 (4170) and HER2 (2064), or the nontargeting control (mock) siRNA (Upstate Cell Signaling Solutions/Dharmacon RNA Technologies, Lafayette, CO, USA) using Lipofectamine® 2000 according to the manufacturer's instructions (Invitrogen, Darmstadt, Germany). Nontargeting control (mock) siRNA is composed of a pool of four siRNAs, which have been characterized by genome-wide microarray analysis and found to have minimal off-target signatures.
Cell death assays
The inhibitory effect of hypoxia, siMCL1, or siHER2 on BC cell lines was assessed using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma Aldrich) as described previously [33], or AlamarBlue® assay (Invitrogen) according to the manufacturer's instructions.
Cell lysis, immunoblotting, and immunoprecipitation
Treated or untreated cells were washed three times with phosphate-buffered saline and lysed with either lysis buffer (10 mm Tris, 50 mm NaCl, 1 % Triton, 30 mM sodium pyrophosphate, pH 7.05) or radioimmune precipitation assay lysis buffer (150 mM NaCl, 10 mM Tris, pH 7.2, 0.1 % SDS, 1 % Triton X-100, 1 % deoxycholate, 5 mM ethylenediamine tetraacetic acid) supplemented with Halt Protease and Phosphatase Inhibitor Cocktail (Pierce, Darmstadt, Germany). Insoluble material was removed by centrifugation (15,000 rpm for 30 minutes at 4 °C). Immunocomplexes were collected following overnight incubation at 4 °C with 10–20 μl of 100 % protein A-Sepharose® CL-4B beads (Amersham, Arlington Heights, USA). For western blotting, cell lysates (10–100 μg/lane) or immunoprecipitates (300–700 μg total proteins) were separated by 8 or 10 % SDS-PAGE prior to electrophoretic transfer onto Hybond™-C super nitrocellulose membranes (Amersham). After blocking with 5 % nonfat milk in phosphate-buffered saline-Tween®20 buffer at room temperature for 1 hour, membranes were sequentially blotted with the indicated specific primary antibodies and then with horseradish peroxidase-conjugated secondary mouse or rabbit Abs (Santa Cruz Biotechnology (Heidelberg, Germany)) and were developed using chemiluminescence (Amersham, Arlington Heights, USA) [33].
Reverse transcription PCR
Reverse transcription PCR was performed as described previously [34]. Specifically, total RNA was purified with the RNeasy® Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions, and then reverse transcribed and synthesized to cDNA using Omniscript® reverse transcriptase (Qiagen). PCR amplification was performed using Taq DNA polymerase (Qiagen). The primers used to amplify human MCL1 were 5′-ATCTCTCGGTACCTTCGGGAGC-3′ (sense) and 5′-CCTGATGCCACCTTCTAGGTCC-3′ (antisense). The primers for human β-ACTIN were 5′-CTGGGACGACATGGAGAAAA-3′ (sense) and 5′-AAGGAAGGCTGGAAGAGTGC-3′ (antisense). The thermal cycle profile consisted of denaturing at 94 °C for 45 seconds, annealing at 60 °C for 45 seconds, and extension at 72 °C for 40 seconds. The samples were amplified for 30 cycles. We used 8 pmol of primers. The integrity of mRNA of all samples was confirmed by amplification of β-actin. PCR products were separated on a 1 % agarose gel and photographed.
Mitochondrial isolation
Extracts of cytoplasmic and mitochondrial fractions were prepared using the Qproteome Mitochondria Isolation Kit (Qiagen), according to the manufacturer’s instructions.
Proliferation assay
Proliferation was measured by the incorporation of 0.5 μCi/well [3H]-thymidine (Perkin Elmer, Baesweiler, Germany) during the last 8 hours of 72-hour experiments. Radioactive labeling was determined by harvesting the cells onto glass-fiber filtermates (Perkin Elmer) with an automatic cell harvester (Harvester 96, Tomtec Inc., Hamden, CT, USA) and counting using the Wallac Trilux Betaplate scintillation counter (Perkin Elmer).
Annexin V apoptosis detection
Cells were treated as indicated, then washed with phosphate-buffered saline and costained with fluorescein isothiocyanate (FITC)-labeled Annexin V and propidium iodide (PI) using the FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, San Diego, CA, USA) following the manufacturer’s instructions. Apoptosis was analyzed on a FACS Canto II flow cytometer (BD Biosciences).
Spheroid formation assay
Single multicellular BC spheroids were formed as described previously [35]. Briefly, 4 × 103–6 × 103 BC cells were seeded on agarose-coated (Sigma Aldrich, Steinheim, Germany) 96-well plates. At the end of the experiments, spheroids were stained with Calcein AM (green) (Sigma Aldrich, Steinheim) and PI (red) (Sigma Aldrich, Steinheim). For image capturing, the Olympus CKX41 microscope using 4× and 10× magnification was connected to an Olympus XC30 digital camera and exported to the cellSens standard software (version 1.4) (Olympus Corporation, Shinjuku, Tokyo, Japan). Based on the automatic measurement of the radius, volumes of the spheroids were calculated (V = 4/3 πr3).
Isobologram analysis
The interaction between EU-5346 and trastuzumab was analyzed using the CompuSyn software program (ComboSyn, Inc., http://www.combosyn.com/), which is based on the Chou-Talalay method [36]. When combination index (CI) = 1, this equation represents the conservation isobologram and indicates an additive effect. CI <1 indicates synergism; CI >1 indicates antagonism.
Statistical analysis
Statistical significance of differences observed in treated versus control cultures was determined by means of an unpaired Student t test. Statistical analyses were performed using SigmaPlot version 12.5 software (Systat Software GmbH, D-40699 Erkrath, Germany). The minimal level of significance was p <0.05.
