
Abstract
Background
WNT signaling is pivotal in embryogenesis and tissue homeostasis. Aberrant WNT signaling, due to mutations in components of this pathway, contributes to the development and progression of human cancers, including colorectal cancer. AXIN2, encoded by the AXIN2 gene, is a key negative regulator and target of the canonical WNT signaling pathway. Germline mutations in AXIN2 are associated with absence of permanent teeth (hypo- and oligodontia) and predisposition to gastrointestinal polyps and cancer. The limited number of patients makes an accurate genotype–phenotype analysis currently challenging.
Case presentation
We present the case of a 55-year-old male with colorectal polyposis and hypodontia. Genetic testing confirmed a novel frameshift germline mutation in exon 8 of the AXIN2 gene. In addition, we provide an updated overview of germline AXIN2 mutations reported in literature.
Conclusions
Although the number of missing teeth is less severe in our patient than in some previously reported cases, our findings provide additional evidence that missing teeth and gastrointestinal neoplasia are associated with rare pathogenic AXIN2 germline mutations.
Similar content being viewed by others


Background
The canonical WNT signal transduction pathway (also denoted as WNT/β-catenin pathway) is an ancient evolutionary conserved pathway that has a prominent role in embryogenesis and diverse physiological processes, including vascular survival and proliferation, maintenance of the intestinal stem cell niche, odontogenesis, and bone tissue remodelling [1]. In basal conditions, cytosolic β-catenin is continuously subjected to phosphorylation and subsequent proteolytic degradation by action of a multisubunit destruction complex [2]. This destruction complex is composed of the tumour suppressors adenomatosis polyposis coli (APC) and axin inhibition protein 1 (AXIN1) or its homologue AXIN2 (also known as conduction), and the kinases casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β) [2]. Upon stimulation by WNT-ligands, the cell-surface receptors Frizzled and co-receptor LRP5 or LRP6 recruit the cytosolic protein Dishevelled (DVL) that subsequently attracts the multisubunit destruction complex and thereby releases β-catenin [2]. This leads to nuclear translocation of β-catenin where it promotes the transcription of WNT target genes via physical interactions with T cell factor/lymphoid enhancer-binding factor (TCF/LEF) transcription factors [2].
Aberrant WNT signaling, due to germline and somatic mutations in components of this pathway, is a frequent driver in human cancer development and progression [2, 3]. For instance, germline loss-of-function mutations in the APC gene underlie familial adenomatous polyposis (FAP) [4, 5], a hereditary condition resulting in numerous colorectal polyps and a very high lifetime risk of colorectal cancer approaching 100% in absence of surveillance and prophylactic proctocolectomy or colectomy. Somatic APC mutations are present in approximately 75% of the apparently sporadic colorectal cancers [6].
Although rare, loss-of-function germline mutations in the gene AXIN2 (OMIM 604025; Fig. 1), encoding for a scaffolding component in the β-catenin destruction complex, also predispose to gastrointestinal polyps and cancer [7,8,9,10,11,12,13,14,15,16,17]. So far, approximately 30 patients with germline mutations in the AXIN2 gene have been described. The limited number of patients makes an accurate genotype-phenotype analysis currently challenging. The clinical phenotype is variable and in addition to gastrointestinal polyps, individuals with AXIN2 mutations tend to have hypo- and oligodontia [7,8,9,10,11,12,13,14,15,16,17]. Hypodontia, encountered in 2.2% to 10.1% in the general white population, denotes the congenital absence of one to five elements—excluding third molars—in the permanent dentition [18]. Oligodontia, which is very rare with an estimated prevalence of 0.14%, represents a more severe form of dental agenesis with the congenital missing of at least six elements—apart from the third molars—in the permanent dentition [18].

Genomic map of the AXIN2 gene showing an overview of the germline variants that have been reported so far in literature (NM_004655.3 and NP_004646.3) [7,8,9,10,11,12,13,14,15,16,17, 26]. Numbered boxes represent exons and the in-between lines depict introns. Please note that exon 1 and exon 11 are untranslated
In one previously described family the AXIN2 mutation not only segregated with colorectal polyposis and oligodontia, the mutation carriers also displayed mild features of ectodermal dysplasia (abnormal development of ectodermally derived structures—skin, sweat glands, hair, and nails) [8]. Furthermore, a single case study more recently suggested that the AXIN2-related phenotype spectrum might be expanded by neuroblastomas and gastric adenomas [11]. Somatic AXIN2 mutations are prevalent in various tissues with carcinoma, including gastro-intestinal [19], skin [20], and adrenal glands.
