
Abstract
Background
The CRASH-3 trial hypothesised that timely tranexamic acid (TXA) treatment might reduce deaths from intracranial bleeding after traumatic brain injury (TBI). To explore the mechanism of action of TXA in TBI, we examined the timing of its effect on death.
Methods
The CRASH-3 trial randomised 9202 patients within 3 h of injury with a GCS score ≤ 12 or intracranial bleeding on CT scan and no significant extracranial bleeding to receive TXA or placebo. We conducted an exploratory analysis of the effects of TXA on all-cause mortality within 24 h of injury and within 28 days, excluding patients with a GCS score of 3 or bilateral unreactive pupils, stratified by severity and country income. We pool data from the CRASH-2 and CRASH-3 trials in a one-step fixed effects individual patient data meta-analysis.
Results
There were 7637 patients for analysis after excluding patients with a GCS score of 3 or bilateral unreactive pupils. Of 1112 deaths, 23.3% were within 24 h of injury (early deaths). The risk of early death was reduced with TXA (112 (2.9%) TXA group vs 147 (3.9%) placebo group; risk ratio [RR] RR 0.74, 95% CI 0.58–0.94). There was no evidence of heterogeneity by severity (p = 0.64) or country income (p = 0.68). The risk of death beyond 24 h of injury was similar in the TXA and placebo groups (432 (11.5%) TXA group vs 421 (11.7%) placebo group; RR 0.98, 95% CI 0.69–1.12). The risk of death at 28 days was 14.0% in the TXA group versus 15.1% in the placebo group (544 vs 568 events; RR 0.93, 95% CI 0.83–1.03). When the CRASH-2 and CRASH-3 trial data were pooled, TXA reduced early death (RR 0.78, 95% CI 0.70–0.87) and death within 28 days (RR 0.88, 95% CI 0.82–0.94).
Conclusions
Tranexamic acid reduces early deaths in non-moribund TBI patients regardless of TBI severity or country income. The effect of tranexamic acid in patients with isolated TBI is similar to that in polytrauma. Treatment is safe and even severely injured patients appear to benefit when treated soon after injury.
Trial registration
ISRCTN15088122, registered on 19 July 2011; NCT01402882, registered on 26 July 2011.
Similar content being viewed by others


Background
The acute management of traumatic brain injury (TBI) aims to avoid secondary brain damage and optimise conditions for recovery [1]. The day of the injury is the most hazardous, accounting for one third of in-hospital deaths [2]. Some TBI victims have brain damage that is incompatible with life and die shortly after admission. In many patients, intracranial bleeding starts soon after impact and continues for several hours, with the majority of haematoma expansion occurring within 1–1.5 h of injury [3, 4]. The accumulating blood can increase intracranial pressure, causing cerebral herniation and death. Tranexamic acid reduces bleeding in surgery and reduces death from bleeding in traumatic and post-partum haemorrhage [5,6,7]. The therapeutic premise of the CRASH-3 trial was that timely tranexamic acid treatment might curtail intracranial bleeding and prevent some of the early bleeding-related deaths. A 1-g bolus started within 3 h of injury was followed by an infusion of 1 g over 8 h. Tranexamic acid has a half-life of 2 h and by the second day is almost completely eliminated. By this time, the bleeding will have stopped, but other pathological processes, likely unaffected by tranexamic acid, will continue to cause deaths. Those who survive the first day run the risk of cerebral oedema, diffuse axonal injury, organ failure, sepsis, pneumonia and many other threats, some iatrogenic, that make up the remaining two thirds of in-hospital deaths.
The management of TBI is only partly based on results from randomised trials. In practice, doctors draw on pathophysiological knowledge, the available evidence and their clinical experience to identify mechanisms of brain damage and target physiologically based treatment accordingly [3]. Large randomised trials can reduce our therapeutic uncertainty, but to categorise them as positive or negative based on arbitrary p value thresholds is inappropriate [8,9,10]. The CRASH-3 trial results have variously been described as ‘negative’, ‘neutral’ or ‘a win for patients with head injury’ that will benefit patients [11,12,13]. We argue that randomised trials can deepen our understanding of pathophysiology and that mechanistic insights should inform their interpretation. To explore the mechanism of action of tranexamic acid in TBI patients, we examined the timing of its effect on death. We also set the results of our analysis in the context of other trials of tranexamic acid in TBI and polytrauma patients, taking into consideration current treatment guidelines that exclude patients with isolated TBI.
Methods
The background to the CRASH-3 trial, the methods, baseline characteristics and main results were previously reported [2, 6, 14]. Briefly, adults with TBI who were within 3 h of injury and had a Glasgow coma scale score (GCS) ≤ 12 or any intracranial bleeding on CT scan and no significant extra-cranial bleeding were eligible. The time window for eligibility was originally 8 h, but in 2016, the protocol was changed to limit recruitment to within 3 h of injury. Between July 2012 and January 2019, we randomly allocated 12,737 patients with TBI to receive tranexamic acid or placebo, of whom 9202 patients were treated within 3 h. Patients were assigned by selecting a numbered treatment pack from a box containing eight packs that were identical apart from the pack number. Patients, care givers and those assessing outcomes were masked to treatment allocation.
Based on previous research on the mechanism of tranexamic acid in bleeding trauma patients, we hypothesised that tranexamic acid would have a greater effect on deaths soon after injury, since early bleeding-related deaths have the most potential to be reduced by tranexamic acid [15]. We pre-specified this hypothesis in the statistical analysis plan that we published before un-blinding [14]. We also anticipated that the treatment effect would be diluted by the inclusion of patients with a GCS score of 3 or unreactive pupils who have a very poor prognosis regardless of treatment [14]. The trial results were consistent with both of these hypotheses [2]. The pre-specified primary outcome in the CRASH-3 trial was death due to head injury within 28 days among patients treated within 3 h of injury. Although our scientific reasons for pre-specifying head injury death as the primary outcome were given in the statistical analysis plan and presented in detail elsewhere [16], there has been strong interest in the effects of tranexamic acid on all-cause mortality. As such, this analysis focusses on early deaths from any cause, excluding patients with a GCS score of 3 or bilateral unreactive pupils. Analyses of head injury deaths and analyses including patients with a GCS score of 3 or bilateral unreactive pupils are presented in the Additional file 1 for comparison with the results presented below and for cross-reference with the main trial results.
