
Abstract
Sepsis is characterized by a dysregulated immune response to infection leading to life-threatening organ dysfunction. Sepsis-induced liver injury is recognized as a powerful independent predictor of mortality in the intensive care unit. During systemic infections, the liver regulates immune defenses via bacterial clearance, production of acute-phase proteins (APPs) and cytokines, and metabolic adaptation to inflammation. Increased levels of inflammatory cytokines and impaired bacterial clearance and disrupted metabolic products can cause gut microbiota dysbiosis and disruption of the intestinal mucosal barrier. Changes in the gut microbiota play crucial roles in liver injury during sepsis. Bacterial translocation and resulting intestinal inflammation lead to a systemic inflammatory response and acute liver injury. The gut-liver crosstalk is a potential target for therapeutic interventions. This review analyzes the underlying mechanisms for the gut-liver crosstalk in sepsis-induced liver injury.
Similar content being viewed by others

Introduction
Sepsis is a life-threatening condition caused by dysregulated host response to infection [1]. Uncontrolled inflammation leads to loss of cellular and organ function, multiple organ dysfunction syndrome (MODS), and death [2]. Annually, more than 1.5 million patients suffer from sepsis in the USA, and the estimated global mortality rate is 25% [3, 4].
During systemic infections, the liver regulates immune defenses via bacterial clearance, production of acute-phase proteins (APPs) and cytokines, and metabolic adaptation to inflammation. Liver injury has a critical effect on the severity and outcome of sepsis. The incidence of liver failure in sepsis is lower than the incidence of failure of other organs because of the high regenerative capacity of the liver and its ability to withstand assaults [5]. However, liver dysfunction and failure are associated with grave complications in sepsis. Mortality rates among sepsis patients with liver dysfunction or failure range from 54 to 68%, which is higher than the mortality rates of sepsis patients with lung dysfunction or failure (the organ most commonly affected in sepsis) [6,7,8,9,10]. Although the pathophysiology of sepsis-induced liver injury is complex, the inflammatory response plays a major role in this process. Furthermore, mitochondrial and endoplasmic reticulum (ER) dysfunctions during the acute phase response elicited by systemic inflammation lead to liver failure in sepsis [11]. In critically ill patients, the clinical manifestations include hypoxic hepatitis, sepsis-induced cholestasis, and secondary sclerosing cholangitis [7].
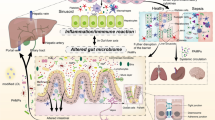
Intestinal barrier failure accompanied by systemic bacterial translocation was the predominant theory for the pathophysiology of MODS [12]. Bacterial pathogens frequently translocate from the gut lumen into the bloodstream and enter the liver via the portal vein [13]. Sepsis-induced epithelial apoptosis increases intestinal permeability (IP) and results in leakage of gut flora. Bacteria and their metabolites enter the lymphatic system through co-transport with chylomicrons, and dietary fat promotes the intestinal absorption of lipopolysaccharides (LPS) produced by intestinal bacteria [14]. These phenomena may exacerbate inflammation and lead to hepatic failure and death [15]. Gut dysbiosis and disruption of the intestinal barrier are caused by liver dysfunction, increased levels of inflammatory cytokines, impaired bacterial clearance, and metabolic disruption (Fig. 1). Changes in tight junctions and associated proteins due to liver dysfunction may increase gut permeability and damage distant organs. Therefore, a better understanding of the gut-liver crosstalk in sepsis-induced liver injury may help elucidate these complex disorders and form the basis for novel therapies.

Effect of increased levels of inflammatory cytokines and decreased bacterial clearance in sepsis-induced liver injury on the gut
Effect of liver dysfunction on the gut
The liver is a major site for the regulation of immune and inflammatory responses [16]. During the acute phase of sepsis, inflammatory cytokines, mainly IL-6 and IL-1, trigger the production of acute-phase proteins (APPs) in hepatocytes. Activated macrophages or Kupffer cells (KCs) and natural killer T (NKT) cells produce inflammatory mediators, including interferons (IFNs), IL-1β, IL-8, and tumor necrosis factor (TNF). Neutrophils and monocytes accumulate in the liver and contribute to antimicrobial defense (via neutrophil extracellular trap [NET] formation or phagocytosis [17]) and immune-mediated hepatocyte injury (through TNF-induced apoptosis) [7, 16].
