
Abstract
Background
The most frequently identified strong cancer predisposition mutations for colorectal cancer (CRC) are those in the mismatch repair (MMR) genes in Lynch syndrome. Laboratory diagnostics include testing tumors for immunohistochemical staining (IHC) of the Lynch syndrome-associated DNA MMR proteins and/or for microsatellite instability (MSI) followed by sequencing or other techniques, such as denaturing high performance liquid chromatography (DHPLC), to identify the mutation.
Methods
In an ongoing project focusing on finding Mendelian cancer syndromes we applied whole-exome/whole-genome sequencing (WES/WGS) to 19 CRC families.
Results
Three families were identified with a pathogenic/likely pathogenic germline variant in a MMR gene that had previously tested negative in DHPLC gene variant screening. All families had a history of CRC in several family members across multiple generations. Tumor analysis showed loss of the MMR protein IHC staining corresponding to the mutated genes, as well as MSI. In family A, a structural variant, a duplication of exons 4 to 13, was identified in MLH1. The duplication was predicted to lead to a frameshift at amino acid 520 and a premature stop codon at amino acid 539. In family B, a 1 base pair deletion was found in MLH1, resulting in a frameshift and a stop codon at amino acid 491. In family C, we identified a splice site variant in MSH2, which was predicted to lead loss of a splice donor site.
Conclusions
We identified altogether three pathogenic/likely pathogenic variants in the MMR genes in three of the 19 sequenced families. The MLH1 variants, a duplication of exons 4 to 13 and a frameshift variant, were novel, based on the InSiGHT and ClinVar databases; the MSH2 splice site variant was reported by a single submitter in ClinVar. As a variant class, duplications have rarely been reported in the MMR gene literature, particularly those covering several exons.
Similar content being viewed by others
Background
Familial cancer, here defined as two or more first-degree relatives diagnosed with the same cancer, accounts for some 15% of colorectal cancer (CRC) [1]. The most frequently identified strong cancer predisposition mutations for CRC are those in mismatch repair (MMR) genes in Lynch syndrome, which account for approximately 1% of CRCs in the population (depending on the population) [2]. A number of other high-risk genes are known but variants in these are very rare [3]. In addition, ever-increasing numbers (> 100) of low-risk gene variants have been described for CRC [4]; yet combined, the high and low-risk variants explain only a small proportion of the known familial risk and even less of the heritability estimated in twin studies [5, 6].
Clinical diagnostics of Lynch syndrome usually first considers family history based on the Amsterdam and Bethesda criteria [7]. These are not perfect as half of germline-confirmed Lynch syndrome patients fail to meet the Amsterdam II criteria and, although the Bethesda guidelines are sensitive, their specificity is low [7]. Diagnostic laboratory tests include testing tumors for immunohistochemical (IHC) staining of the Lynch syndrome-associated DNA MMR proteins and/or for microsatellite instability (MSI) [7]. While these tests alone have a sensitivity ranging from 55 to 90% of predicting Lynch syndrome, combining the two will reach a sensitivity over 90% [7]. The identification of mutations is done by sequencing, or by other techniques, such as denaturing high performance liquid chromatography (DHPLC) or multiplex ligation dependent probe amplification (MLPA) for structural variants [8]. More recently, next generation sequencing panels have become the golden standard in identification of pathogenic germline variants in hereditary cancer syndromes. In a recent study, a universal 83-gene next generation sequencing panel identified nearly double as many pathogenic germline variants related to hereditary cancer syndromes as the guideline-directed targeted testing in unselected cancer patients, leading to a treatment change for nearly 30% of these patients [9]. This highlights the usefulness of next generation sequencing in the clinical praxis and compensates the limitations of the clinical and guideline-based risk assessment.
