
Abstract
Background
Pancreatic cancer (PC) is an aggressive disease with a dismal 5-year survival rate. Surveillance of high-risk individuals is hoped to improve survival outcomes by detection of precursor lesions or early-stage malignancy.
Methods
Since 2011, a national high-risk cohort recruited through St Vincent’s Hospital, Sydney, has undergone prospective PC screening incorporating annual endoscopic ultrasound, formal genetic counselling and mutation analysis as appropriate. PancPRO, a Bayesian PC risk assessment model, was used to estimate 5-year and lifetime PC risks for familial pancreatic cancer (FPC) participants and this was compared to their perceived chance of pancreatic and other cancers. Genetic counselling guidelines were developed to improve consistency. Follow-up questionnaires were used to assess the role of genetic counselling and testing.
Results
We describe the Australian PC screening program design and recruitment strategy and the results of the first 102 individuals who have completed at least one-year of follow-up. Seventy-nine participants met the FPC criteria (≥ two first-degree relatives affected), 22 individuals had both a BRCA2 pathogenic variant and a close relative with PC and one had a clinical diagnosis of Peutz-Jeghers syndrome. Participants reported a high perceived chance of developing PC regardless of their genetic testing status. PancPRO reported FPC participants’ mean 5-year and lifetime PC risks as 1.81% (range 0.2–3.2%) and 10.17% (range 2.4–14.4%), respectively. Participants’ perceived PC chance did not correlate with their PancPRO 5-year (r = − 0.17, p = 0.128) and lifetime PC risks (r = 0.19, p = 0.091). Two-thirds felt that current genetic testing would help them, and 91% of tested participants were glad to have undergone genetic testing. Overall, 79% of participants found genetic counselling to be helpful, and 88% reported they would recommend counselling to their relatives.
Conclusions
Participants reported multiple benefits of genetic counselling and testing but continue to seek greater clarification about their individual PC risk. Extension of PancPRO is required to enable personalised PC risk assessment for all high-risk sub-groups. More detailed discussion of PC risk for BRCA2 pathogenic variant carriers, providing a written summary in all cases and a plan for genetics review were identified as areas for improvement.
Similar content being viewed by others


Background
Pancreatic cancer (PC) is an aggressive and devastating disease. Over 3000 new PC diagnoses were predicted in Australia in 2018 [1]. Whilst this represents only 2.4% of new cancers, PC disproportionately accounts for 6.2% of cancer deaths [1], a trend that is mirrored worldwide [2]. Typically, patients present with advanced disease, making surgical interventions impossible and treatments ineffective [3]. Most PC patients die within months of diagnosis [4], with only 7.7% surviving 5 years [1]. Early identification remains the only successful approach to longer term survival [5].
PC is most commonly sporadic, however 5–10% of PC cases are due to a genetic predisposition [6, 7]. This includes pathogenic variants in cancer predisposition genes, e.g. BRCA2 and PALB2; Lynch Syndrome, Peutz-Jeghers Syndrome (PJS), Familial Atypical Multiple Mole Melanoma and Hereditary Pancreatitis [7, 8]. In addition, many families have apparently autosomal dominant transmission of PC without an identified pathogenic variant (“Familial Pancreatic Cancer (FPC))” [9]. The lifetime risk of PC within FPC families varies according to the number of affected first degree relatives (FDR), with a relative risk of 6.4 (lifetime risk 8–12%) for 2FDR and a relative risk of 32 (lifetime risk 40%) for ≥3FDR [10, 11]. Although screening for PC is not feasible nor recommended in general population due to the low incidence of PC, there is increasing evidence high-risk individuals ((HRI)) may benefit from PC screening in a research setting [12].. The Cancer of the Pancreas Screening (CAPS) consortium recommends PC screening for individuals with > 5% lifetime risk of PC [13]. The rationale of screening asymptomatic HRI is to diagnose precursor lesions or early PC when still resectable and hence, potentially curable to improve survival. Endoscopic ultrasound (EUS) and magnetic resonance imaging (MRI) are the screening methods of choice [13, 14]. EUS is minimally invasive and studies report high sensitivity and specificity for detecting PC lesions less than 2 cm and can also collect biopsy samples [14]. A systematic review showed that screening in HRI led to a higher diagnostic rate of pancreatic tumours than in controls (34% vs 7.2%, p < 0.001) [15]. PC screening resulted in a significantly higher curative resection rate (60% versus 25%, p = 0.001) and a significantly longer median survival time (14.5 month versus 4 months, p < 0.001) [15]. A recently published long-term (16-year) follow-up study of HRI reported that 90% of tumours identified during their PC surveillance program were resectable with a median time from baseline screening to PC diagnosis of 4.8 years and 85% of patients with resected cancers were alive at 3 years [16]. Importantly, these benefits occur in the absence of psychological harms [17, 18].