AXIN2 itself is a major target gene of WNT signalling. It acts in a negative feedback loop to limit and fine-tune the canonical WNT-signaling [21, 22]. The human AXIN2 gene is located on chromosome 17q24.1 and encompasses 10 coding exons that generate a protein of 843 amino acids (Fig. 1) [23]. AXIN2 harbours two highly conserved functional domains. The RGS (regulator of G protein signaling; amino acid 81–200) domain in the N-terminus, which mediates physical interaction with APC, and the DIX (Dishevelled/Axin; amino acid 761–843) domain that includes a binding site for DVL. Furthermore, AXIN2 contains binding motifs for interaction with Tankyrase (amino acid 21–30), GSK3β (amino acid 327–413) and β-catenin (amino acid 413–476).
Here, we report a family with a novel frameshift germline mutation in exon 8 of AXIN2 associated with colorectal polyposis and hypodontia. Furthermore, we provide an updated overview of germline AXIN2 mutations reported in the literature.
Case presentation
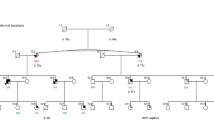
The proband and family members gave consent for the publication of non-identifiable details. The proband (Fig. 2), is a 55-year-old male of mixed European and Chinese descent who was referred by his gastroenterologist to the department of Clinical Genetics in the Amsterdam University Medical Center for genetic evaluation of colonic polyps. A first colono-scopy at the age of 44, was conducted because of changes in the defecation pattern. Two tubular adenomas with low grade dysplasia were identified. The gastroscopy was normal. A subsequent surveillance coloscopy at the age of 54 identified nine tubular adenomas with low grade dysplasia and one sessile serrated lesion without dysplasia. Dental records confirmed that the patient misses four premolars since childhood. The patient has rather scarce body hair which might be due to his partly Asian descent. He had no other possible features of ectodermal dysplasia. The proband has two children. His 15-year-old son is missing two second molars. In addition, agenesis of the third molars seems likely. His daughter has no missing teeth. Given their young age no genetic testing was yet performed in the proband’s children. The proband’s brother has no medical history of gastrointestinal polyps or cancer and has a full dentition. The proband’s father is from Chinese descent. He passed away at the age of 76. He did not have a history of gastrointestinal polyps or cancer. According to the proband, his father required multiple dental prosthesis, which is suggestive of hypo- or oligodontia. No paternale relatives are available for testing of the AXIN2 gene. The proband’s mother is from European descent. At the age of 90, there is no history of gastrointestinal polyps and she has a full dentition.

Family pedigree of the proband with hypodontia and polyposis. The proband is indicated by the arrow. Squares and circles represent males and females, respectively. Deceased individuals are indicated by a diagonal line through the symbol. Phenotypes are elaborated by segments showing the presence of hypodontia and polyposis. Schematic odontograms indicate the missing permanent teeth (in black) in the affected individuals
Methodology
A next-generation sequencing (NGS) test has been performed in the proband, utilizing a gene panel covering 23 relevant genes implicated in polyposis and colorectal cancer (ACVRL1, APC, AXIN2, BMPR1A, ENG, EPCAM, GREM1, MLH1, MLH3, MSH2, MSH3, MSH6, MUTYH, NTHL1, PMS2, POLD1, POLE, PTEN, RNF43, SMAD4, STK11, TSC1, and TSC2). Genomic DNA was extracted from peripheral-blood leukocytes in venous blood samples. DNA libraries for Illumina sequencing were generated using standard protocols from 200 ng to 1 µg of genomic DNA. The libraries were sequenced with paired-end (2 × 150 bp) runs using Illumina MiSeq with a target depth of 30 base coverage. In short, the sequencing reads were aligned to the human reference genome GRCh38 using BWA-MEM (0.7.5). GATK2.8 HaplotypeCaller was used to call SNPs and indels to create gVCF files. Variant Cartagenia Bench Lab V5.0 was used for variant interpretation and prioritization. All exons and 20 flanking intronic nucleotides of the above indicated genes have been analyzed. After gene prioritizing, we were left with one genetic variant in AXIN2.