We have been involved in a whole-exome/whole-genome sequencing (WES/WGS) project aimed at identifying Mendelian type cancer syndromes in families referred to the Hereditary Cancer Center, Szczecin. In three families fulfilling the Amsterdam II criteria of Lynch syndrome with negative results in DHPLC mutation screening of the Lynch syndrome-related MMR genes we identified a mutation in these genes using whole genome sequencing. Here, we report these variants, particularly a large duplication in the MLH1 gene, as these types of large structural variants, particularly insertions are rarely described in Lynch syndrome [10,11,12,13,14].
Patients and methods
In several regions of Poland, population screening was performed mainly in years 2000–2014, in which questionnaires on cancer family history were collected systematically. Individuals with a positive CRC family history were invited to genetic outpatient clinics all over Poland and their more detailed family histories were taken through detailed face-to-face interviews. Nineteen families with strong CRC aggregation compatible with an autosomal dominant pattern of inheritance were recruited to the study. Each family had at least three pathologically confirmed CRC cases; 17 families had at least one case diagnosed below the age of 55 years. All 19 families had undergone DHPLC analysis for MMR variants with negative test results [15]. The ethical approval for this study design was obtained from the Bioethics Committee of the Pomeranian Medical Academy in Szczecin No: BN-001/174/05. Sample collection was performed following the guidelines proposed by this Committee. A written informed consent was signed by each participant in accordance with the Helsinki declaration.
WES on CRC patients and healthy family members of 5 families and WGS on 14 families was performed in the Illumina X10 platform using DNA extracted from the blood samples. WGS was carried out as paired-end sequencing with a read length of 150 bp. Sequences were mapped to the reference human genome (build hg19, assembly hs37d5) using BWA mem (version 0.7.8) and duplicates were marked using Picard (version 1.125). Single nucleotide variants and small indels were called by using Platypus (version 0.8.1) and annotated using ANNOVAR [16], dbSNP [17], 1000 Genomes phase III [18], dbNSFP v.2.9 [19], and ExAC [20], respectively. Variant filtering was carried out by considering a minimum of 5 reads coverage and a minimum QUAL score of 20. To check for family relatedness, a pairwise comparison of variants among the cohort was performed.
GATK gCNV module (version 4.1.7.0) was used to call germline copy number variants (gCNVs) from the WES/WGS samples individually against a background of 200 WGS samples sequenced from the sample platform. The gCNVs were called based on the best practice recommended by the GATK (https://gatk.broadinstitute.org/hc/en-us/articles/360035531152%2D%2DHow-to-Call-common-and-rare-germline-copy-number-variants). The major deviation from the above best practice was that the gCNVs cohort models were created only for the Gencode v19 exonic regions of WGS data by considering them as the target regions. The sequences of the samples from the CRC families were compared against this model. This decreased the turnaround time for the analysis of gCNVs from the WGS data.
The resulting gCNV segments with QS score above 30 were selected and annotated with the subset of gnomAD structural variant (SV) data (version 2.1, variants with ‘PASS’ filter tags and ‘DUP’ or ‘DEL’ SV types) using vcfanno [21]. The segments with at least 80% overlap with a common gnomAD SV (popmax MAF > 0.1%) of same SV subtype were considered as common and removed. In addition, to consider a gCNV as rare, at least 50% of the targets (exons here) in the gCNV segments should have the denoised ploidies among the bottom (in the case of deletion) or top (in the case of duplication) 5% of denoised cohort ploidies from the background cohort samples. Subsequently, the candidate rare gCNVs were selected if they followed the disease inheritance pattern in the family. For the candidate gCNVs the genomic breakpoints were manually reviewed using the Integrative Genomic Viewer (IGV) [22] to determine the genomic coordinates of the gCNVs.
Sequencing data were visually inspected using IGV to exclude false positive variants. For variants causing a frameshift, we used the Translate tool (https://web.expasy.org/translate/) to translate the nucleotide sequence to a protein sequence. The effect of splice site variants on splicing was analyzed using NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/). Combined Annotation-Dependent Depletion (CADD) score was used to evaluate the deleteriousness of the variants; the scores > 20 and > 30 are indicative of the top 1% and top 0.1% of deleterious variants, respectively [23]. The InSiGHT database available at the Leiden Open Variation Database (LOVD) v.3.0 [24, 25], ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) [26], gnomAD database (https://gnomad.broadinstitute.org/) and the recent publication on Chinese MMR variants were used as a reference [11].