Results
Mcl-1 expression correlates with improved adaptation of Her2-positive BC cell lines to hypoxia
Given the prognostic adverse role of hypoxia in BC, we first evaluated whether there are differences in the adaptation between cell lines representing different BC subtypes [37, 38] to hypoxia. Our results demonstrate improved adaptation of Her2-positive BC cells (BT-474, HCC-1954, JIMT-1) versus TNBC (MDA MB 231, MDA MB-468, MDA MB-157), Her2-negative cells (MCF-7), and a nonmalignant breast cell line (MCF-10A) to hypoxic conditions utilizing both MTT (Fig. 1a) and [3H]-thymidine uptake assays (data not shown). Previous studies have indicated a key role of the Bcl-2 family of anti-apoptotic proteins, and Mcl-1 in particular, in BC cell survival. Indeed, Her2 and Mcl-1 expression correlates in BC cells under normoxic conditions [29]. Whether Mcl-1 contributes to the adaptation of BC cells to hypoxia in general, and Her2-positive BC cells in particular, is currently unknown. Our results show consistently high Mcl-1, but variable Bcl-2 and Bcl-xL protein levels in a cell line panel of different BC subtypes, including Luminal A-like (MCF-7), Luminal B-like/Her2-positive (BT-474), Her2-positive (HCC-1954, SKBR3), and TNBC cells (MDA MB-231) [37, 38] under hypoxic conditions (Fig. 1b). In contrast, Mcl-1 protein levels were low in the benign MCF-10A BC cell line. Moreover, Mcl-1 protein levels of BC exposed to hypoxia were transiently increased 2.41-fold (standard deviation (SD) ±0.44), 2.57-fold (SD ±0.34), and 3.31-fold (SD ±0.31) in Her2-positive BT474, SKBR3, and HCC-1954 BC cells, respectively. Increases in Mcl-1 protein levels correlated with increases in protein levels of Hif-1α (Fig. 1c). To determine whether changes in Mcl-1 protein levels were due to elevated transcription or post-transcriptional regulation, we next detected MCL1 mRNA levels over a period of 24 hours using semiquantitative reverse transcription PCR. Our results show that mRNA levels of MCL1 did not change over time, indicating a transient hypoxia-mediated increase in Mcl-1 stability (Additional file 1: Figure S1). Taken together, these data suggest a role for Mcl-1 in BC cell adaptation to hypoxia in general, and Her2-positive BC cells in particular.

Mcl-1 expression correlates with improved adaptation of Her2-positive BC cell lines to hypoxia. a Improved adaptation to hypoxic conditions of Her2-positive versus Her2-negative BC cells and the benign MCF-10A cell line. BC cell lines were incubated under hypoxic conditions for the time periods indicated. Cell survival was analyzed by MTT assay. Data represent mean ± SD for triplicate samples. Results shown are representative of three independent experiments. b Significant protein levels of Mcl-1, but not Bcl-2 or Bcl-xL, in BC cells under hypoxic conditions. BC cell lines or the nonmalignant MCF-10A cells were incubated for 4 hours under hypoxic conditions and whole-cell extracts were analyzed by immunoblotting with indicated antibodies. Immunoblotting for Erk2 confirmed equal protein loading. c Upregulation of Mcl-1 in Her2-positive BC cells under hypoxic conditions. BC cell lines were incubated under hypoxic conditions for up to 24 hours. Whole-cell extracts were analyzed by immunoblotting with indicated antibodies. Immunoblotting for Erk2 confirmed equal protein loading. Densitometric measurements (NIH ImageJ software) [59] were used to quantitate Mcl-1 protein levels from western blot images normalized for Erk2. Densitometric data represent mean values ± SD. Data shown are representative of three independent experiments. C normoxic control, Erk2 extracellular signal-regulated kinase 2, Her2 human epidermal growth factor receptor 2, Hif hypoxia-inducible factor, Mcl-1 myeloid cell leukemia-1
Mcl-1 is an upstream regulator of Her2 under hypoxic conditions
To evaluate a potential functional interrelation of Mcl-1 and Her2 in Her2-positive BC cells under hypoxic conditions, we next transfected the Her2-positive BC cell lines BT-474 and SKBR3 with a pool of siRNA directed against the MCL1 gene (siMCL1) and tested for both Her2 and Hif-1α protein levels. Our data demonstrate marked downregulation of Her2 and Hif-1α in Her2-positive BC cells depleted of Mcl-1. Moreover, silencing of MCL1 induced cell death as evidenced by PARP cleavage (Fig. 2a). Surprisingly, in contrast to previous reports which show Her2-dependent Mcl-1 regulation under normoxic conditions [24, 25, 29], siRNA-mediated knockdown of the HER2 gene (siHER2) in Her2-positive BC cells did not affect Mcl-1 protein levels under hypoxic conditions (Fig. 2b). CoCl2 binds to the iron center of the Hif-specific hydroxylase. It thereby inhibits the hydroxylation of Hif, its ubiquitination by pVHL, and proteasomal degradation. In agreement with our hypothesis that Mcl-1 regulates Her2 and downstream Hif-1α protein levels in BC cells under hypoxic conditions, treatment with CoCl2 increased Hif-1α protein levels in a dose-dependent (Fig. 2c) and time-dependent (Fig. 2d) manner, without changing Mcl-1 or Her2 levels. Moreover, treatment of Her2-positive BC cells but not Her2-negative BC cells with the Her2 inhibitors trastuzumab (Fig. 2e) and lapatinib (Fig. 2f) under hypoxic conditions induced marked downregulation of Hif-1α, but not Mcl-1. These data correlated with the induction of cell death, as evidenced by PARP cleavage (Fig. 2e,f). Importantly, treatment with the pan-caspase inhibitor Z-VAD-FMK abrogated PARP cleavage induced by siMCL1, but not the downregulation of Her2 and Hif-1α in BC cells. These data exclude a caspase-mediated off-target effect of siMCL1 on Her2 protein levels (Fig. 2g). Taken together, these data confirm a role for Mcl-1 upstream of Her2 and Hif-1α under hypoxic conditions.