During sepsis, liver injury varies from active hepatocellular dysfunction to life-threatening fulminant hepatic failure and induces severe and systemic detrimental inflammatory responses in other organs [18]. Liver dysfunction is accompanied by an increase in inflammatory cytokines and impairment of bacterial clearance and excretion of bile acids, which may cause gut microbiota dysbiosis and disruption of the intestinal barrier.
Effect of cytokines on the gut barrier during sepsis
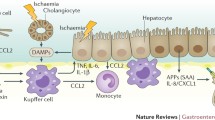
The single layer of epithelial cells in the intestinal tract acts as a selective barrier, allowing paracellular transport of water, solutes, and immune factors while preventing the transfer of potentially harmful pathogens, toxins, and antigens from the lumen to the circulation and mesenteric lymph [19,20,21]. Tight junctions maintain gut barrier function [22] and are linked to the intracellular cytoskeleton by several families of intramembrane proteins (claudin, occludin, tricellulin, junctional adhesion molecule [JAM]) and intracellular connector proteins (zonula occludens [ZO], myosin light chain), which help bridge the intercellular space [23]. During sepsis, increased levels of cytokines can modulate gut permeability by regulating the protein expression of claudin 2, claudin 5, JAM-A, occludin, and ZO-1 [24]. Myosin light chain kinase (MLCK) activation can increase the levels of interleukin (IL)-6, tumor necrosis factor α (TNF-α), and IL-1β. The phosphorylation of myosin regulatory light chain by MLCK can increase paracellular permeability and result in contraction or opening of the apical tight junctions [23, 25], leading to the activation of MLCK in a feed-forward mechanism, in part via altered ZO-1 and occludin [26] (Fig. 2). Increased intestinal permeability leads to increased systemic inflammation via a positive feedback loop [27].

Effect of cytokines on the gut barrier during sepsis. During sepsis, increased levels of cytokines can modulate gut permeability by regulating the protein expression of claudins, JAM-A, occludin, and ZO-1. The phosphorylation of myosin regulatory light chain by myosin light chain kinase (MLCK) can increase paracellular permeability and result in contraction or opening of the apical tight junctions
MLCK inhibition improved survival in a mouse model of sepsis [28] and improved barrier function and tight junction protein assembly in a murine model of burn injury [29]. These results suggest that MLCK inhibition is a potential treatment modality in sepsis and a critical target for reversing intestinal epithelial barrier disruption after severe burn injury.
Toll-like receptor 4 (TLR4) regulates the proliferation and apoptotic death of intestinal stem cells [30]. The increase in the level of cytokines during sepsis inhibits gut cell regeneration and stimulates apoptosis in a TLR4-dependent manner [31]. On a cellular level, the proliferation of crypt cells is decreased, while crypt and villus apoptosis are simultaneously increased [32]. Sepsis decreases intestinal cell proliferation, increases intestinal epithelial apoptosis, and reduces villus height [33]. Decreased villus height is associated with increased intestinal permeability, increased apoptosis of intestinal epithelial cells (IECs), and decreased survival [34]. Moreover, cytokines induce alterations in the mucus layer (reduced thickness, diminished luminal coverage, and poor adherence) and gut barrier function [35]. An increase in the level of inflammatory cytokines results in intestinal apoptosis and hyperpermeability, bacterial translocation, and expansion of the inflammatory responses [36]. Furthermore, increased systemic inflammation leads to liver injury.
The role of bacterial clearance and gut dysbiosis in the gut-liver axis
The liver is an essential site for bacterial phagocytosis and clearance from the bloodstream [37]. KCs, liver sinusoidal endothelial cells (LSECs), and stellate cells are the first line of defense against blood-borne bacteria.
Phagocytic KCs clear bacteria and bacterial metabolic products by pattern-recognition receptors [38] KCs secrete chemokines, leading to neutrophil migration and accumulation in liver sinusoids. Neutrophils and platelets interact with each other and capture and kill bacteria via neutrophil extracellular traps [39]. Liver B cells also phagocytose bacteria [40]. Hepatocytes phagocytose pathogens that cross the sinusoidal cell layer [41]. Hepatocytes use lipoproteins to clear LPS. LPS-binding protein transfers LPS to CD14 and facilitates the interaction between LPS and TLR4 on the surface of phagocytes to remove LPS and initiate the pro-inflammatory cascade [42]. The increased level of inflammatory cytokines results in gut barrier dysfunction.