The Australian PC screening program was established in 2011 at St Vincent’s Hospital, Sydney, in collaboration with the CAPS consortium. To identify high-risk individuals, the Australian Familial Pancreatic Cancer Cohort (AFPaCC) was established under the Australian Pancreatic Cancer Genome Initiative (APGI) [19] as a registry and biorepository of individuals and families with a strong family history of PC. Efforts by the APGI to profile the genomic landscape of PC, including a subset of FPC patients, have been reported elsewhere [20,21,22,23].
The importance of genetic counselling for cancer predisposition syndromes is well recognised, with numerous studies reporting patient satisfaction, improved risk perception and better psychosocial outcomes [24,25,26,27,28]. Genetic counselling for heterogenous conditions like PC can be challenging as there are both strong environmental and inherited factors, and up until recently a lack of standardised genetic testing options [12, 29]. Increasingly, genomic testing is offered to PC patients, thus the need to ensure genetic counselling practices are effective and evidence-based.
The provision of genetic counselling for PC susceptibility is further hindered by insufficient data to estimate an individual’s PC risk. Several studies have assessed PC incidence within families with BRCA2 pathogenic variants demonstrating a 4.4–5.9 relative risk (lifetime risk 5–8%), with a slightly higher risk for males [30,31,32]. Currently, no risk assessment models exist to clarify the PC risk in BRCA2 carriers with and without a family history of PC. For FPC families, Wang et al. [33] developed a Bayesian risk estimation model, PancPRO, to quantify an individuals’ PC risk based on family history. This program considers the number of PC affected relatives, their age at diagnosis, and the age of unaffected relatives to calculate the likelihood of an individual harbouring a “PC susceptibility gene” and their 5-year and lifetime risks of developing PC [33]. PancPRO is freely available as part of the CancerGene software from http://www4.utsouthwestern.edu/breasthealth/cagene. PancPRO has been evaluated in only a small number of actual and theoretical PC populations [34, 35], and further validation is required.
Methods
The aims of this study are: (1) to describe the recruitment and management of an Australian high-risk PC cohort; (2) to ascertain participant feedback regarding genetic counselling and testing for PC; and (3) to assess the utility of PancPRO to estimate PC risk for FPC participants and to compare it with their perceived cancer risks.
Participants
Recruitment was achieved in one of five ways: 1) self-referral via the AFPaCC website; 2) identification as an FDR of an affected individual(s) meeting FPC criteria on the AFPaCC registry; 3) Clinical genetics service/Familial Cancer Clinic (FCC) referral; 4) General Practitioner (GP) or specialist referral; or 5) referral from collaborative cancer organisations (e.g. PanCare, KconFab, Avner Pancreatic Cancer Foundation). While most FCC referrals were prospective, ethics approvals were also sought at key Sydney clinics to allow for retrospective contact of eligible individuals. Participants were assessed to determine eligibility (Additional file 1) by the clinical research coordinator (TD and SM). Where possible, confirmation of PC diagnosis was obtained by pathology reports or death certificates.
PC screening protocol
All eligible participants provided written consent and completed a baseline personal and family health history questionnaire (Fig. 1). Formal genetic counselling was a prerequisite, with most participants having individual counselling. If a participant was referred from an FCC, this prerequisite was assumed to have been met. One interstate family attended a group counselling appointment, however after negative feedback this was discontinued. To facilitate consistent counselling, genetic counselling guidelines were designed and distributed to FCCs (Additional file 2).

Summary of the PC screening protocol
Participants were scheduled for baseline EUS and blood tests, including CA-19.9, C-reactive protein, blood glucose and Macrophage Inhibitory Cytokine 1 (MIC-1), previously identified as a possible marker for PC [36]. Within a week of the baseline EUS, participants received a follow-up phone call to assess their experiences, note any complications and answer further queries. The screening program gastroenterologist allocated participants to continue annual EUS screening (normal EUS) or more intense surveillance by MRI or EUS at 3 or 6 months (abnormal EUS), depending on findings. Follow-up questionnaires are completed one-month, 1-year and 5-years post baseline EUS to assess both the value of genetic counselling and determine the psychological impact of screening.
Instrumentation
Baseline and follow-up questionnaires were modified from the CAPS protocol with permission. A 5-point Likert scale was administered at baseline to assess perceived chance of developing PC and other cancer(s) (1 = much below others to 5 = much above others). At one-month post baseline EUS a follow-up questionnaire using a total of 19 statements with a 5-point Likert scale (1 = strongly agree to 5 = strongly disagree) and two open-ended questions, was administered to assess the overall experience of genetic counselling. Validated psychological assessment scales were also administered at each timepoint; these will be reported separately.