Mcl-1 is an upstream regulator of Her2 under hypoxic conditions. a Genetic depletion of Mcl-1 in Her2-positive BC cells promotes downregulation of Her2 and Hif-1α followed by inhibition of BC cell survival. b Mcl-1 protein levels do not decrease after genetically downregulating Her2. a, b BC cells were transfected with siMCL1 a or siHER2 b for 2 days and then exposed to hypoxia for 6 hours. c, d CoCl2-mediated stabilization of Hif-1α does not alter Mcl-1 or Her2 levels. BT-474 cells were exposed to CoCl2with indicated doses for 24 hours c and 100 μM CoCl2 for the indicated time periods d. e, f Pharmacologically targeting Her2 decreases Hif-1α but not Mcl-1 protein levels. BC cells were treated with the indicated doses of trastuzumab e or lapatinib f for 2 days and then exposed to hypoxia for 6 hours. g siMCL1-induced downregulation of Her2 and Hif-1α is not triggered by caspase-mediated off-target effects of MCL1 siRNA. Her2-positive BC cells were transfected with siMCL1 for 2 days and then exposed to hypoxia for 6 hours. ZVAD (50 μM) was added 24 hours before collecting samples. a–g Whole-cell extracts were analyzed by immunoblotting with indicated antibodies. Immunoblotting for Erk2 confirmed equal protein loading. Erk2 extracellular signal-regulated kinase 2, Her2 human epidermal growth factor receptor 2, Hif hypoxia-inducible factor, kD kilodalton, Mcl-1 myeloid cell leukemia-1, PARP Poly (ADP-ribose) polymerase
The novel small molecule inhibitor EU-5346 specifically blocks Mcl-1 followed by cell death
Our own and other studies show that blocking the effects of Mcl-1 is a promising approach to slow tumor growth, induce apoptosis, and overcome drug resistance in BC cells, and Her2-positive BC cells in particular. One approach to target Mcl-1 functions is the inhibition of its interaction with pro-apoptotic oncogenes of the Bcl-2 family. Using ultrahigh-throughput screening of 315,000 compounds coupled with the hit optimization strategy, the hydroxyquinoline-derived, small molecule EU-5346 (also ML311; Eutropics Pharmaceuticals, Cambridge, MA, USA) was identified as a potent inhibitor of Mcl-1/Bim interaction that shows selective activity against Mcl-1 primed cells [39]. In contrast, only the Bcl-2-specific BH3-mimetic navitoclax but not EU-5346 caused cell death in highly primed Bcl-2 cells [40]. Our own results demonstrate that EU-5346 induces apoptosis in Mcl-1wt/wt but not in Mcl-1Δ/null MEFs (Fig. 3a). Moreover, EU-5346 induced cell death in Bcl-2/Bcl-xL double knockout cells that were established as having Mcl-1 as the dependent anti-apoptotic Bcl-2 family protein [40]. Having verified the presence of Bcl-2 (Additional file 1: Figure S2a), the potent and highly selective Bcl-2 inhibitor ABT-199 [41] induced apoptosis in both Mcl-1wt/wt and Mcl-1Δ/null MEFs (Fig. 3b). These data indicate the specificity of EU-5346 against Mcl-1. Similar to siRNA-mediated knockdown of Mcl-1, EU-5346 induced marked downregulation of Her2 and Hif-1α in SKBR3 BC cells followed by cell death, as evidenced by PARP cleavage (Fig. 3c) and inhibition of [3H]-thymidine uptake (Fig. 3d). Treatment with the pan-caspase inhibitor Z-VAD-FMK abrogated PARP cleavage induced by EU-5346-mediated inhibition of Mcl-1, but not the downregulation of Her2 and Hif-1α in BC cells (Fig. 3c). Moreover, semiquantitative reverse transcription PCR did not show EU-5346-induced changes of mRNA levels of Mcl-1, supporting its role as a BH3-mimetic (Additional file 1: Figure S2b). Taken together these data strongly indicate a role for EU-5346 as a specific inhibitor of Mcl-1 in Her2-positive BC cells.

The novel small molecule inhibitor EU-5346 specifically blocks Mcl-1 followed by cell death. a, b EU-5346 induces apoptosis in Mcl-1wt/wt but not Mcl-1Δ/null MEFs a. In contrast, ABT-199 induces apoptosis in both Mcl-1wt/wt and Mcl-1Δ/null MEFs b. MEFs were treated with indicated concentrations of EU-5346 a or ABT-199 b for 72 hours prior to Annexin V and PI staining. Data represent mean ± SD for triplicate samples. Results shown are representative of three independent experiments. c EU-5346-induced downregulation of Her2 and associated cell death are not triggered by caspase-mediated off-target effects of Mcl-1 siRNA. Her2-positive BC (SKBR3) cells were treated with EU-5346 and/or ZVAD (50 μM) and exposed to hypoxia during the last 6 hours. d EU-5346 inhibits proliferation of Her2-positive BC cells in a dose-dependent manner. BC cells were treated with EU-5346 for 3 days under hypoxic conditions. [3H]-thymidine was added during the last 8 hours. Data represent mean ± SD for triplicate samples. Results shown are representative of three independent experiments. c, d Whole-cell extracts were analyzed by immunoblotting with indicated antibodies. Immunoblotting for Erk2 confirmed equal protein loading. Erk2 extracellular signal-regulated kinase 2, Her2 human epidermal growth factor receptor 2, Hif hypoxia-inducible factor, kD kilodalton, Mcl-1 myeloid cell leukemia-1, MEF murine embryonic fibroblast, PARP Poly (ADP-ribose) polymerase
Her2 co-localizes with Mcl-1 within the mitochondrial fraction
To further support the proposed functional interrelation of Mcl-1 and Her2, we next explored whether Mcl-1 interacts with Her2 under hypoxic conditions. It is well established that Her2 localizes to the cell membrane where it phosphorylates downstream substrates on their tyrosine residues in response to extracellular stimulation. Surprisingly, recent studies found that Her2 also localizes to the mitochondrial fraction of BC cells, where it promotes resistance to hypoxia and E-twenty-six (ETS) transcription factor inhibitors [42]. We therefore next sought to investigate whether Her2 and Mcl-1 colocalize in the mitochondrial fraction. Indeed, using western blot analysis our results show colocalization of Mcl-1 and Her2 in the mitochondrial fraction. Probing of western blot membranes with antibodies against prohibitin-1 and α-tubulin secured the high purity of the mitochondrial fraction (Fig. 4a).