Results
A novel heterozygous frameshift variant in exon 8 of the AXIN2 gene was found in the proband (please note that exon 8 was previously also depicted as exon 7 as exon 1 of AXIN2 is untranslated). The variant, cor-responding to c.2011dup (NM_004655.3), is predicted to cause a frameshift starting at position 671 (denoted as R671Pfs*36 in NP_004646.3) and likely results in a premature termination of translation. No (likely) pathogenic variants have been identified in the other 22 genes tested. This genetic variant was absent in the variation databases Single Nucleotide Polymorphism Database (dbSNP; accessed June 2021) [24] and Genome Aggregation Database (GnomAD) [25]. Furthermore, the variant was not present in an in-house database comprising 1662 individuals who underwent NGS panel testing (January 2015 until December 2022) because of a clinical suspicion of juvenile polyposis (n = 14), serrated polyposis (n = 22), familial polyposis (n = 364), Lynch syndrome (n = 518), Cowden syndrome (n = 1). The proband’s brother and mother, who are both healthy and have normal dentition, tested negative for the AXIN2 variant. Relatives of the proband’s father were not available for DNA testing. According on the American College of Medical Genetics and Genomics standards and guidelines for the interpretation of sequence variants, this novel AXIN2 variant is considered to be pathogenic (class 5).
Discussion
Here we describe the novel heterozygous pathogenic frameshift variant R671Pfs*36 in exon 8 of the AXIN2 gene in a 55-year-old man with polyposis and hypodontia. The rather scarce body hair might be due to his partly Asian descent. No other evident features of ectodermal dysplasia were present. Although the number of missing teeth is less severe in our patient than in some previously reported cases, our findings provide additional evidence that missing teeth and gastrointestinal neoplasia are associated with rare pathogenic AXIN2 germline mutations.
The mutation R671Pfs*36 is predicted to cause a premature termination of protein synthesis and as a consequence this genetic alternation is likely to result in loss of the DIX domain. The DIX domain is not only crucial for interaction with the protein DVL, but is also required for AXIN2 homodimerization. Therefore, in line with previously reported frameshift and truncating mutations affecting exon 8, this novel pathogenic variant will likely impair the inhibitory action of AXIN2 on the WNT sig-naling pathway.
In the AXIN2 gene exon 8 can be considered as a mutation hotspot, as it is a common location for germline (Fig. 1), as well as somatic mutations (not indicated). The germline mutations mainly concern frameshift mutations, which is not surprising, as exon 8 contains several mononucleotide repeat tracts. In the first reported family, 11 individuals lacking at least 8 permanent teeth and colonoscopic findings ranging from normal to polyposis and colorectal cancer, harboured the nonsense mutation R656* in exon 8 [7]. In the same study, the authors identified another mutation affecting exon 8 (de novo frameshift N666Qfs*41 mutation) in an unrelated 13-year-old patient with severe oligodontia [7]. Whether or not there is a gastrointestinal phenotype in this patient is unknown because of his young age. However, in the meantime this pathogenic variant has been identified in at least four unrelated families in which mutation carriers in addition to hypo- or oligodontia displayed a gastrointestinal phenotype ranging from polyposis to colorectal cancer [12, 16, 17, 26]. Furthermore, the phenotype of oligodontia and colorectal polyposis associated with an exon 8 mutation has been confirmed in three family members harbouring the frameshift S658A*31 mutation [10]. Interestingly, the exon 8 mutation W663*, not only segregated with an autosomal dominant pattern with oligodontia and a variable gastrointestinal neoplastic phenotype, the affected individuals also displayed mild features of ectodermal dysplasia [10]. It remains to be defined whether ectodermal dysplasia is indeed due to the AXIN2 mutation as this is the only family reported to date with clear features of ectodermal dysplasia.