Assessment of PC risk
For each participant within the FPC cohort, family history from the baseline questionnaire was assessed using PancPRO (CancerGene Version 6, The Bayes Mendel Group, UT Southwestern Medical Center, Dallas, USA), to calculate their likelihood of carrying a PC susceptibility gene and their 5-year and lifetime PC risk. PancPRO was an internal tool, with the participants and genetic counsellors blinded to the results. As PancPRO does not model the impact of pathogenic variants or clinical diagnosis, PancPRO was not used to generate a personalised PC risk estimate for other screening participants.
Data analysis
Data analysis was performed using IBM SPSS Statistics for Windows (Version 25.0. Armonk, NY). Basic descriptive statistics were generated for participant demographics and genetic counselling responses. Spearman’s Rho was used to assess correlations between perceived PC and other cancer(s) chance scores with all continuous variables (e.g. age at enrolment, number of FDR/total number of PC affected relatives, 5-year and lifetime PC risk etc.). Mann-Whitney U tests were also used to assess differences with perceived PC and other cancer(s) chance scores with binomial data (e.g. gender, depression status and personal cancer history).
Results
From September 2011 to March 2017, 1059 individuals residing across all Australian states contacted or were referred to AFPaCC, based on their family history of PC. Seven hundred and seventy-four individuals were not eligible for the high-risk screening program at St Vincent’s Hospital (SVH), Sydney, as they were either affected by PC, had insufficient family history, or met one or more of the exclusion criteria. The remaining 285 eligible individuals were provided with a participant information and consent form and baseline questionnaire. Surprisingly,124 individuals (44%) did not proceed with enrolment.. Some were “passive decliners” meaning they did not return their recruitment paperwork (the coordinator made two repeat contact attempts). Anecdotally, factors influencing dropout were distance and expense of travel, psychosocial factors (e.g. caring for or grieving the loss of a family member with PC), a lack of perceived benefit and the amount of paperwork required. A second PC screening site was established at the Austin Hospital in Melbourne, Victoria, in 2014 and 59 Victorian participants were referred to this additional site and are not described further in this paper. At the time of analysis, a total of 102 participants provided written consent, completed the baseline questionnaire, commenced EUS screening at SVH and have completed at least 1 year of follow-up. Eleven participants (11%) have transferred to Melbourne or withdrawn from the screening protocol (Fig. 2).

Diagram of participant eligibility, recruitment and retention for the PC screening program
Participant demographics
Prospective data was analysed for the first 102 participants (comprising of 62 families). The average number of EUS was 3.3 but all had at least one EUS at the time of analysis. The mean age was 56 years (35–78 years) at enrolment, 69% were female, 99% were Caucasian with 12% reporting Ashkenazi Jewish ancestry (Table 1). The mean age of youngest PC diagnosis in the family was 55.2 years (range 21–84 years). As 56 participants (55%) were referred from an FCC, their genetic counselling took place prior to study recruitment (“off-protocol”) with the remaining 46 participants (45%) being referred to an FCC for genetic counselling after study recruitment (“on-protocol”).
Genetic testing and PC risk assessment using PancPRO
Seventy-one participants (or their close affected relative) underwent genetic investigation(s) during their genetic counselling appointment(s). Of these, 22 (14 females and eight males) were identified as a BRCA2 pathogenic variant carrier. One female BRCA2 carrier also had a second pathogenic variant detected in BRCA1. The family history of 79 participants met the classification of FPC. Genetic testing was performed on an affected relative (n = 40) or the PC screening participant (n = 9), of which 47 had no mutation detected in either BRCA1/2 (n = 42), MLH1/PMS2 (n = 7), STK11 (n = 6), or showed preserved staining of the mismatch repair proteins using IHC (n = 10). Two additional participants were found to have a variant of uncertain significance (VUS) in BRCA2 (considered no mutation identified). A single participant had a clinical diagnosis of PJS but declined genetic testing.
In the FPC cohort (n = 79), PancPRO estimated a mean 5-year PC risk of 1.81% (range 0.2–3.2%) and a mean lifetime PC risk of 10.17% (range 2.4–14.4%) (Fig. 3). The mean probability of having a hypothetical PC susceptibility gene was 0.431 (43.1%, range 0.047–0.501). There was a significant moderate positive correlation with the total number of affected FDR/second degree relative (SDR) and probability of a PC susceptibility gene (r = 0.52, p < 0.001). The PancPRO risk estimates of our FPC cohort were further categorised based on number of affected relatives (Table 2). The mean lifetime PC risks calculated by PancPRO for individuals with 1FDR, 2FDR and 3FDR were 10.9% (range 3.7–14.4%), 9.5% (range 2.7–13.2%) and 9.7% (range 2.4–14.2%), respectively.