Her2 colocalizes with Mcl-1 within the mitochondrial fraction. a Mcl-1 and Her2 colocalize within the mitochondrial fraction. Mitochondrial and cytosolic proteins were isolated using the Qproteome Mitochondria Isolation Kit (Qiagen) according to the manufacturer’s instructions and analyzed by immunoblotting with indicated antibodies. Anti-Prohibitin 1 was used as a mitochondrial marker and anti-tubulin was used as a cytoplasmic marker. BC cells were exposed to hypoxia for 6 hours. b EU-5346 induces ubiquitination of Mcl-1-bound Her2. SKBR3 cells were treated with EU-5346 for 3 days and exposed to hypoxia during the last 6 hours. Whole-cell extracts were immunoprecipitated (IP) with either Mcl-1 antibody or trastuzumab and analyzed by immunoblotting with indicated antibodies. Nonspecific protein binding and detection were excluded by incubating protein A-Sepharose beads with lysis buffer and Mcl-1 antibody only (left panel). Input (right panel). C control, Erk2 extracellular signal-regulated kinase 2, Her2 human epidermal growth factor receptor 2, IgH heavy chain, kD kilodalton, Mcl-1 myeloid cell leukemia-1
Importantly, our results show that Mcl-1 coimmunoprecipitates with Her2 (Fig. 4b) and that Mcl-1 increases the stability of Her2 by modulating the Her2 protein degradation mechanism. Increased ubiquitination of Her2 was more pronounced in siMCL1-treated (data not shown) and EU-5346-treated BC cells than in control cells (Fig. 4b). Receptor internalization is often associated with receptor ubiquitination and targeting to proteasomes for degradation [43]. Indeed, besides increased Her2 ubiquitination, treatment of SKBR3 cells with EU-5346 (Fig. 4b) as well as siMCL1 (data not shown) resulted in the formation of a 125–130 kD fragment of Her2 that was associated with a reduction in full-length Her2.
Taking these results together, the specific Mcl-1 inhibitor EU-5346 decreases Her2 protein levels. Functionally, our data show that the interaction of Mcl-1 and Her2 mediates protection of Her2 ubiquitination and subsequent degradation. Conversely, blockade of Mcl-1 activity increases Her2 ubiquitination and degradation.
Synergistic effects of combining trastuzumab with Mcl-1-targeting approaches in trastuzumab-sensitive Her2-positive BC cells
In Her2 inhibitor-sensitive Her2-positive BC cells, siMCL1 induced significantly higher rates of cell death when compared with siHER2, indicating the existence of Mcl-1-dependent survival pathways in Her2-positive BC cells, which are independent of the Mcl-1–Her2 axis (Fig. 5a). In agreement with these data, siMCL1 significantly reduced spheroid formation in both BT-474 (Fig. 5b) and SKBR3 (data not shown) cells. Moreover, the combination therapy of trastuzumab with EU-5346 synergistically triggered cell death in BT-474 (Fig. 5c), as determined by [3H]-thymidine uptake and subsequent analysis according to the Chou-Talalay method.

Synergistic effects of combining trastuzumab with Mcl-1-targeting approaches in trastuzumab-sensitive Her2-positive BC cells. a Genetic depletion of Mcl-1 or Her2 induces cell death in Her2-positive BC cells, but not benign breast cells (MCF-10A). BC and MCF-10A cells were transfected with siMCL1 (filled bars) or siHER2 (open bars) for 30 hours and then exposed to hypoxia for 2 days. Cell survival was determined by AlamarBlue® assay. Data represent mean ± SD for triplicate samples. Results shown are representative of three independent experiments. b Synergistic effects of combining trastuzumab with siMCL1 in Her2-positive BC cells. BT-474 cells (5.5 × 103 cells) were seeded per well in agar-coated 96-well plates prior to siMCL1 transfection. Spheroid formation was assessed in siMCL1-treated and/or trastuzumab-treated and control cells using an inverted fluorescence light microscope. Photographs (10× magnification) of spheroid formation are representative of each group and three independent experiments (left). Spheroid volumes were calculated as described in Materials and methods. Data represent mean ± SD (right). c Synergistic growth inhibition of trastuzumab and EU-5346 in trastuzumab-sensitive Her2-positive BC cells under hypoxic conditions. BT-474 cells were pretreated with trastuzumab or dimethyl sulfoxide (DMSO) for 24 hours under normoxic conditions followed by EU-5346 treatment or DMSO for 72 hours under hypoxic conditions. [3H]-thymidine was added during the last 8 hours. Data represent mean ± SD for triplicate samples. Results shown are representative of three independent experiments. Trastuzumab (white bars), EU-5346 (gray bars), and drug combination (black bars). Combination index (CI) was calculated using the Combosyn software as described in Materials and methods. a, b *p <0.05 and **p <0.001 by Student’s t test
Taken together, our results indicate the existence of Mcl-1-dependent survival pathways in Her2-positive BC cells, which are independent of the Mcl-1–Her2 axis; and support the therapeutic benefit of combining Her2 and Mcl-1 inhibitors.
Genetically and pharmacologically targeting Mcl-1 induces cell death also in trastuzumab-resistant Her2-positive BC cells
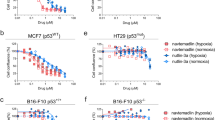
All Her2-positive BC patients eventually develop resistance to trastuzumab and other Her2 inhibitors. The precise mechanism of resistance against these inhibitors remains elusive. We next investigated whether genetically and pharmacologically targeting Mcl-1 is also active in trastuzumab-resistant HCC-1954 cells. Similar to trastuzumab-sensitive BT-474 and SKBR3 BC cells (Figs. 2 and 3), both siMCL1 and EU-5346 treatment induced cell death in HCC-1954 cells, as evidenced by the induction of PARP cleavage (Fig. 6a), inhibition of [3H]-thymidine uptake (Fig. 6b), and inhibition of spheroid formation (Fig. 6c, d). As expected, in contrast to BT-474 cells no synergistic effects of combining trastuzumab with siMCL1 (Fig. 6d) or EU-5346 (data not shown) were observed on spheroid formation (Fig. 6d) or [3H]-thymidine uptake followed by the Cou-Talalay analysis (data not shown). Similar results were obtained in primary BC cells isolated from a patient with advanced disease and resistant against multiple agents including Her2 inhibitors, capecitabine, paclitaxel, eribulin, and everolimus. Specifically, siMCL1 (Fig. 6e) and EU-5346 (Fig. 6f,g) but not siHER2 (Fig. 6e) induced cell death as evidenced by PARP cleavage (Fig. 6e,f) and dose-dependent inhibition of [3H]-thymidine uptake (Fig. 6g).