We examine the temporal distribution of deaths from any cause in the CRASH-3 trial. We explore the effects of tranexamic acid on deaths due to any cause within 24 h of injury and on deaths due to any cause within 28 days, stratified by severity and country income level. We use the baseline GCS score to define severity—mild to moderate (GCS 9–15) and severe (GCS 3–8)—and World Bank definitions to determine country income level (LMIC vs HIC). Because a subgroup analysis demonstrated effect modification by severity, we explore this further. Because most patients were from LMICs, the generalisability of the results to HICs has been questioned and so we explore how the treatment effects vary by country income level. To check if the effect on early deaths could be explained by undiagnosed extra-cranial bleeding, we conducted a sensitivity analysis excluding patients with hypotension (SBP < 90 mmHg). We also examined the effects of tranexamic acid on vascular occlusive events (fatal and non-fatal) in all patients irrespective of time to treatment because theoretically the potential risk of vascular occlusive events would be greater with late treatment as there is a shift from a fibrinolytic to a coagulopathic state. We report risk ratios, 95% confidence intervals and heterogeneity p values. We excluded 98 patients with missing outcome data.
We prespecified an analysis setting the results of the CRASH-3 trial in the context of other evidence, including the CRASH-2 trial, in which 40% of deaths were due to head injury [14]. The CRASH-3 trial essentially represents a subgroup of patients with isolated TBI who were excluded from the CRASH-2 trial. Here, to set our results in the context of tranexamic acid in polytrauma patients, we pooled the data from the CRASH-2 and CRASH-3 trials in a one-step fixed effects individual patient data meta-analysis using a Poisson regression model with sandwich variance estimation, adjusted for time to treatment. In the main CRASH-3 trial publication, we updated a systematic search for randomised trials of tranexamic acid in TBI. We searched PubMed, Science Citation Index, National Research Register, Zetoc, SIGLE, Global Health, LILACS, Current Controlled Trials, the Cochrane Injuries Group Specialised Register, CENTRAL, MEDLINE and EMBASE. We identified three trials in addition to the CRASH-3 trial including the CRASH-2 intracranial bleeding study, a randomised trial of 283 TBI patients sponsored by Khon Kaen University [17] and a randomised trial of pre-hospital tranexamic acid in 967 TBI patients sponsored by the University of Washington (NCT01990768). The CRASH-2 intracranial bleeding study was omitted as this is already contained within the CRASH-2 trial dataset, and the small Thai study was omitted due to a lack of data on timing of death, cause of death and GCS score, and limitations in methodological quality including an unclear risk of selection bias from allocation concealment.
The model for the one-step meta-analysis was as follows:
where trial = 0 for CRASH-2 and 1 for CRASH-3, group = 0 for placebo and 1 for TXA, ttt is time to treatment and β2 is the summary effect estimate across both trials.
We also consider the CRASH-3 trial results in the context of the CRASH-2 trial and the trial of pre-hospital tranexamic acid (NCT01990768) using an aggregate data meta-analysis with fixed effects to assess the effect of tranexamic acid on death at 28 days excluding patients with a GCS score of 3 or bilateral unreactive pupils, and on vascular occlusive events in all patients. An aggregate data meta-analysis was used because we did not have access to the individual patient data for trial NCT01990768.
Results
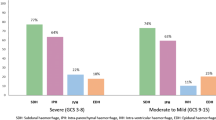
Among the 12,639 randomised patients with outcome data available, 9127 were treated within 3 h of injury. A total of 1490 patients had GCS score of 3 or bilateral unreactive pupils at baseline (16.3%), leaving 7637 patients for analysis. There were 1112 deaths from all causes within 28 days, of which 259 (23.3%) occurred within 24 h of injury (early deaths) and 853 (76.7%) were beyond 24 h of injury. Figure 1 shows the time interval from injury to death in placebo-treated patients overall and according to severity and country income. Overall, the proportion of early deaths was larger in severe head injury (28.1%) and in LMICs (24.1%).

Days from injury to death in placebo group patients randomised within 3 h of injury overall and by severity (GCS) and country income, excluding those with GCS 3 or bilateral unreactive pupils
Effect of tranexamic acid on early deaths
The risk of early death was lower in patients with mild-to-moderate head injury compared to severe head injury (1.1% vs 9.9%) and in HICs compared to LMICs (2.0% vs 3.8%). The risk of early death was reduced with tranexamic acid (112 (2.9%) deaths in the tranexamic acid group vs 147 (3.9%) deaths in the placebo group; risk ratio [RR] RR 0.74, 95% CI 0.58–0.94; see Table 1). There was no evidence that the effect of tranexamic acid on early deaths varied by severity (heterogeneity p = 0.64) or by country income (heterogeneity p = 0.68). When 114 (1.5%) patients with hypotension (SBP < 90 mmHg) at baseline were excluded from the analyses, the results were essentially the same (106 (2.8%) deaths in the tranexamic acid group vs 143 (3.9%) deaths in the placebo group; RR 0.72, 95% CI 0.56–0.92). The effect of tranexamic acid on early death was smaller (261 vs 315 events; RR 0.81, 95% CI 0.69–0.95) when we included patients who had a GCS score of 3 or bilateral unreactive pupils at baseline (see Appendix Table 1). The effect was larger when the analysis was restricted to head injury-related deaths only (Appendix Tables 2 and 3).
Effect of tranexamic acid on deaths after 24 h
The risk of death more than 24 h after injury was lower in patients with mild-to-moderate head injury compared to severe head injury (6.3% vs 25.2%) and in HICs compared to LMICs (8.2% vs 12.1%). The risk of death from all causes beyond 24 h of injury was similar in the tranexamic acid and placebo groups (432 (11.5%) deaths in the tranexamic acid group vs 421 (11.7%) deaths in the placebo group; RR 0.98, 95% CI 0.69–1.12; see Table 1). The effect on deaths beyond 24 h was similar by severity (heterogeneity p = 0.088) and country income (heterogeneity p = 0.36).