a) The 5-year and b) lifetime PC risk of the FPC cohort as calculated by PancPRO. Each bar represents the number of participants within the specified range of PC risk. The corresponding genetic testing status (no mutation identified or not tested) is indicated for each participant
Perceived chance of developing PC and other cancers
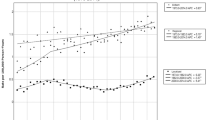
Participants’ perceived chance scores for developing PC and other cancer(s) are shown in Fig. 4. Participants reported a significantly higher perceived chance of developing PC compared to other cancer(s) (mean = 4.18 vs 3.47, p < 0.01, n = 102). There was no significant difference in perceived chance of developing PC between BRCA2 carriers and those with either no mutation identified or those not tested (mean = 4.16 vs 4.23, p = 0.679). Appropriately, BRCA2 carriers had a significantly higher perceived chance of developing other cancer(s) compared to the FPC cohort (mean = 4.18 vs 3.28, p < 0.001). Similarly, participants with a personal history of cancer had a significantly higher perceived chance of developing other cancer(s) compared to those without previous malignancy (mean = 4.15 vs 3.30, p < 0.001), but only a trend towards a higher perceived chance of developing PC (mean = 4.45 vs 4.11, p = 0.055). There was a strong significant correlation in BRCA2 carriers between personal cancer history and perceived chance of PC (r = 0.67, p = 0.001, n = 22) and other cancer(s) (r = 0.69, p < 0.001, n = 22). Additional statistical analyses for perceived chance of PC and other cancer(s) are shown in Table 3. Interestingly, there were no significant correlations between participants’ perceived chance of PC and the objective 5-year PC risk (r = − 0.17, p = 0.128), lifetime PC risk (r = 0.19, p = 0.091), and probability of a PC susceptibility gene (r = 0.21, p = 0.067) as calculated by PancPRO.

a) Participants’ perceived chance of developing PC and b) Participants’ perceived chance of developing other cancers. For each graph, bars represent the number of participants who selected the specified Likert value (1 = much below others, 3 = neutral, 5 = much above others) to indicate their extent of perceived chance. The corresponding genetic testing status is also shown for each participant
Genetic counselling experience
Eighty-eight participants returned the 1-month questionnaire (86.3% response rate) but only 64 participants (73%) responded to all statements, resulting in variability in response numbers. Overall, participants provided positive feedback regarding their genetic counselling. Most (89%, n = 77) wanted to undergo genetic testing to clarify their PC risk, two-thirds (n = 54) thought current genetic testing would help them, and of those tested, 91% (n = 48) were glad they had testing. Participants reported utility of counselling even without genetic testing, with 61% (n = 52) strongly disagreeing that genetic counselling should be delayed until more genes are identified, and 68% (n = 58) wanted another appointment when more information was available. Overall, 79% (n = 67) found the genetic counselling appointment to be helpful, and 88% (n = 75) reported they would recommend counselling to their relatives.
Free text responses describing the “most useful” and “least useful” aspects of genetic counselling were further categorised into main themes, with illustrative quotations provided (Table 4 and 5). Fifty-seven participants (65%) included free-text responses about the most useful part of genetic counselling, with reported benefits including: gaining knowledge and understanding of PC risk; feeling empowered to proactively monitor PC risk; receiving support; and the potential to help future generations.
In contrast, only 14 participants (16%, n = 9 not tested, n = 4 no mutation identified, n = 1 BRCA2 carrier) provided comments about the least useful part of genetic counselling. Of these, 10 comments (71%) reflected inadequate information provision, limited understanding of PC susceptibility genes and/or ability to estimate an individual’s PC risk, and insufficient follow-up. Twelve participants (14%) reported they did not receive a written summary. This included four siblings who attended a one-off group genetic counselling session, to accommodate for interstate travel and to enable attendance for their baseline EUS together. Additionally, one BRCA2 carrier indicated that counselling induced worry and one FPC participant commented that counselling does not resolve the grief caused by losing multiple family members.
Discussion
We evaluate the role of genetic counselling and testing in a national high-risk PC screening cohort and assess the utility of PancPRO to provide a personalised PC risk assessment. We also provide invaluable insights into the recruitment and management of the Australian PC screening program, including barriers to screening uptake. Due to the high mortality of PC and uncertain benefit of screening at this stage [37], the genetic counselling needs of these high-risk individuals may differ from those with other hereditary cancer syndromes and data obtained from our cohort provide important insights in this area. Through the development of formal genetic counselling guidelines, we describe our genetic counselling recommendations for HRI, which in combination with participant feedback, are hoped to guide practice improvements.