Genetically and pharmacologically targeting Mcl-1 induces cell death also in trastuzumab-resistant Her2-positive BC cells. a Both genetic depletion of Mcl-1 (siMCL1) and EU-5346 induces apoptosis in trastuzumab-resistant HCC-1954 BC cells. BC cells were transfected with siMCL1 (left) for 2 days or treated with EU-5346 (right) for 3 days and then exposed to hypoxia for 6 hours. b EU-5346 inhibits proliferation in Her2 inhibitor-sensitive and inhibitor-resistant BC cells in a dose-dependent manner. c EU-5346 inhibits spheroid formation in both trastuzumab-sensitive (BT-474) and trastuzumab-resistant (HCC-1954) BC cells. d Combining trastuzumab with siMCL1 in Her2 inhibitor-resistant Her2-positive BC cells does not induce synergistic effects. HCC-1954 cells (5.5 × 103 cells) were seeded per well in agar-coated 96-well plates prior to siMCL1 transfection. c, d Spheroid formation was assessed in EU-5346-treated or siMCL1-treated and/or trastuzumab-treated and control cells using an inverted fluorescence light microscope. Photographs of spheroid formation (10× magnification) are representative of each group and three independent experiments (left). Spheroid volumes were calculated as described in Materials and methods. Data represent mean ± SD (right). *p <0.05 and **p < 0.001 by Student’s t test. e, f siMCL1 e and EU-5346 f but not siHER2 e induces apoptosis in multidrug/Her2 inhibitor-resistant patient BC cells. BC cells were transfected with siMCL1 or siHER2 e for 2 days or treated with EU-5346 f for 3 days and then exposed to hypoxia for 6 hours. a, e, f Whole-cell extracts were analyzed by immunoblotting with indicated antibodies. Immunoblotting for Erk2 confirmed equal protein loading. g EU-5346 inhibits proliferation in multidrug/Her2 inhibitor-resistant patient BC cells in a dose-dependent manner. b, g BC cells were treated with EU-5346 for 3 days under hypoxic conditions. [3H]-thymidine was added during the last 8 hours. Data represent mean ± SD for triplicate samples. Results shown are representative of three independent experiments. Erk2 extracellular signal-regulated kinase 2, Her2 human epidermal growth factor receptor 2, Hif hypoxia-inducible factor, kD kilodalton, Mcl-1 myeloid cell leukemia-1, PARP Poly (ADP-ribose) polymerase
Taken together these data demonstrate that targeting Mcl-1 has therapeutic potential not only in Her2 inhibitor-sensitive but also in Her2 inhibitor-resistant BC cells.
Genetically and pharmacologically targeting Mcl-1 induces cell death in brain-primed Her2 inhibitor-resistant Her2-positive BC cells
Brain metastasis is an end stage in BC progression. Compared with hormone receptor (HR)-positive BC patients, Her2-positive BC patients show a high incidence of brain metastasis of more than 20 % [44], with an overall survival of 4-6 months with whole brain radiotherapy to about 18 months with multimodal therapies [45]. Although trastuzumab is effective for systemic BC, its efficacy in brain metastasis remains controversial [46]. Moreover, the incidence of brain metastatic disease after adjuvant trastuzumab is increasing with the improved management of systemic disease and prolongation of survival [11, 47]. We next sought to investigate the anti-BC activity of targeting Mcl-1 in brain-primed (JIMT-1BR3) versus maternal Her2 JIMT-1 cells. Our data demonstrate marked downregulation of Her2 and Hif-1α in both JIMT-1 and JIMT-1BR3 cells after siRNA-mediated Mcl-1 downregulation (Fig. 7a). Moreover, silencing of Mcl-1 induced cell death as evidenced by PARP cleavage (Fig. 7a), and AlamarBlue® assay (Fig. 7b). Importantly, similar to maternal JIMT-1 cells, EU-5346 but not trastuzumab and lapatinib induced cell death in brain-primed JIMT-1 BR3 cells, as evidenced by PARP cleavage (Fig. 7c) and [3H]-thymidine uptake (Fig. 7d). Taken together, these data indicate a therapeutic role of targeting Mcl-1 in BC brain metastasis.

Genetically and pharmacologically targeting Mcl-1 induces cell death in brain-primed Her2 inhibitor-resistant Her2-positive BC cells. a, b Genetic depletion of Mcl-1 induces apoptosis in both maternal Her2 inhibitor-resistant Her2-positive JIMT-1 cells as well as in brain-primed JIMT-1 BR3 cells. a BC cells were transfected with siMCL1 for 2 days and exposed to hypoxia for 6 hours. Whole-cell extracts were analyzed by immunoblotting with indicated antibodies. Immunoblotting for Erk2 confirmed equal protein loading. b BC cells were transfected with siMCL1 for 30 hours and then exposed to hypoxia for 2 days. Cell survival was determined by AlamarBlue® assay. Data represent mean ± SD for triplicate samples. Results shown are representative of three independent experiments. c EU-5346 overcomes Her2 inhibitor resistance in brain-primed Her2-positive BC cells. BC cells were treated with indicated drugs for 3 days and then exposed to hypoxia during the last 6 hours. Whole-cell extracts were analyzed by immunoblotting with indicated antibodies. Immunoblotting for Erk2 confirmed equal protein loading. d EU-5346 inhibits proliferation of brain-primed Her2-positive BC cells in a dose-dependent manner. BC cells were treated with EU-5346 for 3 days under hypoxic conditions. [3H]-thymidine was added during the last 8 hours. Data represent mean ± SD for triplicate samples. Results shown are representative of three independent experiments. C control (dimethyl sulfoxide), E EU-5346, Erk2 extracellular signal-regulated kinase 2, Her2 human epidermal growth factor receptor 2, Hif hypoxia-inducible factor, kD kilodalton, L lapatinib, Mcl-1 myeloid cell leukemia-1, T trastuzumab
Discussion
The present study demonstrates a critical role of Mcl-1 predominantly in Her2-positive BC cell survival and proliferation under hypoxic conditions. Under normoxic conditions, Her2 regulates Mcl-1 upregulation. Conversely, trastuzumab treatment reduces Mcl-1 levels and renders cells sensitive to chemotherapy [29]. Surprisingly, in contrast to normoxia our data demonstrate a role for Mcl-1 upstream of Her2 in BC cells under hypoxic conditions. Functionally, we show that under hypoxic conditions Her2 is detected also in the mitochondrial fraction, where it binds Mcl-1, strongly indicating a functional interrelation of these two proteins in BC pathogenesis.