Effect of tranexamic acid on deaths at 28 days
The risk of death at 28 days was lower in mild-to-moderate head injury compared to severe head injury (7.4% vs 35.1%) and in HICs compared to LMICs (10.1% vs 15.9%). The risk of death from any cause at 28 days was 14.0% in the tranexamic acid group versus 15.1% in the placebo group (544 vs 568 events; RR 0.93, 95% CI 0.83–1.03; see Table 1). The effect of tranexamic acid on all-cause mortality at 28 days was similar by severity (heterogeneity p = 0.11) and country income (heterogeneity p = 0.35).
Effect of tranexamic acid on vascular occlusive events
Among the 12,639 randomised patients with outcome data, there were 203 (1.6%) fatal or non-fatal vascular occlusive events. The absolute risk of vascular occlusive events in all patients was lower in mild-to-moderate head injury than in severe head injury (1.2% vs 2.4%) and in LMICs compared to HICs (1.0% vs 3.0%). The risk of vascular occlusive events was 1.6% in both the tranexamic acid and placebo groups (101 vs 102 events; RR 0.98, 95% CI 0.74–1.28; see Table 2).
The results of the CRASH-3 trial in context
When the CRASH-2 and CRASH-3 trial data were pooled in a one-stage individual patient data meta-analysis, early tranexamic acid reduced death within 24 h of injury (RR 0.78, 95% CI 0.70–0.87) and death within 28 days (RR 0.88, 95% CI 0.82–0.94), with no evidence of heterogeneity by trial (death within 24 h p = 0.60; death within 28 days p = 0.18; see Fig. 2). Adjusting for time to treatment made no difference to the results. For deaths with 24 h of injury, the adjusted RR = 0.78 (95% CI 0.70–0.87), and for death within 28 days the adjusted RR = 0.88 (95% CI 0.82–0.94). When a US trial of pre-hospital tranexamic acid for isolated TBI was included in an aggregate data meta-analysis on death from any cause at 28 days, the results were identical (RR 0.88, 95% CI 0.82–0.94), with no evidence of heterogeneity by trial (p = 0.41). There was no difference in the risk of vascular occlusive events between treatment groups (RR 0.87, 95% CI 0.74–1.02), again with no heterogeneity by trial (p = 0.42).

Evidence on the effect of early tranexamic acid on all-cause mortality within 24 h and 28 days of injury, excluding patients with a GCS score of 3 or bilateral unreactive pupils at baseline
Discussion
Based upon this post hoc, exploratory analysis of the CRASH-3 trial, tranexamic acid reduces deaths on the day of the injury regardless of TBI severity and country income but has no apparent effect on deaths beyond the day of the injury. The effect of tranexamic acid on all-cause mortality at 28 days is a weighted average of these early and late effects and, although diluted toward the null, is similar to the results of the CRASH-2 trial and indicative of a survival benefit.
Because a larger proportion of deaths in the CRASH-3 trial occurred after 24 h (69% in CRASH-3 versus 43% in CRASH-2), the effect on mortality at 28 days is smaller (more diluted) in the CRASH-3 trial, although there is no evidence of heterogeneity. As anticipated in the statistical analysis plan, the effect is smaller when including patients with un-survivable injuries prior to treatment. Tranexamic acid did not increase the risk of adverse vascular occlusive events in trauma patients.
Because our choice of head injury death as the primary outcome measure was criticised, these analyses report all-cause mortality. The trial inclusion criteria were clinical and reflect the situation that doctors face in practice. We enrolled TBI patients within 3 h of injury if they had no significant extra-cranial bleeding. The effect of tranexamic acid on early deaths is not explained by undiagnosed extra-cranial bleeding. Only 1.5% of patients had hypotension (SBP < 90 mmHg) at baseline and only 11 of the 1112 deaths (six in the tranexamic acid group and five in the placebo group) were classified as extracranial bleeding deaths. When patients with hypotension are excluded, the results are the same. The reduction in all-cause mortality within 24 h strongly suggests that tranexamic acid reduces intracranial bleeding deaths.
We conducted the CRASH-3 trial because there was reason to believe that tranexamic acid could reduce bleeding-related head injury deaths. Increased fibrinolysis is common in TBI patients and worsens intracranial bleeding. The CRASH-2 trial in 20,211 polytrauma patients (extra-cranial and intra-cranial injury) with significant bleeding found that tranexamic acid reduces mortality, primarily by reducing bleeding deaths on the day of the injury [15]. Because the CRASH-2 trial was large, this early benefit was still apparent at 28 days, although ‘diluted’ by non-bleeding deaths. The CRASH-3 trial was smaller than the CRASH-2 trial, and so despite the higher mortality rate, there were fewer deaths and less statistical power to detect the diluted effect on all-cause mortality at 28 days. A non-significant difference between two groups in a randomised trial can be real difference that is not significant due to a lack of power, or it can be a difference that has occurred by chance. In this case (Table 1), there is a large reduction in deaths within 24 h with tranexamic acid (RR = 0.74) that is highly statistically significant and consistent with the expected biological effects of tranexamic acid but no apparent reduction in deaths beyond 24 h (RR = 0.98). Because the relative risk at 28 days is a weighted average of these effects, the modest reduction in death at 28 days (RR = 0.93) is not statistically significant. We believe the reduction in deaths at 28 days is a real reduction that is not significant due to a lack of statistical power. This interpretation is consistent with biology (intracranial bleeding occurs early, and there is little or no tranexamic acid in the body beyond 24 h) and as shown in the next paragraph is mathematically consistent with dilution. The reduction in deaths at 28 days in the CRASH-3 trial is similar to that seen in the larger (and more powerful) CRASH-2 trial, and when the results are pooled, the reduction in deaths at 28 days with tranexamic acid is highly significant. However, we accept that can never rule out chance as a potential explanation.