Recruiting high-risk PC individuals
Establishing a national registry to capture PC kindreds was a successful approach to target HRI, with over 66% of the enquiries received via the AFPaCC website. Once initial contact was made, additional at-risk relatives were identified and invited to join the registry and/or PC screening program (n = 134). Moreover, the registry enables easy identification and contact of individuals who may become eligible over time (i.e. with age; an incident PC diagnosis in the family). Although clear inclusion criteria were available online, approximately half of those who made contact had only 1FDR with PC, and it was necessary for the clinical research coordinator to provide personalised clarification around sporadic and familial PC. Referrals from FCC identified eligible and motivated individuals who were ready to attend their baseline EUS, resulting in a streamlined recruitment process.
Nearly half of the eligible individuals who expressed interest in screening, chose not to pursue participation. Accessibility (e.g. financial and logistical challenges of travel), time constraints and the amount of paperwork required were reported as the main barriers for at-risk individuals. The Australian population is widely dispersed across a large land mass, and participants must self-fund transport and/or accommodation. These reported barriers align with a German PC screening study, which found the cost of travel and the time required for participation as factors impacting screening uptake [38]. Many eligible individuals did not actively decline participation, suggesting screening uptake may increase should the perceived benefits improve and the perceived or actual barriers to participation be overcome. Strategies to address these challenges include: expansion of the screening program into other states with concomitant funding for a full-time national coordinator, funding for travel arrangements, subsidised local accommodation and simplification of the questionnaire.
Genetic counselling experience
Participants believed genetic counselling to be helpful even without genetic testing, would recommend it and were interested in genetics follow-up as more PC genes are discovered. Participants felt strongly that their family history was due to a pathogenic variant, though were split on whether they had inherited it. The current study reflects the increasing expectation that genetic testing will clarify an individual’s PC predisposition. Ninety-one percent of our tested participants were glad to have undergone testing and two-thirds felt that current genetic testing would help them, compared to approximately 40% previously reported [39]. Key differences between our cohort and that of Axilbund et al. [39], were 1) genetic counselling in our study was conducted by multiple genetic counsellors (employed in numerous services across four Australian states), compared to a single genetic counsellor, and 2) our cohort contained individuals with variable family histories, whilst participants in the earlier study had at least three affected relatives. These data suggest that the perceived benefits of genetic counselling are not influenced by patients having FPC or a known pathogenic variant.
Our participants reported that genetic counselling provided essential information to aid understanding of risks (both inherited and environmental) and was empowering. This correlates with previous reports on patient perception following genetic counselling for PC, which indicate improved understanding and psychological function [27, 28]. Consistent with Underhill et al. [37], participants were motivated to prevent PC mortality and felt reassured by PC surveillance. Disappointment with genetic counselling was present in a minority, with comments reflecting distress at the limitations in current knowledge of PC predisposition. Some participants indicated that their counselling focused on their BRCA2 carrier status, rather than providing information regarding their PC risk (understandable given almost two-thirds of the BRCA2 cohort were female, and nine had a personal history of breast cancer). Emphasizing the PC risk associated with hereditary breast and ovarian cancer (HBOC) and Lynch syndrome is not appropriate for all families, as the incidence of PC is lower in these syndromes [40, 41] though should be tailored for those with a family history of PC. As all BRCA2 carriers completed counselling off-protocol, some many years prior to the screening program enrolment, it is conceivable that some participants received counselling prior to their relatives’ diagnosis with PC. New screening participants who previously received genetic counselling, may therefore benefit from a review appointment to obtain current information on PC screening, risk management and genetic testing options.
Several participants commented that they did not receive a written summary of their counselling appointment(s), despite it being common genetic counselling practice [42]. Written summaries are reported to reduce cancer anxiety and improve accuracy in risk perception [25], assist patient understanding and recall, and facilitate accurate family communication [26, 43] Importantly, participants in the current study who received summaries, found them easy to understand and almost 80% either partially or strongly agreed that they would share it with relatives. These data suggest that documenting counselling discussions for PC is important due to current limitations with informative genetic testing, accurate risk assessments and a need to individualise recommendations based on family history and environmental risk factors.
Participants with the highest perceived chance of developing other cancer(s), were either BRCA2 carriers, had a personal history of cancer, or a family history of multiple cancer types, consistent with results from Rantala et al. [24]. Our data suggest these participants may be most vulnerable to increased cancer worry and perceived risk, which may warrant exploration during counselling. Genetic testing where appropriate, might help clarify an individual’s cancer susceptibility, may alleviate worry of developing other cancer(s) and contribute to better psychological outcomes. Participants’ interest in receiving additional counselling further demonstrates the importance of establishing a plan for genetics review to facilitate genetic testing as more information about FPC becomes available.