To date, therapeutic BH3 mimetics that abrogate anti-apoptotic activity have been predominantly developed to target Bcl-2 and Bcl-xL. They function by slotting into the hydrophobic groove on the surface of Bcl-2 and Bcl-xL, thereby blocking their capacity to inhibit apoptosis [48]. Based on their promising preclinical activities, Bcl-2/Bcl-xL inhibitor ABT-737 [48] and its oral derivate navitoclax/ABT-263 [49], as well as the highly selective Bcl-2 inhibitor ABT-199 [41], are currently validated in advanced clinical trials in solid tumors, acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), and non-Hodgkin lymphoma (NHL) (https://clinicaltrials.gov). Similar to previous data, our own results show consistently high Mcl-1 but variable Bcl-2 and Bcl-xL protein levels in cell lines of different BC subtypes. It is therefore clear that inhibitors of Mcl-1 hold great promise as a new class of targeting agents and are the current focus of widespread cancer drug development efforts. However, the clinical development of Mcl-1 inhibitors has been challenged by structural differences between the BH3-binding grooves of Mcl-1 and Bcl-2. Indeed, because of low affinities, ABT-737, navitoclax, and ABT-199 do not block Mcl-1 activity [28, 48, 49]. The high interest towards the identification of Mcl-1 inhibitors has resulted in the recent development of several small molecule BH3 mimetics, which disrupt Mcl-1 function with higher affinity and overcome resistance to available Bcl-2 family inhibitors [30, 50]. To date there are no viable clinical candidates. Here we evaluated the novel hydroxychinoline-derived small molecule inhibitor EU-5346 for its activity in Her2-positive BC cells under hypoxic conditions. Our data demonstrate EU-5346-induced cell death of Her2-positive BC cells and that Mcl-1 is required for cell killing. In addition we have found that, functionally, EU-5346 triggers ubiquitination of Mcl-1-bound Her2, indicating a previously unknown role for Mcl-1 to stabilize Her2 protein levels. Studies which delineate detailed molecular mechanisms by which Mcl-1 protects Her2 against ubiquitination and concomitant degradation are currently ongoing. In addition to Mcl-1/Her2-dependent survival pathways, our results indicate the existence of Mcl-1-dependent survival pathways in Her2-positive BC cells, which are independent of the Mcl-1–Her2 axis (Fig. 8). Importantly, siRNA-mediated knockdown of Mcl-1 and EU-5346 induced cell death also in BC cell lines resistant against Her2 inhibitors, trastuzumab and lapatinib in particular (Fig. 8); as well as in brain-primed Her2-positive BC cells resistant to Her2 inhibitors.

Summary: Mcl-1 confers protection of Her2-positive BC cells to hypoxia—therapeutic implications. Homodimerization of Her2 with Her2 or heterodimerization of Her2 with Her3 enhances phospho-inositol 3-kinase (PI3K)/AKT and RAS/mitogen-activated protein kinase (MAPK) pathways, which regulate BC cell proliferation, survival, and migration, as well as angiogenesis [60, 61]. The Her2 inhibitor trastuzumab [62] binds to the C-terminal portion of Her2. Another Her2 inhibitor, lapatinib [63], binds to the ATP binding site of Her2, but also Her1. Similar to siHER2, trastuzumab and lapatinib inhibit downstream signaling events, induce apoptosis, and inhibit proliferation of BC cells (red lines). Here we show a novel role for the antiapoptotic Bcl-2 family member Mcl-1 in Her2-positive BC cell adaptation to hypoxia. Specifically, our results show that Mcl-1 forms a protein complex with Her2 at the mitochondrial membrane and stabilizes Her2 by inhibiting its ubiquitination. Conversely, genetically (siMCL1) or pharmacologically (EU-5346) targeting Mcl-1 triggers ubiquitination and proteosomal degradation of Her2, thereby inducing apoptosis, and inhibiting proliferation and spheroid formation under hypoxic conditions. In addition, our results indicate the existence of a Mcl-1-dependent survival pathway in Her2-positive BC cells, which is independent of the Mcl-1–Her2 axis supporting the therapeutic benefit of combining Her2 (red lines) and Mcl-1 inhibitor (blue lines). Importantly, based on these findings, targeting Mcl-1 is also active in Her2-positive BC cells resistant to Her2 inhibitors, including a brain-primed Her2-positive cell line. Her human epidermal growth factor receptor, Mcl-1 myeloid cell leukemia-1. (Color figure online)
Mcl-1 mediates inherent resistance against widely used anticancer therapies including paclitaxel [26] and gemcitabine [27], as well as BH3 mimetics. Moreover, Mcl-1 may also contribute to the acquired resistance to specific BH-3 mimetics [51]. Silencing of Mcl-1 resensitized tumor cells to ABT-737 and decreased their resistance to anoikis [52, 53]. Owing to their functional redundancy, pan-active Bcl-2 inhibitors with at least some activity against Mcl-1 or the combination of Mcl-1 with other Bcl-2/Bcl-xL inhibitors are likely to achieve higher response rates than targeting one individual member of the Bcl-2 subfamily.
The use of multiprotein indexes has been proposed to predict response rates to single-agent Mcl-1 inhibitors or their combination with other inhibitors of the Bcl-2 family [54, 55]. However, in vivo studies are needed to confirm the utility of these indexes. Currently we are evaluating the in vitro anti-BC activity of EU-5346 in combination with ABT-199, and navitoclax, as well as with paclitaxel and gemcitabine.
Finally, our own and other data demonstrate high Mcl-1 protein levels also in Her2-negative BC cells, Luminal A-like BC and TNBCs in particular [25, 55–57]. Based on this protein profile, we evaluated whether siMCL1 and EU-5346 induce cell death also in these BC subtypes. Indeed, our results demonstrate anti-BC activity in Luminal A-like (MCF-7) and TNBC (MDA MB-231) cells upon both siRNA-mediated downregulation of Mcl-1 (Additional file 1: Figure S3a,b) as well as treatment with EU-5346 (Additional file 1: Figure S3c,d), as evidenced by PARP cleavage (Additional file 1: Figure S3a,c), AlamarBlue® assays (Additional file 1: Figure S3b), and [3H]-thymidine uptake (Additional file 1: Figure S3d). Molecular mechanisms by which Mcl-1 induces cell death in these BC subtypes need to be further explored (Additional file 1: Figure S3e). Importantly, Balko et al. [58] recently demonstrated that TNBC cells residual after neoadjuvant chemotherapy express high levels of Mcl-1; and that Mcl-1 protects TNBCs from chemotherapy-induced apoptosis. These data support a therapeutic role of targeting Mcl-1 also in TNBC cells. Future investigations into whether a similar role for Mcl-1 also exists in other BC subtypes are of high interest.
