
Abstract
Targeting the function of epidermal growth factor receptor (EGFR) has failed as an effective clinical option for breast cancer. Understanding the drivers of inherent resistance has been a challenge. One possible mechanism is the acquisition of stem-like properties through the process of epithelial-mesenchymal transition. A recent study by Seguin and colleagues adds to our understanding of this process by demonstrating a functional role for unligated αvβ3 integrin in mediating a stem-like phenotype and facilitating resistance to EGFR-targeted therapy via enhanced downstream coupling to a KRAS:RalB:NF-κB pathway. Importantly, the identified mechanism may reveal a possible strategy for sensitizing breast cancer cells to EGFR-targeted therapies.
Similar content being viewed by others


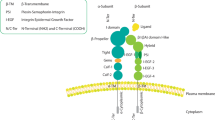
Background
Expression of epidermal growth factor receptor (EGFR) serves as a robust marker for the basal-like subtype of breast cancer (BC) and a biomarker for decreased survival times of BC patients [1],[2]. Moreover, numerous studies have established functional roles for epidermal growth factor/EGFR in facilitating the systemic dissemination of BC cells [3]–[5]. However, clinical trials targeting EGFR for the treatment of BC have produced disappointing results [6],[7]. These contrasting findings suggest that while BC cells utilize EGFR signaling to facilitate certain aspects of tumor progression, they become inherently resistant to inhibition of EGFR function during the later stages of disease progression. Defining the drivers of this type of inherent resistance has been difficult because, as opposed to acquired resistance, these cells are resistant prior to therapeutic intervention.
Integrin receptors are vital conduits that mediate cell-cell and cell-extracellular matrix interactions, as well as initiating intracellular signaling cascades promoting survival and migration. The complexity of signaling events that emanate from these receptors has prevented effective targeting of their function for the treatment of BC. β3 integrin is a well established marker of highly aggressive and metastatic BC [8]. Furthermore, aberrant expression of β3 integrin is strongly related to epithelial-mesenchymal transition [9], a process involved in several aspects of tumor metastasis [10], generation of a stem-like state [11] and resistance to EGFR-targeted therapies [12]. However, the functional role of β3 integrin in mediating a stem-like state and resistance to EGFR-targeted therapies has remained undefined.
The article
In a recent article published in Nature Cell Biology, Seguin and colleagues [13] demonstrate the importance of an αvβ3:KRAS:RalB:NF-κB signaling pathway in driving a stem-like state and resistance to EGFR-targeted therapies. In this work, the authors first demonstrate that patient-derived tumor tissues can be enriched in tumor-initiating potential based on expression of β3 integrin. Correspondingly, depletion of β3 integrin in tumor cell lines decreases stem-like cells as measured by several assays. Importantly, use of the nonadherent tumor sphere assay was the first indication that the role of β3 integrin in modulating tumor initiating capacity was independent of its role in cell adhesion. Next, the authors demonstrate that β3 integrin is necessary and sufficient to impart growth resistance to the EGFR inhibitor erlotinib. Interestingly, expression of β3 integrin had no impact on the response to nucleoside analogues or DNA crosslinking drugs, suggesting this result is restricted to targeted therapies. Moving downstream of β3 integrin the authors expand upon their previous findings [14] and show that the ability of β3 integrin to facilitate drug resistance does not function through the more traditional focal adhesion complex, but instead via a KRAS:RalB:TBK1:NF-κB pathway that is facilitated by galectin-3. Finally, the authors demonstrate that combining erlotinib with the proteasome inhibitor bortezomib can overcome β3 integrin-mediated resistance to EGFR inhibition (EGFRi).
Viewpoint
With the enhanced specificity and reduced toxicity of targeted molecular therapies comes the drawback of inherent and acquired resistance. As stated above, several lines of evidence point to EGFRi as an effective therapeutic strategy for BC, but these predications have not been borne out in the clinic. Together with previous findings, the current study suggests that targeting of galectin-3, RalB [15] or TBK1 [16] may actually be capable of inducing sensitivity to EGFRi therapies. Of particular importance is the treatment of triple-negative BCs, which currently lack any effective targeted therapeutic option. With regard to the other BC subtypes, whether this β3:NF-κB mechanism of resistance takes place during acquisition of resistance to HER2-targeted or endocrine-targeted therapies also remains to be determined [17]. Similar to EGFRi, vascular endothelial growth factor (VEGF)-targeted therapy has only had limited success in BC treatment. Several clinical trials currently underway are combining the VEGF-targeted agent bevacizumab with bortezomib, but further investigations into the role of β3 integrin:NF-κB in this resistance process are clearly warranted.
Mechanistically, the finding that β3 integrin induces stemness during the unligated state is very intriguing. Indeed, cilengitide is a cyclic peptide antagonist of αv binding that is currently in clinical trials for the treatment of multiple cancers. However, the current study demonstrates that this compound is unable to target the stem-like cells that drive drug resistance, suggesting it may not be an effective therapy for BC. Specificity remains a major drawback to compounds targeting the NF-κB pathway. Despite this, several ongoing clinical trials are exploring the combination of bortezomib with erlotinib and other targeted therapies [18]. Using β3 integrin as a histological marker, these clinical approaches can be evaluated for eradication of the stem-like cell population in the hopes of unlocking the potential of erlotinib as an effective BC therapy.