Although these data support the value of genetic counselling despite current limitations in testing utility, the genomic era will likely transform genetic counselling practices for FPC [44]. Studies are demonstrating that family history alone is a poor predictor of mutation status [45, 46] and further genomic characterisation of genetic modifiers in PC will improve understanding of FPC and positively impact genetic counselling processes [44]. Elucidation of causative pathogenic variants may further clarify the cancer risk(s) for unaffected blood relatives and assist appropriate stratification into high-risk screening programs [47].
Utility of PancPRO for FPC risk-assessment
PancPRO is not commonly used in Australia during the provision of genetic counselling. Using hypothetical scenarios, Leonardi et al. [35] found PancPRO risk estimates to be a valid method to stratify high-risk FPC families to PC screening protocols. Our study prospectively assesses the utility of PancPRO to provide personalised PC risk estimates in a true cohort of participants. Overall, our mean lifetime PC risk of 10.17% is consistent with that reported by Barnes et al. [34], the only other study to generate PancPRO risk estimates for FPC screening participants. Further comparison is not possible, as their cohort consisted of only 33 individuals and no subgroup analysis was published. The mean PancPRO lifetime PC risk values calculated for our participants with two affected FDR was 9.5%, which is comparable to those reported following prospective analysis of incident pancreatic cancers from the National Familial Pancreatic Tumor Registry (lifetime risk 8–12%) [10, 11]. However, the lifetime PC risk previously reported for those with ≥3FDR (lifetime risk 40%) [10, 11] were higher than the calculated PancPRO lifetime risks for our cohort (9.7%). Potentially, this is due to the small number of participants with 3FDR (n = 11) and it must be noted that 2/11 participants with at least 3FDR had both parents and a sibling affected. PancPRO reported a large range in lifetime PC risk estimates for participants with 1FDR (3.7–14.4%), yet the overall mean was equivalent to the 2FDR and 3FDR subgroups. This is likely due to the variable family history and age of diagnosis in these subgroups, or the unreliability of PancPRO. Further studies with subgroup analysis are needed to clarify this observed issue.
Incorporation of pathogenic variant status and environmental risk factors in future versions of PancPRO (or alternate PC risk estimation programs), once reliable PC penetrance data exist would be beneficial. Due to the genetic heterogeneity of PC, developing a comprehensive risk estimation model would aid genetic counselling and facilitate informed decision-making regarding risk-modification behaviours. Personalised risk assessments may also improve the uptake of screening for the subset of eligible participants who failed to see benefit.
Study limitations
Inherited forms of PC are rare and recruitment of HRI is challenging and time-consuming. Participants in our high-risk PC cohort had variable personal and family history of cancer, and the cohort is biased toward younger, healthy participants, resulting in low 5-year PC risk estimates. More detailed information about barriers to screening uptake would have been valuable. Another limitation is the lack of histopathologically confirmed pancreatic malignancies in all probands, with reliance on self-reports and death certificates for some kindreds; however, Fiederling et al. [48] found self-reported PC family histories are valid for preventative counselling.
As participants received genetic counselling at different genetics services and were recruited across a period of six years, the information provided and genetic testing practices may have varied. Although the genetic counselling guidelines were circulated to improve consistency, some completed counselling several years prior to screening program recruitment. Their views on the genetic counselling process are likely to exhibit recall bias due to the greater time since counselling occurred.
Conclusions and future directions
This study is the first to describe results from the Australian pancreatic cancer screening program. Participants reported positive genetic counselling experiences with feedback indicating improved knowledge and feelings of empowerment. Our data indicate that participants want genetic testing and clarification of their mutation status in relation to PC predisposition. Tailored discussion of PC risk in patients with BRCA2 mutations, provision of a written summary in all cases and setting a plan for genetics review were identified as areas for improvement. Updating the genetic counselling guidelines to include participant feedback may further support exploration of participants’ perceived cancer chance within the context of their estimated risk and improved risk communication strategies. PancPRO has the potential to become a useful, personalised risk assessment and communication tool for FPC for use in clinical practice. We highlight that extension of PancPRO to model PC risk for individuals with pathogenic variants and incorporation of environmental factors would provide greater clarity regarding individualised PC risk and its’ development should be a research priority.
Strategies to overcome barriers to recruitment in Australia to improve program accessibility are needed. We aim to establish a uniform screening program across Australia, provide consistent genetic testing and counselling and facilitate a centralised, national database to promote efficient access for PC researchers and close international collaborations.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article and its additional files.