The gut and the associated local immune system work together to prevent translocation of intestinal bacteria into the portal vein. After the gut intestinal epithelial barrier, the liver constitutes the second line of defense in eliminating invading bacteria and bacterial products, inhibiting a spread of bacteria into the body [13, 37]. During the liver injury, the impairment of the reticuloendothelial system, ability of neutrophils to phagocytose and kill bacteria, complement production, and antigen presentation (downregulation of monocyte human leukocyte antigen DR expression) impair bacterial clearance [43]. Accordingly, patients with liver cirrhosis have an increased risk of bacterial infections, which is often due to bacterial translocation from the intestine [44, 45].
The altered gut microflora plays an important role in the gut-liver axis. Gut dysbiosis, may damage the gut barrier and increase permeability [46]. Schnabl has shown that gut dysbiosis seems to participate in the disruption of intestinal epithelial tight junctions and imbalance in the proliferation and apoptosis of IECs [13]. Recent research has found that gut microbial dysbiosis leads to serious liver injury in mice after cecal ligation and puncture [47]. Several mechanisms may answer this phenotype. First, liver anatomy allows close interaction with the gut, where nutrients and microbial products contribute to the maintenance of healthy metabolism and liver homeostasis. Second, through the portal circulation, intestinal products are transported to the liver [48]. Therefore, gut dysbiosis directly aggravates sepsis-induced liver injury.
Another clinically relevant consequence of gut dysbiosis is the induction of inflammation with hemodynamic derangement, which is caused by the translocation of intestinal bacteria to the peritoneal cavity and the systemic circulation [49].
In summary, the alteration of gut microbial communities and the translocation of bacteria and toxins increase the systemic inflammatory response and contribute to multiple organ failure and death [50]. The interplay between the gut and the liver may amplify the inflammatory response and contribute to the development of sepsis and MODS.
Role of bile acids in the gut-liver axis
The liver is an endocrine gland that secretes bile acids (BAs) into the intestine and regulates the composition of the intestinal flora [51]. BAs are synthesized from cholesterol in the liver and chemically modified by the gut microbiota in the distal small intestine and colon (Fig. 3) [52]. The majority (95%) of BAs are reabsorbed in the distal ileum and return to the liver through the portal circulation in a process known as the enterohepatic cycle [53].

The role of bile acids in the gut-liver axis. During sepsis, the liver receives microbial input and influences intestinal microbes via bile acids. The gut microbiota can contribute to the pathogenesis of sepsis-induced liver injury by altering bile acids metabolism and its signaling pathways
During liver dysfunction, the increased BA levels activate inflammatory and oxidative stress and result in apoptosis or necrosis, fibrosis, and cirrhosis [54, 55]. BAs are key signaling molecules [56]. The farnesoid X receptor (FXR) and G protein-coupled receptors such as TGR5 mediate the regulatory actions of BA [57]. The microbiota modify signaling through receptors via its metabolism of BAs [58]. BAs activate FXR in the intestine and induce the expression of fibroblast growth factor 15 (FGF15). FGF15 decreases BA levels through a gut-microbiota-liver feedback loop by suppressing the expression of cholesterol 7α-hydroxylase (CYP7A1) in the liver—the rate-limiting step in BA synthesis [52]. In addition, FXR activation controls hepatic de novo lipogenesis, triglyceride (TG) transport by very-low-density lipoprotein (VLDL), and gluconeogenesis [59]. Secondary BAs activate TGR5, which improves glucose tolerance and liver function by inducing the release of intestinal glucagon-like peptide-1 (GLP-1) from intestinal enteroendocrine L cells [60]. Therefore, BAs regulate lipid and glucose homeostasis in the liver [59].
In the intestine, bacteria metabolize BAs to hydrophobic species through 7α-dehydroxylation and/or deconjugation of hydrophilic groups, resulting in the formation of the secondary BAs deoxycholic acid and lithocholic acid [61]. Moreover, BAs have bactericidal activity and reduce the intestinal permeability to endotoxins [62]. However, increased BA deconjugation reduces the bactericidal properties of the bile, causing the growth of bacteria that promote further BA deconjugation, and ultimately, bacterial translocation and endotoxemia in homeostasis conditions [62]. Higher levels of unconjugated primary BAs in the stool are correlated with intestinal dysbiosis [63]. Gut dysbiosis can disrupt intestinal epithelial tight junctions and cause an imbalance between the proliferation and apoptosis of IECs [13].