Because ‘dilution’ is key to understanding the CRASH-3 results, it is best considered quantitatively. Figure 3 shows results from a hypothetical trial in which the treatment reduces the risk of early bleeding deaths (red circles) by one quarter (relative risk = 0.75), but has no effect (relative risk = 1.00) on later non-bleeding deaths (blue circles). The relative risk at the end of the follow-up period is a weighted average of these relative risks: relative risk = 0.75(4/12) + 1.0(8/12) = 11/12 = 0.92, where (4/12) and (8/12) are the proportions of deaths in the untreated group that are early or late. Because the relative risk at the end of follow-up is closer to the null (0.92 versus 0.75), and smaller effects are harder to detect, the treatment effect is less visible and, in this sense, is diluted. But the biological effect did not change. It was not offset by any harm but was simply obscured by deaths unrelated to its mechanism of action. Deaths that are inevitable before randomisation also dilute treatment effects. Many patients with a GCS score of 3 or unreactive pupils have un-survivable injuries and will die soon after admission regardless of treatment. Errors in the estimation of the time of injury could result in the inclusion of patients outside the eligibility time window, and because late treatment is less effective, this will also cause dilution. This is most relevant in LMICs where patients are often taken to hospital by bystanders or family members in private vehicles with no recording of the time of injury.

Hypothetical trial in which the effect on all-cause mortality is a weighted average of the effect on cause-specific mortality. The trial treatment reduces the risk of early (bleeding) deaths by one quarter (RR = 0.75) but has no effect on late (non-bleeding) deaths (RR = 1.00). The overall relative risk for all-cause mortality at the end of follow-up is a weighted average of these relative risks (RR = 0.92)
Because no treatment has effects on all causes of death, all-cause mortality at 28 days is a composite outcome that combines deaths affected by the trial treatment with those that are unaffected by it [16]. Using all-cause mortality to assess the ‘true’ effect of a treatment has counterintuitive consequences since it means that the effect of any given treatment depends on the effects of all the others. An antibiotic that reduces pneumonia deaths in week 2, by reducing the proportion of late deaths, will appear to increase the effectiveness of a treatment for early bleeding. Because the proportion of late deaths varies with injury severity and in different locations, all-cause mortality is not generalisable. The only generalisable measure is the undiluted biological effect of the trial treatment.
Conclusions
Tranexamic acid safely reduces early deaths in non-moribund TBI patients regardless of TBI severity or country income. The effect of tranexamic acid in patients with isolated TBI is similar to that in polytrauma, reducing deaths on the day of the injury by over 20% in the CRASH-2 and the CRASH-3 trials. Tranexamic acid is included in treatment guidelines for the pre-hospital care of bleeding trauma patients, but patients with isolated TBI were excluded. The CRASH-3 trial data support the reconsideration of tranexamic acid for administration in isolated TBI, and even severely injured patients appear to benefit when treated soon after injury.
Availability of data and materials
Following publication of the primary and secondary analyses, individual de-identified patient data from the CRASH-3 trial will be made available via our data sharing portal, The Free Bank of Injury and Emergency Research Data (freeBIRD) website (http://freebird.Lshtm.ac.uk) indefinitely. The CRASH-2 trial data is already available. The trial protocols, statistical analysis plans and trial publications will be freely available online. The trial protocol, statistical analysis plan and trial publications will be freely available at http://www.txacentral.org/.
References
Stocchetti N, Carbonara M, Citerio G, et al. Severe traumatic brain injury: targeted management in the intensive care unit. Lancet Neurol. 2017;16:452–64.
CRASH-3 Collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet. 2019;394:1713–23.
Oertel M, Kelly DF, McArthur D, et al. Progressive hemorrhage after head trauma: predictors and consequences of the evolving injury. J Neurosurg. 2002;96:109–16.
Collaborators C-3 IBMS. Tranexamic acid in traumatic brain injury: an explanatory study nested within the CRASH-3 trial. Eur J Trauma Emerg Surg. 2020. https://doi.org/10.1007/s00068-020-01316-1.
Ker K, Roberts I. Tranexamic acid for surgical bleeding. BMJ. 2014;349:g4934.
CRASH-2 trial collaborators, Shakur H, Roberts I, et al. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet (London, England). 2010;376:23–32.
WOMAN Trial Collaborators. Effect of early tranexamic acid administration on mortality, hysterectomy, and other morbidities in women with post-partum haemorrhage (WOMAN): an international, randomised, double-blind, placebo-controlled trial. Lancet. 2017;389:2105–16.
Amrhein V, Greenland S, McShane B. Scientists rise up against statistical significance. Nature. 2019;567:305–7.
Gardner MJ, Altman DG. Confidence intervals rather than P values: estimation rather than hypothesis testing. Br Med J (Clin Res Ed). 1986;292:746–50.
Heidet M. Tranexamic acid for acute traumatic hemorrhage in emergency medicine. Eur J Emerg Med. 2020;1:85–86.
Taccone FS, Citerio G, Stocchetti N. Is tranexamic acid going to CRASH the management of traumatic brain injury? Intensive Care Med. 2019. https://doi.org/10.1007/s00134-019-05879-5.
Heymann EP. Tranexamic acid in traumatic intracranial bleeding: recognizing the limit of results (of the CRASH-3 trial). Eur J Emerg Med. 2019; published online Dec 18. https://doi.org/10.1097/MEJ.0000000000000657.
Cap AP. CRASH-3: a win for patients with traumatic brain injury. Lancet. 2019;394:1687–8.
Roberts I, Belli A, Brenner A, et al. Tranexamic acid for significant traumatic brain injury (The CRASH-3 trial): statistical analysis plan for an international, randomised, double-blind, placebo-controlled trial. Wellcome Open Res. 2018;3:86.
Roberts I, Prieto-Merino D, Manno D. Mechanism of action of tranexamic acid in bleeding trauma patients: an exploratory analysis of data from the CRASH-2 trial. Crit Care. 2014;18:685.
Brenner A, Arribas M, Cuzick J, et al. Outcome measures in clinical trials of treatments for acute severe haemorrhage. Trials. 2018;19:533.
Yutthakasemsunt S, Kittiwatanagul W, Piyavechvirat P, Phuenpathom N, Lumbiganon P. Tranexamic acid for patients with traumatic brain injury: a randomized, double-blinded, placebo-controlled trial. 2013 http://www.biomedcentral.com/1471-227X/13/20 (accessed 1 July 2019).