IHC and MSI analyses were performed as reported previously [8, 27] in CRC samples from individuals with a MMR gene variant detected through WES or WGS.
Results
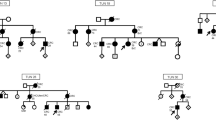
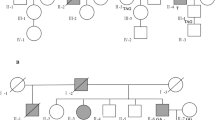
In three of the 19 families sequenced, pathogenic/likely pathogenic MMR gene variants were identified. The pedigree of family A is shown in Fig. 1. Several patients diagnosed with CRC were present in three generations. We sequenced the affected father (diagnosed at age 70 years) and his son (diagnosed at age 32 years). Additionally, two unaffected individuals were sequenced. The pedigrees of families B and C are found in Additional file 1: Fig. 1.

Pedigree of the colorectal cancer family with MLH1 exon 4–13 duplication
The detected variants are listed in Table 1. In family A, a structural variant, a duplication of chr3:37045366–37,071,869 covering exons 4 to 13 of MLH1 was identified. It was predicted to lead to a frameshift at amino acid 520 and a premature stop codon at amino acid 539. The duplication was identified in both patients and in an unaffected female relative who was 9 years older than her affected brother. In family B, a one base pair deletion was found in MLH1 which resulted in a frameshift and a stop codon at amino acid 491. In family C, the three affected individuals carried a splice site variant in MSH2, with a CADD score of 23.4 (Table 1). According to NetGene2, the MSH2 variant c.792 + 1G > C lead to a loss of a splice donor site.
The IHC and MSI results of the tumor samples from the Lynch syndrome patients are shown in Table 2. Tumor samples from patients from families A and B did not express MLH1 and PMS2 proteins while in family C the tumor sample was negative for MSH2 and MSH6 proteins. The results are in line with the mutation analysis as MLH1 and PMS2 as well as MSH2 and MSH6 form heterodimers. Further in line, the MSI analysis showed MSI-high for families A and C; the analysis for family B failed. Capillary electrophoresis MSI diagrams for samples from families A and C are shown with pattern shifts for the monomorphic markers (Additional file 1: Fig. 2). The identical migration of the pentanucleotide markers confirms the sample identity.
The consequences of the MLH1 variants on the gene and protein structure are shown in Fig. 2. In family A, the large duplication of exons 4 to13 covered a small section of the ATP binding domain (HATPase C domain) and the entire mismatch repair domain (MutL, i.e., MSH2-MLH1 heterodimer binding domain) as well as a small part of the MLH1 C-terminal domain (Fig. 2a). The duplication was predicted to lead to a frameshift at amino acid 520 and a premature stop codon at amino acid 539. Figure 2b shows the MLH1 frameshift variant at amino acid 425 in family B leading to a premature stop codon at amino acid 491. Both variants were predicted to lead to the deletion of the MLH1 C-terminal domain, which is needed for the MLH1-PMS2 heterodimerization.

Graphic representation of the MLH1 structure describing the consequences and location of the MLH1 duplication and the frameshift variant. (a) MLH1 duplication of exons 4–13 in family A leads to a frameshift at amino acid 520 and a premature stop codon at amino acid 539. (b) MLH1 frameshift variant at amino acid 425 in family B leads to a premature stop codon at amino acid 491. Both variants lead to the deletion of the MLH1 C-terminal domain, which is needed for the MLH1-PMS2 heterodimerization
Discussion
The present sequencing effort in families with a CRC family history suggestive of autosomal dominant inheritance identified two families with a pathogenic variant in the MLH1 gene and one family with a likely pathogenic variant in the MSH2 gene. In Poland, over 100 MMR gene point mutations have been identified, most of which are either frameshift or nonsense mutations leading to a truncated protein [28]. In over 60% of all Polish Lynch syndrome families a recurrent mutation is present. Two of the most frequent alterations are a substitution of A to T at the splice donor site of intron 5 of MSH2 and a missense change (A681T) of MLH1 [8]. In Polish patients, large deletions have been described particularly in the MSH2 gene [8].