In summary, the liver may play a crucial regulatory role in controlling microbial populations by receiving microbial input and influencing intestinal microbes via BAs [64]. The gut microbiota contributes to the pathogenesis of sepsis-induced liver injury by altering BA metabolism and its signaling pathways. In the gut-liver axis, changes in BA metabolism cause an imbalance of the intestinal microflora and liver inflammation [65]. The interplay of BA between gut microbiota and the liver may promote organ inflammation and injury during sepsis.
Effect of sepsis-related gut injury on the liver
The gut contains 70–80% of the body’s immune cells and more than 100 trillion microorganisms [66]. Inflammation and hypoperfusion play an important role in the pathophysiology of gut injury in sepsis patients [67]. Sepsis increases apoptosis, decreases proliferation, and reduces migration of the epithelium. Furthermore, changes in tight junctions result in intestinal hyperpermeability and gut barrier dysfunction. Several other changes occur in the gut physiology in septic patients, due to extrinsic factors (antibiotics and parenteral nutrition) and intrinsic factors (systemic inflammation). These changes, in turn, influence the composition of the enteric flora [68]. Damage to the gut barrier causes the translocation of bacteria and toxins from the intestinal lumen to the mesenteric lymph nodes and systemic circulation. Bacterial translocation may increase systemic inflammation, leading to multiple organ failure and death [50].
Monocytes/macrophages play a vital role in the initiation or progression of inflammatory diseases [69]. KCs are involved in the regeneration of liver homeostasis by serving as gatekeepers for the clearance of pathogens [70]. KCs are crucial for host defense against bacteremia by clearing 70–80% of bloodstream bacteria [71].
After gut injury, bacteria and their products enter the liver from the gastrointestinal tract via the portal circulation and lymphatic system. Evidence suggests that the principal route of bacterial translocation is the lymphatic system.
Pathogen-associated molecular patterns (PAMPs) are exposed to the liver and recognized by pattern-recognition receptors (PRRs), which belong to the TLR and NOD-like receptor families [72]. PAMPs bind to KCs or stellate cells and induce the transcriptional upregulation of pro-inflammatory genes coding for cytokines such as IL6, IL-1β, and TNFα. The production of APPs is initiated when the liver switches from tolerogenic towards immunogenic responses [73]. Many of the APPs contribute to systemic immunity by acting as opsonins, activating neutrophils or macrophages via PRRs, and antimicrobial functions of the complement system [74,75,76].
Since the polarization of KCs plays an important role in liver injury, modulating KC function is essential for protection against liver injury during sepsis [77]. KCs are polarized into distinct phenotypes, M1 (proinflammatory) and M2 (alternative), depending on the local microenvironment. During sepsis, as the gut barrier function is impaired, increased bacterial translocation and inflammation change the immune microenvironment, and KCs are polarized into an M1 phenotype. Studies showed that enhancing M2 polarization in macrophages could protect the liver against injury [78, 79]. However, further investigation is needed to identify the signaling pathways involved in inhibiting the M1 phenotype in macrophages.
Bacterial metabolites such as short-chain fatty acids (SCFAs) maintain immune homeostasis [80]. SCFAs enhance the intestinal barrier function and promote the maturation of regulatory T (Treg) cells [80]. A recent study found that SCFA-producing bacteria improved colonization resistance against enteric pathogens in animals and humans. Notwithstanding, additional controlled studies are required to assess whether dietary fiber can modulate the gut microbiome and benefit patients in the intensive care unit [81].
The liver immune response contributes to bacterial clearance but may lead to organ damage due to an overwhelming systemic inflammatory response. Mitochondrial dysfunction and ER stress-related inflammation during the acute phase response culminate in liver failure.
Future prospects
As the gut plays an important role in the progression of sepsis-induced liver injury, targeting microbiota can have preventive and therapeutic potential in liver dysfunction. Some observations suggest that microbiome depletion is related to the association between antibiotic exposure and subsequent sepsis [82]. Moreover, modulation in target microbiota in at-risk patients before sepsis onset may decrease the incidence of sepsis. Treatments with fecal microbiota transplantation and microbiota-targeted metabolites, including butyrate or other SCFAs, may use to treat dysbiosis during sepsis. But the benefits of fecal microbiota transplantation should be balanced with the associated risks such as infectious complications [83]. Based on these findings, probiotics and synbiotic preparations can decrease the incidence of infections in the ICU [84,85,86,87].