Acknowledgements
The CRASH-3 trial collaborators
Writing Committee—Prof Ian Roberts and Professor Haleema Shakur-Still (co-chairs), Amy Aeron-Thomas, Prof Antonio Belli, Amy Brenner, Prof Muhammad Anwar Chaudary, Prof Rizwana Chaudhri, Sabariah Faizah Bt Jamaluddin, Lauren Frimley, Kiran Javaid, Prof Rashid Jooma, Aasia Kayani, Caroline Leech, Prof Khalid Mahmood, Raoul Mansukhani, Julina Md Noor, Jorge Mejia-Mantilla, Phil Moss, Jason Pott, Prof Peter Sandercock, Prof Temitayo Shokunbi, and Liliana Vallecilla. Trial Steering Committee—Peter Sandercock (Chair), Henry Benjamin Hartzenberg, Manjul Joshipura (2011–2016), Amy Aeron-Thomas (patient representative [trial steering committee]; advocacy and justice manager, RoadPeace), Ian Roberts, Pablo Perel, and Haleema Shakur-Still. Data Monitoring and Ethics Committee—Michael J Clarke (chair), Samuel C Ohaegbulam, Anthony Rodgers, and Tony Brady (independent statistician). Protocol Committee—Ian Roberts, Haleema Shakur-Still, Yashbir Dewan, Phil Edwards, Edward O Komolafe, Jorge Mejia-Mantilla, and Pablo Perel. Clinical Trials Unit (CTU)—Monica Arribas (trial manager and research assistant), Emma Austin (assistant trial manager), Eni Balogun (trial manager), Lin Barneston (data manager 2011–2012), Collette Barrow (trial administrator), Danielle Beaumont (senior trial manager and research fellow), Myriam Benyahia (CTU administrator), Amy Brenner (research fellow), Imogen Brooks (trial assistant 2016–2018), Madeleine Cargill (data assistant), Laura Carrington (assistant trial administrator), Lisa Cook (assistant trial manager 2011), Beatrice Cornu-Hewitt (trial assistant), Phil Edwards (statistician 2011–2016), Lauren Frimley (trial manager and research assistant), Amber Geer (assistant data manager), Daniel Gilbert (data assistant 2012–2013), Catherine Gilliam (trial administrator), Julio Gil-Onandia (trial assistant), Daniel Hetherington (trial assistant 2012–2013), Courtenay Howe (CTU administrator 2015–2017), Carolyn Hughes (data assistant 2016–2017), David I’anson (assistant trial manager 2016–2017), Rob Jackson (data manager 2012–2014), Miland Joshi (statistician 2016–2017), Sneha Kansagra (assistant trial manager 2016–2018), Taemi Kawahara (senior trial manager 2011–2015), Katharine Ker (lecturer), Sergey Kostrov (systems officer), Abda Mahmood (PhD candidate), Raoul Mansukhani (medical statistician), Hakim Miah (IT systems manager), Bernard Ndungu (assistant trial manager 2016–2017), Kelly Needham (medical statistician), Cecilia Okusi (data assistant 2014), Aroudra Outtandy (trial assistant 2013–2015), Raul Pardinaz-Solis (assistant trial manager 2012–2013), Daniel Pearson (data assistant), Tracey Pepple (acting senior data manager), Claude Pisani (assistant trial manager 2013), Jason Pott (lead UK research nurse 2018), David Prieto-Merino (statistician 2012–2015), Danielle Prowse (assistant data manager), Nigel Quashi (data manager 2013–2016), Anna Quinn (data assistant 2013–2015), Maria Ramos (senior project administrator 2011–2015), Mia Reid (clerical assistant 2016–2018), Ian Roberts (chief investigator and CTU co-director), Chris Roukas (trial administrator 2013–2015), Giulia Scrapa (assistant trial manager 2018), Haleema Shakur-Still (project director and CTU co-director), Chelci Squires (trial assistant 2014–2016), Jemma Tanner (clinical trials associate 2013–2016), Andrew Thayne (data assistant), Lesley Vidaurre (assistant trial manager 2012), and Elizabeth Woods (assistant trial manager 2012–2015). Nigeria Coordinating Team—Bukola Fawole (coordinating centre director), Olusade Adetayo (assistant trial coordinator), Olujide Okunade (assistant trial coordinator), and Temitayo Shokunbi (clinical lead). Pakistan Coordinating Team—Rizwana Chaudhri (coordinating centre director), Kiran Javaid (assistant research coordinator), Rashid Jooma (clinical lead), and Aasia Kayani (research coordinator). National Coordinators—Rizwana Chaudhri (Pakistan), Rashid Jooma (Pakistan), Sabariah Faizah Bt Jamaluddin (Malaysia), Julina Md Noor (national coordinators assistant, Malaysia), Tamar Gogichaishvili (Georgia), Maria de los Angeles Munoz-Sanchez (Spain), Bukola Fawole (Nigeria), Temitayo Shokunbi (Nigeria), Jorge Mejia-Mantilla (Colombia), Liliana Vallecilla (Colombia), Fatos Olldashi (Albania), Satish Krishnan (United Arab Emirates), Vincent Djientcheu (Cameroon), Jorge Loria Castellanos (Mexico), Frank Rasulo (Italy), Qadamkhear Hama (Iraq), Yakub Mulla (Zambia), Prof Ioan Stefan Florian (Romania), Juan Tobar (El Salvador), Hussein Khamis (Egypt), Conor Deasy (Ireland), Bobby Wellsh (Papua New Guinea), Jean Williams-Johnson (Jamaica), Susilo Chandra (Indonesia), and Vincent Mutiso (Kenya).