Abbreviations
- BC:
-
Breast cancer
- EGFR:
-
Epidermal growth factor receptor
- EGFRi:
-
Epidermal growth factor receptor inhibition
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Tischkowitz M, Brunet J-S, Bégin LR, Huntsman DG, Cheang MCU, Akslen LA, Nielsen TO, Foulkes WD: Use of immunohistochemical markers can refine prognosis in triple negative breast cancer. BMC Cancer. 2007, 7: 134-10.1186/1471-2407-7-134.
Livasy CA, Karaca G, Nanda R, Tretiakova MS, Olopade OI, Moore DT, Perou CM: Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod Pathol Off J U S Can Acad Pathol Inc. 2006, 19: 264-271.
Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J, Condeelis J: A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64: 7022-7029. 10.1158/0008-5472.CAN-04-1449.
Wendt MK, Smith JA, Schiemann WP: Transforming growth factor-β-induced epithelial-mesenchymal transition facilitates epidermal growth factor-dependent breast cancer progression. Oncogene. 2010, 29: 6485-6498. 10.1038/onc.2010.377.
Huang M, Anand S, Murphy EA, Desgrosellier JS, Stupack DG, Shattil SJ, Schlaepfer DD, Cheresh DA: EGFR-dependent pancreatic carcinoma cell metastasis through Rap1 activation. Oncogene. 2012, 31: 2783-2793. 10.1038/onc.2011.450.
Dickler MN, Cobleigh MA, Miller KD, Klein PM, Winer EP: Efficacy and safety of erlotinib in patients with locally advanced or metastatic breast cancer. Breast Cancer Res Treat. 2009, 115: 115-121. 10.1007/s10549-008-0055-9.
Dickler MN, Rugo HS, Eberle CA, Brogi E, Caravelli JF, Panageas KS, Boyd J, Yeh B, Lake DE, Dang CT, Gilewski TA, Bromberg JF, Seidman AD, D’Andrea GM, Moasser MM, Melisko M, Park JW, Dancey J, Norton L, Hudis CA: A phase II trial of erlotinib in combination with bevacizumab in patients with metastatic breast cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2008, 14: 7878-7883. 10.1158/1078-0432.CCR-08-0141.
Sloan EK, Pouliot N, Stanley KL, Chia J, Moseley JM, Hards DK, Anderson RL: Tumor-specific expression of alphavbeta3 integrin promotes spontaneous metastasis of breast cancer to bone. Breast Cancer Res. 2006, 8: R20-10.1186/bcr1398.
Galliher AJ, Schiemann WP: Beta3 integrin and Src facilitate transforming growth factor-beta mediated induction of epithelial-mesenchymal transition in mammary epithelial cells. Breast Cancer Res. 2006, 8: R42-10.1186/bcr1524.
Wendt MK, Tian M, Schiemann WP: Deconstructing the mechanisms and consequences of TGF-β-induced EMT during cancer progression. Cell Tissue Res. 2012, 347: 85-101. 10.1007/s00441-011-1199-1.
Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA: The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008, 133: 704-715. 10.1016/j.cell.2008.03.027.
Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong K-K, Brandstetter K, Wittner B, Ramaswamy S, Classon M, Settleman J: A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010, 141: 69-80. 10.1016/j.cell.2010.02.027.
Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, Yebra M, Mielgo A, Lowy AM, Husain H, Cascone T, Diao L, Wang J, Wistuba II, Heymach JV, Lippman SM, Desgrosellier JS, Anand S, Weis SM, Cheresh DA: An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition.Nat Cell Biol 2014, 16:457–468.,
Desgrosellier JS, Barnes LA, Shields DJ, Huang M, Lau SK, Prévost N, Tarin D, Shattil SJ, Cheresh DA: An integrin alpha(v)beta(3)-c-Src oncogenic unit promotes anchorage-independence and tumor progression. Nat Med. 2009, 15: 1163-1169. 10.1038/nm.2009.
Chien Y, Kim S, Bumeister R, Loo Y-M, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M, Yeaman C, Camonis JH, Zhao Y, White MA: RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006, 127: 157-170. 10.1016/j.cell.2006.08.034.
Li J, Huang J, Jeong J-H, Park S-J, Wei R, Peng J, Luo Z, Chen YT, Feng Y, Luo J-L: Selective TBK1/IKKi dual inhibitors with anticancer potency. Int J Cancer. 2014, 134: 1972-1980. 10.1002/ijc.28507.
Bailey ST, Miron PL, Choi YJ, Kochupurakkal B, Maulik G, Rodig SJ, Tian R, Foley KM, Bowman T, Miron A, Brown M, Iglehart JD, Biswas DK: NF-κB activation-induced anti-apoptosis renders HER2-positive cells drug resistant and accelerates tumor growth. Mol Cancer Res. 2014, 12: 408-420. 10.1158/1541-7786.MCR-13-0206-T.
Combination therapy of bortezomib with novel targeted agents: an emerging treatment strategy. []., [http://clincancerres.aacrjournals.org]
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Brown, W.S., Wendt, M.K. Integrin-mediated resistance to epidermal growth factor receptor-targeted therapy: an inflammatory situation. Breast Cancer Res 16, 448 (2014). https://doi.org/10.1186/s13058-014-0448-0
Published:
DOI: https://doi.org/10.1186/s13058-014-0448-0