The present three variants have so far not been reported in InSiGHT [24, 25]; only the MSH2 variant has been reported once in ClinVar [26]. However, the InSiGHT database lists similar MLH1 variants causing a frameshift and leading to a protein truncation at approximately the same position as our variants, both have a classification “pathogenic” [24, 25] (Table 3). The MSH2 variant is reported in ClinVar by a single submitter (accession number VCV000951452.1) and predicted to be “likely pathogenic” [26]. InSiGHT reports another nucleotide change at the same position, a c.792 + 1G > A variant, with a classification “pathogenic” (Table 3).
The present MLH1 variants were predicted to cause protein truncation and to be pathogenic, while the MSH2 splice site variant was predicted to be likely pathogenic. They add to the large collection of (likely) pathogenic variants in the MMR genes. Although all of these were unique, the duplication is of special interest as large duplications have rarely been reported for MMR genes. In the European literature somewhat over 10 exon level duplications have been reported, most of them in MSH2 and fewer in MLH1 [10, 12]. Similarly, in the recent Chinese literature survey on 34,000 individuals including both cancer cases and individuals without cancer, 540 MMR variants were found, but only 3 single exon duplications were reported for MLH1 and one for MSH2 [11]. In one of these papers the breakpoints implicated Alu mediated recombination as a mechanism and the duplication was predicted to create a premature stop codon and the formation of a truncated protein [12]. In the InSiGHT database, only 9 exon-level duplications in MLH1 are reported compared to 77 deletions; in MSH2 the numbers are 7 duplications and 84 deletions [24, 25] (Additional file 1: Table 1). While all the deletions had clinical classification “pathogenic”, only 2 duplications in MLH1 and 3 in MSH2 were classified as “pathogenic”. While large deletions most likely lead to non-functional proteins, the effect of large duplications may depend on whether the duplication is in-frame or not. The duplication in MLH1 we present here is predicted to cause a frameshift and a truncated protein.
The present duplication of exons 4 through 13 covered a small section of the ATP binding domain (HATPase C domain), the entire mismatch repair domain (MutL, i.e., MSH2-MLH1 heterodimer binding domain) and part of the MLH1 C-terminal domain [29,30,31]. The out-of-frame change at amino acid 520 was predicted to cause a stop codon further down-stream at amino acid 539. Thus, the resulting truncated protein is probably degraded by non-sense mediated decay as supported by the IHC results of lack of MLH1 protein in the tumor. The C-terminal end of MLH1 contains important binding sites for heterodimeric MMR proteins that contribute to the various key functions such as endonuclease activity [30, 31].
The fact that these three mutations were missed in the previous screening early 2000s may be due to the methodology used at that time, DHPLC. The DHPLC primers were designed to cover all exons and approximately 30–60 bp upstream and downstream of each exon. As the breakpoints of the large duplication in MLH1were located 526 bp downstream of exon 4 and 1446 bp upstream of exon 13, it was missed. Also the splice site variant in MSH2 may have been missed, because its distance to the upstream primer for detecting exon 4 was only 2 bp. Only the frameshift variant in MLH1 was located in the middle of exon 12 and might have been possible to detect. This calls for the recommendation that historically negative cases, assessed by inferior methods, should be re-considered for testing using up-to-date methodologies.
Conclusions
We identified three novel MMR gene variants that were predicted to lead to truncated proteins. The variants segregated with the disease and are expected to predispose to Lynch syndrome phenotypes, including CRC.