Conclusions
A large amount of literature supporting the possibility of the strong interplay between gut and liver in sepsis has been reviewed. The translocation of bacteria or metabolites and the resulting intestinal inflammation lead to liver dysfunction. Furthermore, sepsis-induced liver injury can promote gut dysbiosis and increase intestinal permeability. Increased permeability of the mesenteric vasculature causes intestinal edema and gut barrier dysfunction, leading to systemic inflammation and acute liver injury. Long-term inflammation leads to devastating consequences. Further understanding of the gut-liver crosstalk may provide insights on homeostatic regulation in sepsis and help develop effective therapies to prevent sepsis-induced liver damage.
Availability of data and materials
Not applicable.
Abbreviations
- APP:
-
Acute-phase proteins
- BAs:
-
Bile acids
- DAMPs:
-
Damage-associated molecular patterns
- ER:
-
Endoplasmic reticulum
- FGF15:
-
Fibroblast growth factor 15
- FMT:
-
Fecal microbiota transplantation
- FXR:
-
Farnesoid X receptor
- GLP-1:
-
Glucagon-like peptide-1
- IL:
-
Interleukin
- IP:
-
Intestinal permeability
- JAM:
-
Junctional adhesion molecule
- LSECs:
-
Liver sinusoidal endothelial cells
- MLCK:
-
Myosin light chain kinase
- MODS:
-
Multiple organ dysfunction syndrome
- NET:
-
Neutrophil extracellular trap
- PAMP:
-
Pathogen-associated molecular patterns
- PRR:
-
Pattern-recognition receptors
- SCFAs:
-
Short-chain fatty acids
- TLR4:
-
Toll-like receptor
- TNF-α:
-
Tumor necrosis factor α
- VLDL:
-
Very-low-density lipoprotein
- ZO:
-
Zonula occludens
References
Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–10.
Harrison C. Sepsis: calming the cytokine storm. Nat Rev Drug Discov. 2010;9(5):360–1.
Seymour CW, Gesten F, Prescott HC, Friedrich ME, Iwashyna TJ, Phillips GS, Lemeshow S, Osborn T, Terry KM, Levy MM. Time to treatment and mortality during mandated emergency care for sepsis. N Engl J Med. 2017;376(23):2235–44.
Vincent JL, Marshall JC, Namendys-Silva SA, Francois B, Martin-Loeches I, Lipman J, Reinhart K, Antonelli M, Pickkers P, Njimi H, et al. Assessment of the worldwide burden of critical illness: the intensive care over nations (ICON) audit. Lancet Respir Med. 2014;2(5):380–6.
Weiss YG, Bellin L, Kim PK, Andrejko KM, Haaxma CA, Raj N, Furth EE, Deutschman CS. Compensatory hepatic regeneration after mild, but not fulminant, intraperitoneal sepsis in rats. Am J Physiol Gastrointest Liver Physiol. 2001;280(5):G968–73.
Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. 2017;14(1):55–66.
Yan J, Li S, Li S. The role of the liver in sepsis. Int Rev Immunol. 2014;33(6):498–510.
Cheng B, Xie G, Yao S, Wu X, Guo Q, Gu M, Fang Q, Xu Q, Wang D, Jin Y, et al. Epidemiology of severe sepsis in critically ill surgical patients in ten university hospitals in China. Crit Care Med. 2007;35(11):2538–46.
Brun-Buisson C, Meshaka P, Pinton P, Vallet B, Group ES. EPISEPSIS: a reappraisal of the epidemiology and outcome of severe sepsis in French intensive care units. Intensive Care Med. 2004;30(4):580–8.
Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–10.
Lelubre C, Vincent JL. Mechanisms and treatment of organ failure in sepsis. Nat Rev Nephrol. 2018;14(7):417–27.
Klingensmith NJ, Coopersmith CM. The gut as the motor of multiple organ dysfunction in critical illness. Crit Care Clin. 2016;32(2):203–12.
Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146(6):1513–24.
Ghoshal S, Witta J, Zhong J, de Villiers W, Eckhardt E. Chylomicrons promote intestinal absorption of lipopolysaccharides. J Lipid Res. 2009;50(1):90–7.
Mittal R, Coopersmith CM. Redefining the gut as the motor of critical illness. Trends Mol Med. 2014;20(4):214–23.
Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14(10):996–1006.
McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. 2012;12(3):324–33.
Gustot T, Durand F, Lebrec D, Vincent JL, Moreau R. Severe sepsis in cirrhosis. Hepatology. 2009;50(6):2022–33.
Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9(11):799–809.
Odenwald MA, Turner JR. Intestinal permeability defects: is it time to treat? Clin Gastroenterol Hepatol. 2013;11(9):1075–83.