CRASH trial sites and investigators (number of patients randomly assigned)
Pakistan (4567)—Lahore General Hospital Neurosurgery Unit I (1178): Rizwan Butt, Muhammad Hammad Nasir, Salman Ahmad, Farwah Aslam, Khurram Ishaque, Faheem Usmani, Shahrukh Rizvi, Farhad Ali, Omair Sajjad, and Ali Zunair; Jinnah Postgraduate Medical Centre (700): Lal Rehman, Raza Rizvi, Farrukh Javeed, Shakeel Ahmed, Asad Abbas, Ali Afzal, and Ali Mikdad; Lahore General Hospital Neurosurgery Unit III (648): Asif Bashir, Anwar Chaudary, Tariq Salahuddin, Bashir Ahemed, Shahrukh Rizvi, Faheem Usmani, and Amir Aziz; Jinnah Hospital Lahore (619): Naveed Ashraf, Shahzad Hussain, Usman Ahmad, Muhammad Asif, Muhammad Adil, and Adeel Rauf; Lahore General Hospital Neurosurgery Unit II (607): Khalid Mahmood, Rizwan Khan, Bilal Ahmad, Umair Afzal, Hassan Raza, and Quratul Ain; DHQ Hospital Narowal (303): Sajjad Yaqoob, Qaiser Waseem, Muffasser Nishat, Suneel Semvel, and Javed Iqbal; Services Hospital Lahore (226) Samra Majeed, Sana Zulfiqar, Madeeha Iqbal, Nazia Majeed, and Manzoor Ahmed; DHQ Rawalpindi (137): Nadeem Akhtar, Mohammad Malik, Yasir Shehzad, and Muhammad Yousaf; DHQ Hospital Khuzdar (65): Abdul Wahid, Abdul Samad, and Saifullah Shah; Lady Reading Hospital (31): Mumtaz Ali and Jehan Zeb; Shifa International Hospital (29): Abdus Salam Khan and Adeela Irfan; Liaquat National Hospital and Medical College (14): Salman Sharif; Liaquat University Hospital (7): Riaz Memon; Aga Khan University Hospital (3): Rashid Jooma. UK (3143)—Royal London Hospital (501): Ben Bloom, Tim Harris, Jason Pott, Imogen Skene, Geoffrey Bellhouse, and Olivia Boulton; University Hospital Coventry (312): Caroline Leech, Geraldine Ward, Catherine Jarvis, Carly Swann, and Sathananathan Ratnam; Queen Elizabeth Hospital Birmingham (302): Antonio Belli, Ronald Carrera, Kamal Yakoub, David Davies, and Emma Fellows; St George’s Hospital (280): Phil Moss, Heather Jarman, Sarah Rounding, Elizabeth Johnson, and Catherine Loughran; Salford Royal Hospital (176): Fiona Lecky, Kate Clayton, Angiy Michael, and Angela Coumbarides; Southmead Hospital (156): Jason Kendall, Beverley Faulkner, Ruth Worner, and Emma Gendall; King’s College Hospital (155): Philip Hopkins, Paul Riozzi, Hannah Cotton, and Raine Astin-Chamberlain; St Mary’s Hospital, London (117): Mark Wilson, Jan Bodnar, Rachel Williams, and Alberto Rigoni; Aintree University Hospital (108): Abdo Sattout, John Fletcher, Calum Edge, and Nina Maryanji; Addenbrooke’s Hospital (103): Adrian Boyle, Susie Hardwick, Ellen Nichols, and Catherine Hayhurst; Queen’s Medical Centre (100): Frank Coffey, Chris Gough, Philip Miller, and Lucy Ryan; John Radcliffe Hospital (76): Melanie Darwent, Alexis Espinosa, and Sally Beer; Royal Stoke University Hospital (71): Julie Norton, Holly Maguire, and Kay Finney; Derriford Hospital (67): Anthony Kehoe, Rosalyn Squire, and Alison Jeffery; Queen Alexandra Hospital (60): Christiane Vorwerk, Denise Foord, and Eliot Wilkinson; Northern General Hospital (57): Avril Kuhrt, Shammi Ramlakhan, and Stuart Reid; Royal Preston Hospital (41): Andy Curran and Sean McMullan; Leeds General Infirmary (39): Tajek Hassan and Stuart Nuttall; Great Western Hospital (32): Stephen Haig and Saif Al-Nahhas; Southampton General Hospital (31): Diederik Bulters and Ardalan Zolnourian; Dorset County Hospital (27): Tamsin Ribbons and Ian Mew; Gloucestershire Royal Hospital (27): Tanya de Weymarn and Victoria Hughes; Royal Liverpool Hospital (21): Jane McVicar; Queen Elizabeth University Hospital (20): Cieran McKiernan; Royal Berkshire Hospital (20): Liza Keating; Poole Hospital (17): Henrik Reschreiter; James Cook University Hospital (16): Judith Wright; Basingstoke and North Hampshire Hospital (13): Louisa Chan; Whiston Hospital (13): Himanshu Kataria; Glasgow Royal Infirmary (12): Alastair Ireland; Manchester Royal Infirmary (12): Richard Body; Royal Alexandra Hospital (12): Alasdair Corfield; Milton Keynes University Hospital (11): Shindo Francis; Hull Royal Infirmary (10): William Townend; Leicester Royal Infirmary (10): Timothy Coats; Musgrove Park Hospital (10): James Gagg; Wexham Park Hospital (10): Sarah Wilson; Royal Sussex County Hospital (8): Rowley Cottingham; Blackpool Victoria Hospital (7): Simon Tucker; Norfolk and Norwich University Hospital (7): Frank Sutherland; North Devon District Hospital (7): Louisa Mitchell; Whipps Cross University Hospital (7): Tim Harris; Whittington Hospital (7): Lucy Parker; Darlington Memorial Hospital (6): Ola Afolabi; Monklands Hospital (6): Fiona Hunter; Royal Cornwall Hospital (6): Mark Jadav; University Hospital of North Tees (6): Kayode Adeboye; Worthing Hospital (5): Mandy Grocutt; Royal Oldham Hospital (4): Gabrielle May; Royal United Hospitals Bath (4): David Watson; Arrowe Park Hospital (3): Andrea Wootten; Pinderfields General Hospital (3): Sarah Robertshaw; Birmingham Heartlands Hospital (2): Susan Dorrian; Gwynedd Hospital, Bangor (2): Rob Perry; Newham University Hospital (2): Tim Harris; University Hospital Lewisham (2): Hyun Choi; Western Infirmary (2): Claire McGroarty; Worcestershire Royal Hospital (1): Paul Shone; Yeovil District Hospital (1): David Maritz. Malaysia (1567)—Hospital Sungai Buloh (410): Sabariah Jamaluddin, Julina Noor, Norizan Rosli, Leonard Leong Sang Xian and Yong De Jun; Hospital Sultanah Bahiyah (241): Fatahul Mohamed, Cheng Hee Song, Arman Hawari, Leong Yuen Chin, and Hardawani Mohd