Availability of data and materials
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CRC:
-
colorectal cancer
- MMR:
-
mismatch repair
- IHC:
-
immunohistochemical
- MSI:
-
microsatellite instability
- DHPLC:
-
denaturing high performance liquid chromatography
- MLPA:
-
multiplex ligation dependent probe amplification
- WES/WGS:
-
whole-exome/whole-genome sequencing
- gCNV:
-
germline copy number variant
- SV:
-
structural variant
- IGV:
-
Integrative Genomic Viewer
- CADD:
-
Combined Annotation-Dependent Depletion
References
Frank C, Sundquist J, Yu H, Hemminki A, Hemminki K. Concordant and discordant familial cancer: familial risks, proportions and population impact. Int J Cancer. 2017;140:1510–6.
Chubb D, Broderick P, Frampton M, Kinnersley B, Sherborne A, Penegar S, et al. Genetic diagnosis of high-penetrance susceptibility for colorectal cancer (CRC) is achievable for a high proportion of familial CRC by exome sequencing. J Clin Oncol. 2015;33(5):426–32.
Valle L, de Voer RM, Goldberg Y, Sjursen W, Forsti A, Ruiz-Ponte C, et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol Asp Med. 2019;69:10–26.
Huyghe JR, Bien SA, Harrison TA, Kang HM, Chen S, Schmit SL, et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet. 2019;51(1):76–87.
Lichtenstein P, Holm N, Verkasalo P, Illiado A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer. N Engl J Med. 2000;343:78–85.
Mucci LA, Hjelmborg JB, Harris JR, Czene K, Havelick DJ, Scheike T, et al. Familial risk and heritability of Cancer among twins in Nordic countries. JAMA. 2016;315(1):68–76.
Cohen SA, Pritchard CC, Jarvik GP. Lynch syndrome: from screening to diagnosis to treatment in the era of modern molecular oncology. Annu Rev Genomics Hum Genet. 2019;20:293–307.
Kurzawski G, Suchy J, Lener M, Klujszo-Grabowska E, Kladny J, Safranow K, et al. Germline MSH2 and MLH1 mutational spectrum including large rearrangements in HNPCC families from Poland (update study). Clin Genet. 2006;69(1):40–7.
Samadder NJ, Riegert-Johnson D, Boardman L, Rhodes D, Wick M. Okuno S, et al. JAMA Oncol: Comparison of Universal Genetic Testing vs Guideline-Directed Targeted Testing for Patients With Hereditary Cancer Syndrome; 2020.
Baert-Desurmont S, Buisine MP, Bessenay E, Frerot S, Lovecchio T, Martin C, et al. Partial duplications of the MSH2 and MLH1 genes in hereditary nonpolyposis colorectal cancer. Eur J Hum Genet. 2007;15(3):383–6.
Zhang L, Bhaskaran SP, Huang T, Dong H, Chandratre K, Wu X, et al. Variants of DNA mismatch repair genes derived from 33,998 Chinese individuals with and without cancer reveal their highly ethnic-specific nature. Eur J Cancer. 2020;125:12–21.
Liccardo R, De Rosa M, Rossi GB, Rigler G, Izzo P, Duraturo F. Characterization of novel, large duplications in the MSH2 gene of three unrelated lynch syndrome patients. Cancer Genet. 2018;221:19–24.
Duraturo F, Cavallo A, Liccardo R, Cudia B, De Rosa M, Diana G, et al. Contribution of large genomic rearrangements in Italian lynch syndrome patients: characterization of a novel alu-mediated deletion. Biomed Res Int. 2013;2013:219897.
Kloor M, Sutter C, Wentzensen N, Cremer FW, Buckowitz A, Keller M, et al. A large MSH2 Alu insertion mutation causes HNPCC in a German kindred. Hum Genet. 2004;115(5):432–8.
Kurzawski G, Safranow K, Suchy J, Chlubek D, Scott RJ, Lubinski J. Mutation analysis of MLH1 and MSH2 genes performed by denaturing high-performance liquid chromatography. J Biochem Biophys Methods. 2002;51(1):89–100.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Smigielski EM, Sirotkin K, Ward M, Sherry ST. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 2000;28(1):352–5.
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37(3):235–41.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91.