Conclusion
In summary, the present study adds another facet to the critical pathophysiologic role of Mcl-1 in BC pathogenesis in general, and Her2-positive BC cells in particular. The study thereby supports the clinical development of therapies, which target Mcl-1 alone or in combination with other therapies, both under normoxic and hypoxic conditions in order to further improve patient outcome in BC.
Change history
02 April 2024
A Correction to this paper has been published: https://doi.org/10.1186/s13058-024-01811-y
Abbreviations
- BC:
-
Breast cancer
- BH3:
-
Bcl-2 homology 3
- CI :
-
Combination index
- DKFZ:
-
German Cancer Research Center
- DMEM:
-
Dulbecco’s modified Eagle’s medium
- ER:
-
Estrogen receptor
- Erk2:
-
Extracellular signal-regulated kinase 2
- FACS:
-
Fluorescence-activated cell sorting
- FBS:
-
Fetal bovine serum
- FITC:
-
Fluorescein isothiocyanate
- Her2:
-
Human epidermal growth factor receptor 2
- Hif:
-
Hypoxia-inducible factor
- HR:
-
Hormone receptor
- Mcl-1:
-
Myeloid cell leukemia-1
- MEF:
-
Murine embryonic fibroblast
- MEM:
-
Minimum essential medium
- MTT:
-
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- PARP:
-
Poly (ADP-ribose) polymerase
- PEST:
-
Proline, glutamic acid, serine, and threonine
- PI:
-
Propidium iodide
- PR:
-
Progesterone receptor
- SD:
-
Standard deviation
- siRNA:
-
Small interfering RNA
- TNBC:
-
Triple-negative breast cancer
References
Sørlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–74.
Inwald EC, Klinkhammer-Schalke M, Hofstädter F, Zeman F, Koller M, Gerstenhauer M, et al. Ki-67 is a prognostic parameter in breast cancer patients: results of a large population-based cohort of a cancer registry. Breast Cancer Res Treat. 2013;139:539–52.
Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013;3:27–34.
Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52.
Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006;7:96.
Eroles P, Bosch A, Pérez-Fidalgo JA, Lluch A. Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat Rev. 2012;38:698–707.
Sahlberg KK, Hongisto V, Edgren H, Mäkelä R, Hellström K, Due EU, et al. The HER2 amplicon includes several genes required for the growth and survival of HER2 positive breast cancer cells. Mol Oncol. 2013;7:392–401.
Yarden Y. Biology of HER2 and its importance in breast cancer. Oncology. 2001;61 Suppl 2:1–13.
Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707–12.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91.
Lin NU, Dieras V, Paul D, Lossignol D, Christodoulou C, Stemmler H-J, et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin Cancer Res. 2009;15:1452–9.
Lundgren K, Holm C, Landberg G. Hypoxia and breast cancer: prognostic and therapeutic implications. Cell Mol Life Sci. 2007;64:3233–47.
Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584:2981–9.
Germain M, Duronio V. The N terminus of the anti-apoptotic BCL-2 homologue MCL-1 regulates its localization and function. J Biol Chem. 2007;282:32233–42.
Akgul C, Turner PC, White MR, Edwards SW. Functional analysis of the human MCL-1 gene. Cell Mol Life Sci. 2000;57:684–91.
Yang T, Kozopas KM, Craig RW. The intracellular distribution and pattern of expression of Mcl-1 overlap with, but are not identical to, those of Bcl-2. J Cell Biol. 1995;128:1173–84.
Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74:609–19.
Del Gaizo MV, Letai A. BH3 profiling--measuring integrated function of the mitochondrial apoptotic pathway to predict cell fate decisions. Cancer Lett. 2013;332:202–5.
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–8.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905.
Ding Q, He X, Hsu J-M, Xia W, Chen C-T, Li L-Y, et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–17.
Placzek WJ, Wei J, Kitada S, Zhai D, Reed JC, Pellecchia M. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis. 2010;1:e40.
Booy EP, Henson ES, Gibson SB. Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene. 2011;30:2367–78.
Schacter JL, Henson ES, Gibson SB. Estrogen regulation of anti-apoptotic Bcl-2 family member Mcl-1 expression in breast cancer cells. PLoS One. 2014;9:e100364.
Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature. 2011;471:110–4.
Wei S-H, Dong K, Lin F, Wang X, Li B, Shen J-J, et al. Inducing apoptosis and enhancing chemosensitivity to gemcitabine via RNA interference targeting Mcl-1 gene in pancreatic carcinoma cell. Cancer Chemother Pharmacol. 2008;62:1055–64.
van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99.
Henson ES, Hu X, Gibson SB. Herceptin sensitizes ErbB2-overexpressing cells to apoptosis by reducing antiapoptotic Mcl-1 expression. Clin Cancer Res. 2006;12(3 Pt 1):845–53.
Belmar J, Fesik SW. Small molecule Mcl-1 inhibitors for the treatment of cancer. Pharmacol Ther. 2015;145:76–84.
Palmieri D, Duchnowska R, Woditschka S, Hua E, Qian Y, Biernat W, et al. Profound prevention of experimental brain metastases of breast cancer by temozolomide in an MGMT-dependent manner. Clin Cancer Res. 2014;20:2727–39.
Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–6.
Podar K, Gouill SL, Zhang J, Opferman JT, Zorn E, Tai Y-T, et al. A pivotal role for Mcl-1 in Bortezomib-induced apoptosis. Oncogene. 2008;27:721–31.
Fan F, Tonon G, Bashari MH, Vallet S, Antonini E, Goldschmidt H, et al. Targeting Mcl-1 for multiple myeloma (MM) therapy: drug-induced generation of Mcl-1 fragment Mcl-1(128-350) triggers MM cell death via c-Jun upregulation. Cancer Lett. 2014;343:286–94.
Friedrich J, Seidel C, Ebner R, Kunz-Schughart LA. Spheroid-based drug screen: considerations and practical approach. Nat Protoc. 2009;4:309–24.
Chou T-C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–6.