Nalle SC, Kwak HA, Edelblum KL, Joseph NE, Singh G, Khramtsova GF, Mortenson ED, Savage PA, Turner JR. Recipient NK cell inactivation and intestinal barrier loss are required for MHC-matched graft-versus-host disease. Sci Transl Med. 2014;6(243):243ra287.
Bischoff SC, Barbara G, Buurman W, Ockhuizen T, Schulzke JD, Serino M, Tilg H, Watson A, Wells JM. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014;14:189.
Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci. 2012;1258:34–42.
Yoseph BP, Klingensmith NJ, Liang Z, Breed ER, Burd EM, Mittal R, Dominguez JA, Petrie B, Ford ML, Coopersmith CM. Mechanisms of intestinal barrier dysfunction in sepsis. Shock. 2016;46(1):52–9.
Zahs A, Bird MD, Ramirez L, Turner JR, Choudhry MA, Kovacs EJ. Inhibition of long myosin light-chain kinase activation alleviates intestinal damage after binge ethanol exposure and burn injury. Am J Physiol Gastrointest Liver Physiol. 2012;303(6):G705–12.
Yu D, Marchiando AM, Weber CR, Raleigh DR, Wang Y, Shen L, Turner JR. MLCK-dependent exchange and actin binding region-dependent anchoring of ZO-1 regulate tight junction barrier function. Proc Natl Acad Sci U S A. 2010;107(18):8237–41.
Alexopoulou A, Agiasotelli D, Vasilieva LE, Dourakis SP. Bacterial translocation markers in liver cirrhosis. Ann Gastroenterol. 2017;30(5):486–97.
Lorentz CA, Liang Z, Meng M, Chen CW, Yoseph BP, Breed ER, Mittal R, Klingensmith NJ, Farris AB, Burd EM, et al. Myosin light chain kinase knockout improves gut barrier function and confers a survival advantage in polymicrobial sepsis. Mol Med. 2017;23:155–65.
Chen C, Wang P, Su Q, Wang S, Wang F. Myosin light chain kinase mediates intestinal barrier disruption following burn injury. PLoS One. 2012;7(4):e34946.
Neal MD, Sodhi CP, Jia H, Dyer M, Egan CE, Yazji I, Good M, Afrazi A, Marino R, Slagle D, et al. Toll-like receptor 4 is expressed on intestinal stem cells and regulates their proliferation and apoptosis via the p53 up-regulated modulator of apoptosis. J Biol Chem. 2012;287(44):37296–308.
Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. 2008;453(7195):620–5.
Neal MD, Sodhi CP, Dyer M, Craig BT, Good M, Jia H, Yazji I, Afrazi A, Richardson WM, Beer-Stolz D, et al. A critical role for TLR4 induction of autophagy in the regulation of enterocyte migration and the pathogenesis of necrotizing enterocolitis. J Immunol. 2013;190(7):3541–51.
Dominguez JA, Vithayathil PJ, Khailova L, Lawrance CP, Samocha AJ, Jung E, Leathersich AM, Dunne WM, Coopersmith CM. Epidermal growth factor improves survival and prevents intestinal injury in a murine model of pseudomonas aeruginosa pneumonia. Shock. 2011;36(4):381–9.
Yang J, Radulescu A, Chen CL, Zhang HY, James IO, Besner GE. Heparin-binding epidermal growth factor-like growth factor improves intestinal barrier function and reduces mortality in a murine model of peritonitis. Surgery. 2013;153(1):52–62.
Chang M, Alsaigh T, Kistler EB, Schmid-Schonbein GW. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS One. 2012;7(6):e40087.
Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE. Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27(7):1230–51.
Protzer U, Maini MK, Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol. 2012;12(3):201–13.
Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, Martin C, van Rooijen N, Ochando JC, Randolph GJ, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62(1):279–91.
Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat Immunol. 2013;14(8):785–92.
Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, Tiglao E, Figueiredo JL, Iwamoto Y, Theurl I, et al. Innate response activator B cells protect against microbial sepsis. Science. 2012;335(6068):597–601.
Soji T, Murata Y, Ohira A, Nishizono H, Tanaka M, Herbert DC. Evidence that hepatocytes can phagocytize exogenous substances. Anat Rec. 1992;233(4):543–6.
Deng M, Scott MJ, Loughran P, Gibson G, Sodhi C, Watkins S, Hackam D, Billiar TR. Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis. J Immunol. 2013;190(10):5152–60.