Pedersen BS, Layer RM, Quinlan AR. Vcfanno: fast, flexible annotation of genetic variants. Genome Biol. 2016;17(1):118.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14(2):178–92.
Kircher M, Witten DM, Jain P, O'Roak BJ. Cooper GM. A general framework for estimating the relative pathogenicity of human genetic variants. 2014;46(3):310–5.
Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557–63.
Plazzer JP, Sijmons RH, Woods MO, Peltomaki P, Thompson B, Den Dunnen JT, et al. The InSiGHT database: utilizing 100 years of insights into lynch syndrome. Familial Cancer. 2013;12(2):175–80.
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062–D7.
Kurzawski G, Suchy J, Debniak T, Kladny J, Lubinski J. Importance of microsatellite instability (MSI) in colorectal cancer: MSI as a diagnostic tool. Ann Oncol. 2004;15 Suppl 4:iv283–4.
Dymerska D, Serrano-Fernández P, Suchy J, Pławski A, Słomski R, Kaklewski K, et al. Combined iPLEX and TaqMan assays to screen for 45 common mutations in lynch syndrome and FAP patients. The Journal of molecular diagnostics : JMD. 2010;12(1):82–90.
Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710.
Gueneau E, Dherin C, Legrand P, Tellier-Lebegue C, Gilquin B, Bonnesoeur P, et al. Structure of the MutLα C-terminal domain reveals how Mlh1 contributes to Pms1 endonuclease site. Nat Struct Mol Biol. 2013;20(4):461–8.
Tamura K, Kaneda M, Futagawa M, Takeshita M, Kim S, Nakama M, et al. Genetic and genomic basis of the mismatch repair system involved in lynch syndrome. Int J Clin Oncol. 2019;24(9):999–1011.
Acknowledgements
We thank the Genomics and Proteomics Core Facility (GPCF) of the German Cancer Research Center (DKFZ), for providing library preparation and sequencing services. We also thank the Omics IT and Data management Core Facility (ODCF) of the DKFZ for the whole genome sequencing data management.
Funding
The study was supported by the EU Transcan Project, the European Union’s Horizon 2020 research and innovation programme, No 856620 and the Sino-German Mobility Programm (No. M-0008). A.K. is a recipient of Ramalingaswami Re-Retry Faculty Fellowship (Grant; BT/RLF/Re-entry/38/2017) from Department of Biotechnology (DBT), Government of India (GOI). This article is based upon work from COSTAction CA17118, supported by COST (European Cooperation in Science and Technology, www.cost.eu). Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization KH, RS, JL, AF; data curation NP, MS; formal analysis AK, NP; funding acquisition KH, RS, JL; methodology AK, NP, MS, DD, KG; project administration KH, AF; resources NP, MS, DD, KG, MK, JL; supervision KH, AF; visualization AK, AF; writing – original draft KH, AF; writing – review & editing all authors. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The ethical approval for this study design was obtained from the Bioethics Committee of the Pomeranian Medical Academy in Szczecin No: BN-001/174/05. Sample collection was performed following the guidelines proposed by this Committee. A written informed consent was signed by each participant in accordance with the Helsinki declaration.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
(Family B) Pedigree of the colorectal cancer family with MLH1 frameshift variant. (Family C) Pedigree of the colorectal cancer family with MSH2 splice site variant. Fig. S2. Microsatellite instability (MSI) analysis of the tumor samples of two family members from Family A and one tumor sample from Family C. For each family, individuals with the tumor samples analyzed are indicated by an arrow and the MSI plots are shown for the corresponding germline and tumor samples. Table S1. Number of large deletions and duplications in the mismatch repair genes reported in the InSiGHT database and their clinical classification according to Mismatch Repair Gene Variant Classification Criteria by the InSiGHT Variant Interpretation Committee.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kumar, A., Paramasivam, N., Bandapalli, O.R. et al. A rare large duplication of MLH1 identified in Lynch syndrome. Hered Cancer Clin Pract 19, 10 (2021). https://doi.org/10.1186/s13053-021-00167-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13053-021-00167-0