Hussein; Hospital Sultanah Nur Zahirah (205): Mohd Lotfi, Hafiq Hamid, Nujaimin Udin, Peck Lian, and See Choo; Penang General Hospital (161): Kwanhathai Wong, Fathiyah Gani, Mardhiah Jusoh, and Darrsini Rajakumar; Miri General Hospital (111): Chia Boon Yang, Nur Shahidah Binti Dzulkiflee, Wong Chok Ky, and Muhaimin Azwan Bin Mohd Azman; Hospital Raja Permaisuri Bainun (101): Adi Bin Osman, Azma Haryaty Ahmad, Ramzuzaman Ismail, and Si Qi Lai; Hospital Sultanah Aminah (94): Mohd Amin Bin Mohidin, Norwani Binti Deraman, and Salliza Binti Selamat; Hospital Tuanku Fauziah (72): Ida Abidin, Nurkhairulnizam Halim, and Zuraini Bakar; Hospital Tengku Ampuan Afzan (41): Zainalabidin Mohamed Ismail, Badrul Hisham, and Ruhaida Kamal; Hospital Sultan Abdul Halim (36): Zainal Effendy and Mashitah Ismail; Hospital Seberang Jaya (30): Noor Azleen and Liu Yeo Seng; Universiti Sains Malaysia (26): Kamarul Aryffin Baharuddin and Regunath Kandasamy; Hospital Langkawi (13): Azlan Kamalludin; Hospital Kulim (8): Shamsul Asmee; Hospital Kemaman (7): Mohd Fadzil; Hospital Segamat (6): Ahmad Basitz; Hospital Pakar Sultanah Fatimah (5): Norhaya Abdullah. Georgia (771)— High Technology Medical Center, University Clinic (751): Tamar Gogichaishvili, Giorgi Ingorokva, Shota Ingorokva, Iamze Agdgomelashvili, Kote Mumladze, Ioseb Maisuradze, and Iulia Kugusheva; Archangel St Michael Multiprofile Clinical Hospital (18): Buba Shalamberidze; City Hospital 1 (2): Gia Tomadze. Spain (425)—Hospital Regional Universitario Carlos Haya (102): Juan Fernandez-Ortega, Raimundo Seara-Valero, Guillermo Ibañez-Botella, Victoria Garcia-Martinez; Hospital Alvaro Cunqueiro VIGO (82): Melida Garcia Martul, Santiago Freita Ramos, Guillermo Lago Preciado; Hospital Universitario Virgen del Rocio (77): Claudio Garcia-Alfaro, Angeles Munoz-Sanchez, Rafael Bellido-Alba; Hospital General Universitario de Ciudad Real (67): Carmen Corcobado, Ana Bueno, Alfonso Ambros; Complejo Hospitalario de Navarra (44): JuanTihista Jimenez, Jose Roldan Ramirez; Hospital Torrecardenas (21): José Martín; Hospital de Lucus Augusti (13): Laura Inés Rodríguez; Hospital Clinico de Barcelona (9): Jaime Fontanals; Hospital Universitario Puerta del Mar de Cadiz (9): José Manuel Jiménez-Moragas; Hospital General Universitario De Albacete (1): Joaquín Paya Berbegal. Nigeria (409)—National Hospital Abuja (64): Olaomi Oluwole, Raji Mahmud, and Nancy Ukwu; Lagos University Teaching Hospital (55): Femi Bankole, Abidemi Oseni, and Bamidele Adebayo; University College Hospital, Ibadan (53): Adefolarin Malomo, Liadi Tiamiyu, and Adefisayo Adekanmbi; Olabisi Onabanjo University Teaching Hospital (38): Lateef Thanni and Ayodeji Olubodun; Federal Medical Centre Abeokuta (36): Fidelis Ojeblenu and Michael Uwaezuoke; Obafemi Awolowo University Teaching Hospitals (31): Edward Komolafe and Oluwafemi Owagbemi; Lagos State Accident and Emergency Centre (22): Fatai Ishola; Bowen University Teaching Hospital Ogbomoso (17): Adewumi Durodola; Federal Medical Centre Lokoja (13): Ukpong Udoffa, Federal Medical Centre Bida (12): Adeniran James; Abubakar Tafawa Balewa University Teaching Hospital (11): Azeez Tella; Irrua Specialist Teaching Hospital (9): Andrew Dongo; Federal Medical Centre Umuahia (8): Uchechi Ekpemiro; Nnamdi Azikiwe University Teaching Hospital (8): Stanley Anyanwu; State Hospital, Ijaiye, Abeokuta (8): Nafiu Aigoro; University of Nigeria Teaching Hospital Enugu (7): Wilfred Mezue; Jos University Teaching Hospital (6): Danaan Shilong; University of Benin Teaching Hospital (6): Abiodun Azeez; Federal Medical Centre Ido-Ekiti (2): Olakunle Babalola; Federal Teaching Hospital, Gombe (2): Mohammed Ibrahim; University of Abuja Teaching Hospital (1): Joseph Obande. Colombia (335)—Hospital Pablo Tobon Uribe (127), Alfredo Constain Franco, Edwin Vasquez Salazar, Sebastian Betancur Londoño, and Viviana Medina Cardona; Hospital Universitario San Vicente Fundacion (112): Carlos Morales; Santiago Upegui; Santiago Naranjo; July Agudelo; Fundacion Valle del Lili (96): Jorge Mejia-Mantilla, Sandra Carvajal, and Yidhira Fajardo-Gaviria. Nepal (255)—Neuro Hospital (103): Yam Roka, Ushma Ghising, Narayani Roka, and Manzil Shrestha; National Institute of Neurological and Allied Sciences (64): Upendra Devkota, Bivek Vaidya, and Pankaj Nepal; Kathmandu Medical College Teaching Hospital (47): Amit Thapa and Bidur KC; Chitwan Medical College Teaching Hospital (24): Ajit Shrestha; Bir Hospital (11): Rajiv Jha; B & B Hospital Ltd. (6): Prabin Shrestha. Albania (214)—University Hospital of Trauma (214): Fatos Olldashi, Irgen Hodaj, Erion Spaho, Asllan Selaj, and Nirian Bendo. Japan (165)—Matsudo City Hospital (64): Tomohisa Shoko, Hideki Endo, and Atsushi Senda; Senshu Trauma and Critical Care Centre (61): Yasushi Hagihara, Takashi Fuse, and Naohisa Masunaga; Tokyo Medical and Dental University (28): Yasuhiro Otomo and Ryuichiro Egashira; Teikyo University Hospital (12): Takahiro Ohnuki. United Arab Emirates (126)—Al Qassimi Hospital (126): Satish Krishnan, Alya AlMazmi, Subrata Saha, and Alexander Suvarov. Myanmar (121)—1000 Bedded Nay Pyi Taw Hospital (121): Than Latt Aung, Kaung Myat Tun, Tint Tint Khaing, and Thinzar Maw. Cameroon (116)—Yaounde Central Hospital (38): Vincent Djientcheu and Orlane Ndome; Hopital General Douala (31): Mireille Moumi and André Mbida; Hopital Laquintinie de Douala (28): Joseph Fondop and N’Diaye; Yaounde General Hospital (19): Mba Sebastien. Afghanistan (87)—Nangarhar University Teaching Hospital (87): Abdul Azim, Jan Adil, and Zabiullah Amiry. Mexico (79)—Hospital Regional 25 IMSS (24): Jorge Loría-Castellanos; Hospital General Jose G Parres (21): Nancy Guevara Rubio; Hospital General de Uruapan, Pedro Daniel Martinez (11): Patricia Ortega Leon; Hospital General Regional No 1 (10): Francisco Estrada; Hospital General de Zona 197 (8): Erandy Montes de Oca-García; Hospital General Regional Bernardo Sepulveda (3): Hafid Sanchez; Hospital General La Perla (2): Angélica Soria. Italy (72)—Azienda Ospedaliera Universitaria Senese (35): Paola Bonucci and Federico Franchi; Fondazione Poliambulanza (19): Alan Girardini; Spedali Civili Di Brescia (18): Frank Rasulo. Iraq (55)—Rozhawa Emergency Hospital (51): Qadamkhear Hama, Himdad Hameed, and Muhammad Basim; Rojhelat Emergency Hospital (3): Qadamkhear Hama; Par Hospital (1): Qadamkhear Hama. Cambodia (45)—World Mate Emergency Hospital (45): Simon Stock and Eap Hourt. Zambia (44)—University Teaching Hospital Lusaka (40): Yakub Mulla and Ali Ilunga; Kitwe Central Hospital (4): Jonathan Mulenga. Romania (35)—Timisoara County Hospital (17): Horia Ples; Spitalul Sf Pantelimon Bucharest (11): Adam Danil; Bagdasar-Arseni Emergency Clinical Hospital (5): Mircea Gorgan; Cluj County Emergency Hospital (2): Ioan Florian. El Salvador (28)—Hospital Nacional Rosales (28): Juan Tobar, Fernandez. Egypt (20)—Mataria Teaching Hospital (20): Hussein Khamis. Slovenia (15)—University Medical Centre Ljubljana (15): Dusan Vlahovic. Ireland (12)—Cork University Hospital (12): Conor Deasy. Papua New Guinea (10)—Port Moresby General Hospital (10): Bobby Wellsh. Canada (7)—Saint John Regional Hospital (7): James French. Jamaica (7)—Cornwall Regional Hospital (5): Jeffrey East; University Hospital of the West Indies (2): Jean Williams-Johnson. Indonesia (6)—Rumah Sakit Sekar Kamulyan (6): Antonius Kurniawan. Kenya (1)—Kenyatta National Hospital, University of Nairobi (1): Julius Kiboi.
Funding
The CRASH-3 trial was funded by the JP Moulton Charitable Foundation, the UK National Institute for Health Research Health Technology Assessment programme (NIHR HTA; 14/190/01), the Joint Global Health Trials, Medical Research Council, Department for International Development, Global Challenges Research Fund, and the Wellcome Trust (MRM0092111). The CRASH-2 trial was funded by the UK National Institute for Health Research Health Technology Assessment programme, JP Moulton Charitable Foundation, the BUPA Foundation and Pfizer (grant-in-aid for tranexamic acid and placebo). The studies were designed, conducted, analysed and interpreted by the investigators, entirely independently of all funding sources. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the funders. The funders had no role in the study design; the collection, analysis or interpretation of the data; the writing of the report; or the decision to submit the paper for publication.
Author information
Authors and Affiliations
Consortia
Contributions
Study conception: IR and HSS. Data collection: ABe, RC, TC, LF, SJ, LF, RJ, PS and TS. Trial management: LF. Data analysis: ABr and RM. Data interpretation: ABr, RM and IR. Drafting the manuscript: ABr and IR. All authors were responsible for reviewing and revising the manuscript and have approved the final version. The corresponding author had full access to all the data and had final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Most patients with TBI are unable to provide informed consent to participate in a clinical trial due to the nature of their injury. As per the Declaration of Helsinki, patients who are incapable of giving consent are an exception to the general rule of informed consent in clinical trials. In the CRASH-3 trial, consent was sought from the patient’s relative or a legal representative unless no such representative was available, in which case the study proceeded with the agreement of two clinicians. If the patient regained capacity, they were told about the trial and written consent was sought to continue participation. If either the patient or their representative declined consent, participation was stopped. If patients were included in the trial but did not regain capacity, consent was sought from a relative or legal representative. We adhered to the requirements of the local and national ethics committees.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Supplementary Table 1. Effect of tranexamic acid on all-cause mortality within 24 hours of injury, after 24 hours and at 28 days stratified by severity and country income in patients randomised within 3 hours of injury. Supplementary Table 2. Effect of tranexamic acid on head injury death within 24 hours, after 24 hours and at 28 days by severity and country income in patients randomised within 3 hours of injury, excluding those with GCS 3 or bilateral unreactive pupils. Supplementary Table 3. Effect of tranexamic acid on head injury death within 24 hours, after 24 hours and at 28 days by severity and country income in patients randomised within 3 hours of injury.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Brenner, A., Belli, A., Chaudhri, R. et al. Understanding the neuroprotective effect of tranexamic acid: an exploratory analysis of the CRASH-3 randomised trial. Crit Care 24, 560 (2020). https://doi.org/10.1186/s13054-020-03243-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-020-03243-4