Wasmuth HE, Kunz D, Yagmur E, Timmer-Stranghoner A, Vidacek D, Siewert E, Bach J, Geier A, Purucker EA, Gressner AM, et al. Patients with acute on chronic liver failure display “sepsis-like” immune paralysis. J Hepatol. 2005;42(2):195–201.
Singal AK, Salameh H, Kamath PS. Prevalence and in-hospital mortality trends of infections among patients with cirrhosis: a nationwide study of hospitalised patients in the United States. Aliment Pharmacol Ther. 2014;40(1):105–12.
Bajaj JS, O'Leary JG, Reddy KR, Wong F, Olson JC, Subramanian RM, Brown G, Noble NA, Thacker LR, Kamath PS, et al. Second infections independently increase mortality in hospitalized patients with cirrhosis: the North American consortium for the study of end-stage liver disease (NACSELD) experience. Hepatology. 2012;56(6):2328–35.
Giannelli V, Di Gregorio V, Iebba V, Giusto M, Schippa S, Merli M, Thalheimer U. Microbiota and the gut-liver axis: bacterial translocation, inflammation and infection in cirrhosis. World J Gastroenterol. 2014;20(45):16795–810.
Liu Z, Li N, Fang H, Chen X, Guo Y, Gong S, Niu M, Zhou H, Jiang Y, Chang P, et al. Enteric dysbiosis is associated with sepsis in patients. FASEB J. 2019;33(11):12299–310.
Bloemen JG, Venema K, van de Poll MC, Olde Damink SW, Buurman WA, Dejong CH. Short chain fatty acids exchange across the gut and liver in humans measured at surgery. Clin Nutr. 2009;28(6):657–61.
Bellot P, Frances R, Such J. Pathological bacterial translocation in cirrhosis: pathophysiology, diagnosis and clinical implications. Liver Int. 2013;33(1):31–9.
Dominguez JA, Coopersmith CM. Can we protect the gut in critical illness? The role of growth factors and other novel approaches. Crit Care Clin. 2010;26(3):549–65 x.
Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141(5):1773–81.
Yuan L, Bambha K. Bile acid receptors and nonalcoholic fatty liver disease. World J Hepatol. 2015;7(28):2811–8.
Dawson PA, Karpen SJ. Intestinal transport and metabolism of bile acids. J Lipid Res. 2015;56(6):1085–99.
Kalhan SC, Guo L, Edmison J, Dasarathy S, McCullough AJ, Hanson RW, Milburn M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metab Clin Exp. 2011;60(3):404–13.
Faubion WA, Guicciardi ME, Miyoshi H, Bronk SF, Roberts PJ, Svingen PA, Kaufmann SH, Gores GJ. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest. 1999;103(1):137–45.
Copple BL, Li T. Pharmacology of bile acid receptors: evolution of bile acids from simple detergents to complex signaling molecules. Pharmacol Res. 2016;104:9–21.
Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11(1):55–67.
Huang F, Zheng X, Ma X, Jiang R, Zhou W, Zhou S, Zhang Y, Lei S, Wang S, Kuang J, et al. Theabrownin from Pu-erh tea attenuates hypercholesterolemia via modulation of gut microbiota and bile acid metabolism. Nat Commun. 2019;10(1):4971.
Claudel T, Staels B, Kuipers F. The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol. 2005;25(10):2020–30.
Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10(3):167–77.
Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–59.
Lorenzo-Zuniga V, Bartoli R, Planas R, Hofmann AF, Vinado B, Hagey LR, Hernandez JM, Mane J, Alvarez MA, Ausina V, et al. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology. 2003;37(3):551–7.
Mouzaki M, Wang AY, Bandsma R, Comelli EM, Arendt BM, Zhang L, Fung S, Fischer SE, McGilvray IG, Allard JP. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS One. 2016;11(5):e0151829.
Brandl K, Kumar V, Eckmann L. Gut-liver axis at the frontier of host-microbial interactions. Am J Physiol Gastrointest Liver Physiol. 2017;312(5):G413–9.
Alaish SM, Smith AD, Timmons J, Greenspon J, Eyvazzadeh D, Murphy E, Shea-Donahue T, Cirimotich S, Mongodin E, Zhao A, et al. Gut microbiota, tight junction protein expression, intestinal resistance, bacterial translocation and mortality following cholestasis depend on the genetic background of the host. Gut Microbes. 2013;4(4):292–305.
Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164(3):337–40.