Although genotype–phenotype correlations are difficult to establish due to the limited number of patients with (likely) pathogenic AXIN2 mutations, missense mutations that are situated substantially away from exon 8 and affect other protein domains have been described in patients with gastrointestinal polyps and without oligodontia (Table 1). For other cancer predisposition genes, including the APC gene, different phenotypes have been described for mutations in different parts of the gene. In light of studying pathogenic AXIN2 variants and their effect on protein function and clinical phenotype, the currently build AXIN2 patient registry will be a unique valuable resource [11]. The AXIN2 patient registry can also contribute to awareness among medical specialists, including dentists that people with oligodontia or families with hypodontia should be referred to a clinical geneticist to discuss AXIN2 analysis. This is important as people with AXIN2 mutations can benefit from regular colonoscopy. By removal of polyps the development of colorectal cancer can possibly be prevented. As no comprehensive guidelines are currently available and patients lack evidence of accelerated adenoma-carcinoma progression, we recommend carriers to receive colono-scopy every three years starting from the age of 25 years. The gastroscopy in our patient was negative. In light of the recently reported co-occurrence of germline AXIN2 mutation with a gastric adenoma, one-off gastroscopy in carriers might gain insight whether gastric adenomas are part of the AXIN2-related phenotype spectrum.
Furthermore, with the recent advances of the organoid technology [27], a patient registry also provides an extraordinary opportunity for additional basic and translational studies as patient-derived intestinal organoids – obtained during surveillance colonoscopy – can gain important insight in WNT signaling in a pathophysiological relevant environment. Application of the CRISPR/CAS9 technology in the recently developed dental pulp-organoids [28] provides an alternative to recapitulate the actions of AXIN2 mutations and study the basics in human tooth organogenesis. In-depth investigation of molecular mechanisms underlying carcinogenesis and tooth development, patient-derived organoids will pave the way for more targeted strategies for personalized cancer medicine and allow for tooth regeneration [29].
Availability of data and materials
Not applicable.
Abbreviations
- APC:
-
Adenomatosis polyposis coli
- AXIN1:
-
Axin inhibition protein 1
- AXIN2:
-
Axin inhibition protein 2
- CK1:
-
Casein kinase 1
- GSK3β:
-
Glycogen synthase kinase 3β
- DVL:
-
Dishevelled
- TCF/LEF:
-
T cell factor/lymphoid enhancer-binding factor
- FAP:
-
Familial adenomatous polyposis
- RGS:
-
Regulator of G protein signaling
References
Steinhart Z, Angers S. Wnt signaling in development and tissue homeostasis. Development. 2018;145:dev146589. Available from: http://dev.biologists.org/content/145/11/dev146589.
Bugter JM, Fenderico N, Maurice MM. Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat Rev Cancer. 2021;21(1):5–21. https://doi.org/10.1038/s41568-020-00307-z.
Nusse R, Clevers H. Wnt/β-Catenin Signaling, Disease, and emerging therapeutic modalities. Cell. 2017;169:985–99. Available from: https://www.sciencedirect.com/science/article/pii/S0092867417305470.
Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science (80-). 1991;253:665-LP−669. Available from: http://science.sciencemag.org/content/253/5020/665.abstract.
Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell Elsevier. 1991;66:589–600. https://doi.org/10.1016/0092-8674(81)90021-0.
Network CGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–7. Available from: https://pubmed.ncbi.nlm.nih.gov/22810696.
Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, et al. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74:1043–50 (The American Society of Human Genetics) Available from: https://pubmed.ncbi.nlm.nih.gov/22810696.
Marvin ML, Mazzoni SM, Herron CM, Edwards S, Gruber SB, Petty EM. AXIN2-associated autosomal dominant ectodermal dysplasia and neoplastic syndrome. Am J Med Genet A. 2011;155A:898–902.
Beard C, Purvis R, Winship IM, Macrae FA, Buchanan DD. Phenotypic confirmation of oligodontia, colorectal polyposis and cancer in a family carrying an exon 7 nonsense variant in the AXIN2 gene. Fam Cancer Netherlands. 2019;18:311–5.
Rivera B, Perea J, Sánchez E, Villapún M, Sánchez-Tomé E, Mercadillo F, et al. A novel AXIN2 germline variant associated with attenuated FAP without signs of oligondontia or ectodermal dysplasia. Eur J Hum Genet. 2014;22:423–6.
Macklin- Mantia SK, Hines SL, Chaichana KL, Donaldson AM, Ko SL, Zhai Q, et al. Case report expanding the germline AXIN2- related phenotype to include olfactory neuroblastoma and gastric adenoma. BMC Med Genet. 2020;21:161. https://doi.org/10.1186/s12881-020-01103-0.
Macklin-Mantia SK, Riegert-Johnson DL. An American patient with polyposis carrying a Scandinavian AXIN2 pathogenic variant. Hered Cancer Clin Pract. 2020;18:14. https://doi.org/10.1186/s13053-020-00149-8.