Subik K, Lee J-F, Baxter L, Strzepek T, Costello D, Crowley P, et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer. 2010;4:35–41.
Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13:215.
Bannister T, Koenig M, He Y, Mishra J, Spicer T, Minond D, Saldanha A, Mercer BA, Cameron M, Lena R, Carlson N, Richard D, Cardone M, Hodder P. ML311: A small molecule that potently and selectively disrupts the protein-protein interaction of Mcl-1 and Bim: a probe for studying lymphoid tumorigenesis. 2012 Apr 16 [Updated 2013 Mar 14]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010. Available from: http://www.ncbi.nlm.nih.gov/books/NBK143557/. Accessed 22 Feb 2016.
Richard DJ, Lena R, Bannister T, Blake N, Pierceall WE, Carlson NE, et al. Hydroxyquinoline-derived compounds and analoguing of selective Mcl-1 inhibitors using a functional biomarker. Bioorg Med Chem. 2013;21:6642–9.
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–8.
Ding Y, Liu Z, Desai S, Zhao Y, Liu H, Pannell LK, et al. Receptor tyrosine kinase ErbB2 translocates into mitochondria and regulates cellular metabolism. Nat Commun. 2012;3:1271.
Tikhomirov O, Carpenter G. Geldanamycin induces ErbB-2 degradation by proteolytic fragmentation. J Biol Chem. 2000;275:26625–31.
Aversa C, Rossi V, Geuna E, Martinello R, Milani A, Redana S, et al. Metastatic breast cancer subtypes and central nervous system metastases. Breast. 2014;23:623–8.
Kocher M, Soffietti R, Abacioglu U, Villà S, Fauchon F, Baumert BG, et al. Adjuvant whole-brain radiotherapy versus observation after radiosurgery or surgical resection of one to three cerebral metastases: results of the EORTC 22952-26001 study. J Clin Oncol. 2011;29:134–41.
Kodack DP, Chung E, Yamashita H, Incio J, Duyverman AMMJ, Song Y, et al. Combined targeting of HER2 and VEGFR2 for effective treatment of HER2-amplified breast cancer brain metastases. Proc Natl Acad Sci U S A. 2012;109:E3119–27.
Bendell JC, Domchek SM, Burstein HJ, Harris L, Younger J, Kuter I, et al. Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer. 2003;97:2972–7.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81.
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8.
Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR, et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512–7.
Vogler M, Butterworth M, Majid A, Walewska RJ, Sun X-M, Dyer MJS, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113:4403–13.
Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88.
Boisvert-Adamo K, Longmate W, Abel EV, Aplin AE. Mcl-1 is required for melanoma cell resistance to anoikis. Mol Cancer Res. 2009;7:549–56.
Al-Harbi S, Hill BT, Mazumder S, Singh K, Devecchio J, Choudhary G, et al. An antiapoptotic BCL-2 family expression index predicts the response of chronic lymphocytic leukemia to ABT-737. Blood. 2011;118:3579–90.
Goodwin CM, Rossanese OW, Olejniczak ET, Fesik SW. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death Differ. 2015;22:2098–106.
Thrane S, Pedersen AM, Thomsen MBH, Kirkegaard T, Rasmussen BB, Duun-Henriksen AK, et al. A kinase inhibitor screen identifies Mcl-1 and Aurora kinase A as novel treatment targets in antiestrogen-resistant breast cancer cells. Oncogene. 2015;6:4199–210.
Petrocca F, Altschuler G, Tan SM, Mendillo ML, Yan H, Jerry DJ, et al. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell. 2013;24:182–96.
Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4:232–45.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5.
Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–54.
Yamaguchi H, Chang S-S, Hsu JL, Hung M-C. Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene. 2014;33:1073–81.
Cuello M, Ettenberg SA, Clark AS, Keane MM, Posner RH, Nau MM, et al. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res. 2001;61:4892–900.
Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene. 2009;28:803–14.
Acknowledgements
The authors thank M Bucur (DKFZ) for technical advice and S Malvestiti for technical assistance. JIMT-1-BR3 cells (brain metastatic BC cell line) were a kind gift from Dr Patricia Steeg (Laboratory of Molecular Pharmacology, NCI’s Women's Malignancies Branch, National Cancer Institute/National Institutes of Health, Bethesda MD, USA). The authors thank Dr Frank Winkler (DKFZ) for helpful discussions. MHB is supported by the DAAD-Indonesian German Scholarship Programme (IGSP) for doctoral fellowship. KP is the recipient of a B. Braun Stiftungs-Grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
MA and MHC are employees of Eutropics, Inc. The remaining authors declare that they have no conflict of interest.
Authors’ contributions
MHB conceived of the study, designed and performed experiments, analyzed data, and wrote the manuscript. FF, SV, MA, CL-M, and FM performed experiments and participated in data analysis and interpretation. MS, AS, MHC, and JTO conceived of the study, and participated in data analysis and interpretation. HS-B, IZ, and DJ made substantial contributions to the acquisition and interpretation of data. KP conceived of the study, designed and coordinated experiments, analyzed and interpreted data, and wrote the manuscript. All authors were involved in revising the manuscript critically for important intellectual content. All authors approved the final version of the manuscript.
The original version of this article was revised: PARP blots of JIMT-1 and JIMT-1-BR3 cells in Figure 7C of the original publication were mistakenly duplicated. It has now been revised. The email address of the Corresponding Author has been updated.
Additional file
Additional file 1: Figure S1.
Showing MCL-1 mRNA levels do not change in Her2-positive BC cells under hypoxic conditions. Figure S2 showing a Bcl-2 is expressed in both Mcl-1wt/wt and Mcl-1Δ/null MEFs. Whole-cell extracts were analyzed by immunoblotting with indicated antibodies. Immunoblotting for Erk2 confirmed equal protein loading. b EU-5346 does not change Mcl-1 mRNA expression in Her2-positive BC cells under hypoxic conditions. Figure S3 showing Mcl-1 is a potential therapeutic target not only in Her2-positive, but also in Luminal A-like and TNBC cells under hypoxic conditions. (PDF 1580 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bashari, M.H., Fan, F., Vallet, S. et al. Mcl-1 confers protection of Her2-positive breast cancer cells to hypoxia: therapeutic implications. Breast Cancer Res 18, 26 (2016). https://doi.org/10.1186/s13058-016-0686-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-016-0686-4