Habes QLM, van Ede L, Gerretsen J, Kox M, Pickkers P. Norepinephrine contributes to enterocyte damage in septic shock patients: a prospective cohort study. Shock. 2018;49(2):137–43.
Defazio J, Fleming ID, Shakhsheer B, Zaborina O, Alverdy JC. The opposing forces of the intestinal microbiome and the emerging pathobiome. Surg Clin North Am. 2014;94(6):1151–61.
Gordon S, Pluddemann A, Martinez Estrada F. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. 2014;262(1):36–55.
Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. 2010;10(11):753–66.
Ashare A, Monick MM, Powers LS, Yarovinsky T, Hunninghake GW. Severe bacteremia results in a loss of hepatic bacterial clearance. Am J Respir Crit Care Med. 2006;173(6):644–52.
Arroyo V, Moreau R, Jalan R, Gines P, Study E-CCC. Acute-on-chronic liver failure: a new syndrome that will re-classify cirrhosis. J Hepatol. 2015;62(1 Suppl):S131–43.
Bode JG, Albrecht U, Haussinger D, Heinrich PC, Schaper F. Hepatic acute phase proteins--regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-kappaB-dependent signaling. Eur J Cell Biol. 2012;91(6–7):496–505.
Shah C, Hari-Dass R, Raynes JG. Serum amyloid A is an innate immune opsonin for Gram-negative bacteria. Blood. 2006;108(5):1751–7.
Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449(7164):819–26.
Ward PA, Gao H. Sepsis, complement and the dysregulated inflammatory response. J Cell Mol Med. 2009;13(10):4154–60.
Wan J, Benkdane M, Teixeira-Clerc F, Bonnafous S, Louvet A, Lafdil F, Pecker F, Tran A, Gual P, Mallat A, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59(1):130–42.
Kang JW, Lee SM. Resolvin D1 protects the liver from ischemia/reperfusion injury by enhancing M2 macrophage polarization and efferocytosis. Biochim Biophys Acta. 2016;1861(9 Pt A):1025–35.
Xie J, Wu X, Zhou Q, Yang Y, Tian Y, Huang C, Meng X, Li J. PICK1 confers anti-inflammatory effects in acute liver injury via suppressing M1 macrophage polarization. Biochimie. 2016;127:121–32.
Kamada N, Seo SU, Chen GY, Nunez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13(5):321–35.
Fu Y, Moscoso DI, Porter J, Krishnareddy S, Abrams JA, Seres D, Chong DH, Freedberg DE. Relationship between dietary fiber intake and short-chain fatty acid-producing bacteria during critical illness: a prospective cohort study. JPEN J Parenter Enteral Nutr. 2020;44(3):463–71.
Baggs J, Jernigan JA, Halpin AL, Epstein L, Hatfield KM, McDonald LC. Risk of subsequent sepsis within 90 days after a hospital stay by type of antibiotic exposure. Clin Infect Dis. 2018;66(7):1004–12.
DeFilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH, Turbett S, Chung RT, Chen YB, Hohmann EL. Drug-resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med. 2019;381(21):2043–50.
Manzanares W, Lemieux M, Langlois PL, Wischmeyer PE. Probiotic and synbiotic therapy in critical illness: a systematic review and meta-analysis. Crit Care. 2016;19:262.
Li Q, Wang C, Tang C, He Q, Zhao X, Li N, Li J. Successful treatment of severe sepsis and diarrhea after vagotomy utilizing fecal microbiota transplantation: a case report. Crit Care. 2015;19:37.
Li Q, Wang C, Tang C, He Q, Zhao X, Li N, Li J. Therapeutic modulation and reestablishment of the intestinal microbiota with fecal microbiota transplantation resolves sepsis and diarrhea in a patient. Am J Gastroenterol. 2014;109(11):1832–4.
Wei Y, Yang J, Wang J, Yang Y, Huang J, Gong H, Cui H, Chen D. Successful treatment with fecal microbiota transplantation in patients with multiple organ dysfunction syndrome and diarrhea following severe sepsis. Crit Care. 2016;20(1):332.
Acknowledgements
We are grateful to J. Z. for the helpful advice.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
YY and JT conceived of the study, participated in coordination, and helped draft the manuscript. JS drafted the manuscript and substantively revised it. JZ and XW designed the figures. CR and FJ contributed to manuscript editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
No competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sun, J., Zhang, J., Wang, X. et al. Gut-liver crosstalk in sepsis-induced liver injury. Crit Care 24, 614 (2020). https://doi.org/10.1186/s13054-020-03327-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-020-03327-1