Hamzaoui N, Alarcon F, Leulliot N, Guimbaud R, Buecher B, Colas C, et al. Genetic, structural, and functional characterization of POLE polymerase proofreading variants allows cancer risk prediction. Genet Med United States. 2020;22:1533–41.
Rohlin A, Rambech E, Kvist A, Törngren T, Eiengård F, Lundstam U, et al. Expanding the genotype-phenotype spectrum in hereditary colorectal cancer by gene panel testing. Fam Cancer. 2017;16:195–203.
Hansen MF, Johansen J, Sylvander AE, Bjørnevoll I, Talseth-Palmer BA, Lavik LAS, et al. Use of multigene-panel identifies pathogenic variants in several CRC-predisposing genes in patients previously tested for Lynch Syndrome. Clin Genet Denmark. 2017;92:405–14.
Jensen JM, Skakkebæk A, Gaustadness M, Sommerlund M, Gjørup H, Ljungmann K, et al. Familial colorectal cancer and tooth agenesis caused by an AXIN2 variant: how do we detect families with rare cancer predisposition syndromes? Fam Cancer Netherlands. 2022;21:325–32.
Chan JM, Clendenning M, Joseland S, Georgeson P, Mahmood K, Walker R, et al. Rare germline variants in the AXIN2 gene in families with colonic polyposis and colorectal cancer. Fam Cancer Netherlands. 2022;21:399–413.
Polder BJ, Van’t MA, Van der Linden FPGM, Kuijpers-Jagtman AM. A meta-analysis of the prevalence of dental agenesis of permanent teeth. Community Dent Oral Epidemiol. 2004;32:217–26. https://doi.org/10.1111/j.1600-0528.2004.00158.x. (Wiley & Sons, Ltd).
Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, Krishnadath KK, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet United States. 2000;26:146–7.
Pedace L, Castiglia D, De Simone P, Castori M, De Luca N, Amantea A, et al. AXIN2 germline mutations are rare in familial melanoma. Genes Chromosomes Cancer. 2011;50(5):370–3. https://doi.org/10.1002/gcc.20855.
Jho E, Zhang T, Domon C, Joo C-K, Freund J-N, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–83.
Lustig B, Jerchow B, Sachs M, Weiler S, Pietsch T, Karsten U, et al. Negative feedback loop of Wnt signaling through upregulation of conductin/axin2 in colorectal and liver tumors. Mol Cell Biol. 2002;22:1184–93.
Dong X, Seelan RS, Qian C, Mai M, Liu W. Genomic structure, chromosome mapping and expression analysis of the human AXIN2 gene. Cytogenet Cell Genet Switzerland. 2001;93:26–8.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. https://doi.org/10.1093/nar/29.1.308.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43. https://doi.org/10.1038/s41586-020-2308-7.
Bergendal B, Klar J, Stecksén-Blicks C, Norderyd J, Dahl N. Isolated oligodontia associated with mutations in EDARADD, AXIN2, MSX1, and PAX9 genes. Am J Med Genet A United State. 2011;155A:1616–22.
Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature England. 2009;459:262–5.
Rosowski J, Bräunig J, Amler A-K, Strietzel FP, Lauster R, Rosowski M. Emulating the early phases of human tooth development in vitro. Sci Rep. 2019;9:7057. https://doi.org/10.1038/s41598-019-43468-0.
Merenda A, Fenderico N, Maurice MM. Wnt signaling in 3d: recent advances in the applications of intestinal organoids. Trends Cell Biol Elsevier. 2020;30:60–73. https://doi.org/10.1016/j.tcb.2019.10.003.
Acknowledgements
We thank the proband and his family for their participation.
Funding
Not applicable. No funding.
Author information
Authors and Affiliations
Contributions
M.B. and L.H. have written the report. L.H. and M.U. were involved in the clinical characterisation. E.R. was involved in the genetic analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent for publication of their clinical details was obtained from the patient.
Consent for publication
Written informed consent for publication of their clinical details was obtained from the patient. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Broekema, M.F., Redeker, E.J.W., Uiterwaal, M.T. et al. A novel pathogenic frameshift variant in AXIN2 in a man with polyposis and hypodontia. Hered Cancer Clin Pract 21, 16 (2023). https://doi.org/10.1186/s13053-023-00260-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13053-023-00260-6