Abbreviations
- AFPaCC:
-
Australian Familial Pancreatic Cancer Cohort
- APGI:
-
Australian Pancreatic Cancer Genome Initiative
- CAPS:
-
Cancer of the Pancreas Screening
- CT:
-
Computed tomography
- EUS:
-
Endoscopic ultrasound
- FCC:
-
Familial cancer clinic
- FDR:
-
First degree relative
- FPC:
-
Familial pancreatic cancer
- GP:
-
General Practitioner
- HBOC:
-
Hereditary Breast and Ovarian Cancer
- HRI:
-
High-risk individuals
- MIC-1:
-
Macrophage inhibitory cytokine 1
- MRI:
-
Magnetic resonance imaging
- PC:
-
Pancreatic cancer
- PJS:
-
Peutz-Jeghers syndrome
- SDR:
-
Second degree relative
- STVH:
-
St Vincent’s Hospital
- VUS:
-
variant of uncertain significance
References
Australian Institute of Health and Welfare. Pancreatic cancer in Australia. 2018 23/07/2018]; Available from: https://pancreatic-cancer.canceraustralia.gov.au/statistics.
Yabar CS, Winter JM. Pancreatic Cancer: a review. Gastroenterol Clin N Am. 2016;45(3):429.
Klein AP. Identifying people at a high risk of developing pancreatic cancer. Nature Rev Cancer. 2013;13(1):66.
Rustgi AK. Familial pancreatic cancer: genetic advances. Genes Dev. 2014;28(1):1–7.
Boursi, B., B. Finkelman, B.J. Giantonio, K. Haynes, A.K. Rustgi, A.D. Rhim, et al., A Clinical Prediction Model to Assess Risk for Pancreatic Cancer Among Patients With New-onset Diabetes. Gastroenterology, 2017. 152(4): p. 840–850.e3.
Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, et al. Recent Progress in pancreatic Cancer. CA Cancer J Clin. 2013;63(5):318–48.
Amundadottir LT. Pancreatic Cancer genetics. Int J Biol Sci. 2016;12(3):314–25.
McKay SH, Humphris JL, Johns AL, Gill AJ, Tucker K. Inherited pancreatic cancer. Cancer Forum. 2016;40(1):30–3.
Bartsch DK, Gress TM, Langer P. Familial pancreatic cancer--current knowledge. Nature Rev Gastroenterol Hepatol. 2012;9(8):445–53.
Klein AP, Brune KA, Petersen GM, Goggins M, Tersmette AC, Offerhaus GJA, et al. Prospective risk of pancreatic Cancer in familial pancreatic Cancer Kindreds. Cancer Res. 2004;64(7):2634.
Grover, S. and S. Syngal, Hereditary Pancreatic Cancer. Gastroenterology, 2010. 139(4): p. 1076–1080.e2.
Brand RE, Lerch MM, Rubinstein WS, Neoptolemos JP, Whitcomb DC, Hruban RH, et al. Advances in counselling and surveillance of patients at risk for pancreatic cancer. Gut. 2007;56(10):1460–9.
Canto MI, Harinck F, Hruban RH, Offerhaus GJ, Poley JW, Kamel I, et al. International Cancer of the pancreas screening (CAPS) consortium summit on the management of patients with increased risk for familial pancreatic cancer. Gut. 2013;62(3):339–47.
Bhutani MS, Koduru P, Joshi V, Saxena P, Suzuki R, Irisawa A, et al. The role of endoscopic ultrasound in pancreatic cancer screening. Endosc Ultrasound. 2016;5(1):8–16.
Lu C, Xu C-F, Wan X-Y, Zhu H-T, Yu C-H, Li Y-M. Screening for pancreatic cancer in familial high-risk individuals: a systematic review. World J Gastroenterol. 2015;21(28):8678–86.
Canto, M.I., J.A. Almario, R.D. Schulick, C.J. Yeo, A. Klein, A. Blackford, et al., Risk of Neoplastic Progression in Individuals at High Risk for Pancreatic Cancer Undergoing Long-term Surveillance. Gastroenterology, 2018. 155(3): p. 740–751.e2.
Konings ICAW, Sidharta GN, Harinck F, Aalfs CM, Poley JW, Kieffer JM, et al. Repeated participation in pancreatic cancer surveillance by high-risk individuals imposes low psychological burden. Psycho-Oncology. 2016;25(8):971–8.
Paiella S, Salvia R, De Pastena M, Pollini T, Casetti L, Landoni L, et al. Screening/surveillance programs for pancreatic cancer in familial high-risk individuals: a systematic review and proportion meta-analysis of screening results. Pancreatology. 2018;18(4):420–8.
AFPaCC Registry. 2011; Available from: http://www.pancreaticcancer.net.au/research-familial/.
Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52.
Johns AL, Miller DK, Simpson SH, Gill AJ, Kassahn KS, Humphris JL, et al. Returning individual research results for genome sequences of pancreatic cancer. Genome Med. 2014;6(5):42.
Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501.
Johns AL, McKay SH, Humphris JL, Pinese M, Chantrill LA, Mead RS, et al. Lost in translation: returning germline genetic results in genome-scale cancer research. Genome Med. 2017;9(1):41.
Rantala J, Platten U, Lindgren G, Nilsson B, Arver B, Lindblom A, et al. Risk perception after genetic counseling in patients with increased risk of cancer. Hered Cancer Clin Pract. 2009;7(1):15.
Lobb EA, Butow PN, Barratt A, Meiser B, Gaff C, Young MA, et al. Communication and information-giving in high-risk breast cancer consultations: influence on patient outcomes. Br J Cancer. 2004;90(2):321–7.
Kausmeyer DT, Lengerich EJ, Kluhsman BC, Morrone D, Harper GR, Baker MJ. A survey of patients’ experiences with the Cancer genetic counseling process: recommendations for Cancer genetics programs. J Genet Couns. 2006;15(6):409–31.
Hart SL, Torbit LA, Crangle CJ, Esplen MJ, Holter S, Semotiuk K, et al. Moderators of cancer-related distress and worry after a pancreatic cancer genetic counseling and screening intervention. Psycho-Oncology. 2012;21(12):1324–30.
Maheu C, Vodermaier A, Rothenmund H, Gallinger S, Ardiles P, Semotiuk K, et al. Pancreatic cancer risk counselling and screening: impact on perceived risk and psychological functioning. Familial Cancer. 2010;9(4):617–24.
Stoffel EM, McKernin SE, Brand R, Canto M, Goggins M, Moravek C, et al. Evaluating susceptibility to pancreatic Cancer: ASCO provisional clinical opinion. J Clin Oncol. 2019;37(2):153–64.
Moran A, O'Hara C, Khan S, Shack L, Woodward E, Maher ER, et al. Risk of cancer other than breast or ovarian in individuals with BRCA1 and BRCA2 mutations. Familial Cancer. 2012;11(2):235–42.
van Asperen CJ, Brohet R, Meijers-Heijboer E, Hoogerbrugge N, Verhoef S, Vasen H, et al. Cancer risks in BRCA2 families: estimates for sites other than breast and ovary. J Med Genet. 2005;42(9):711–9.
Mocci E, Milne RL, Mendez-Villamil EY, Hopper JL, John EM, Andrulis IL, et al. Risk of pancreatic cancer in breast cancer families from the breast cancer family registry. Cancer Epidemiol Biomark Prev. 2013;22(5):803–11.
Wang W, Chen S, Brune KA, Hruban RH, Parmigiani G, Klein AP. PancPRO: risk assessment for individuals with a family history of pancreatic Cancer. J Clin Oncol. 2007;25(11):1417–22.
Barnes CA, Krzywda E, Lahiff S, McDowell D, Christians KK, Knechtges P, et al. Development of a high risk pancreatic screening clinic using 3.0 T MRI. Familial Cancer. 2018;17(1):101–11.
Leonardi, G., S. Marchi, M. Falconi, A. Zerbi, V. Ussia, N. de Bortoli, et al., "PancPro" as a tool for selecting families eligible for pancreatic cancer screening: an Italian study of incident cases. Dig Liver Dis, 2012. 44(7): p. 585.
Koopmann J, Buckhaults P, Brown DA, Zahurak ML, Sato N, Fukushima N, et al. Serum macrophage inhibitory cytokine 1 as a marker of pancreatic and other Periampullary cancers. Clin Cancer Res. 2004;10(7):2386.
Underhill M, Berry D, Dalton E, Schienda J, Syngal S. Patient experiences living with pancreatic cancer risk. Hered Cancer Clin Pract. 2015;13(1):13.
Franke FS, Matthäi E, Slater EP, Schicker C, Kruse J, Bartsch DK. German National Case Collection for familial pancreatic Cancer (FaPaCa) - acceptance and psychological aspects of a pancreatic cancer screening program. Hered Cancer Clin Pract. 2018;16(1):17.
Axilbund JE, Brune KA, Canto MI, Brehon BC, Wroblewski LD, Griffin CA. Patient perspective on the value of genetic counselling for familial pancreas cancer. Hered Cancer Clin Pract. 2005;3(3):115–22.
Kastrinos F, Mukherjee B, Tayob N, Wang F, Sparr J, Raymond VM, et al. The risk of pancreatic Cancer in families with Lynch syndrome. JAMA. 2009;302(16):1790–5.
Iqbal J, Ragone A, Lubinski J, Lynch HT, Moller P, Ghadirian P, et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2012;107(12):2005–9.
VandenBoom E, Trepanier AM, Carmany EP. Assessment of current genetic counselor practices in post-visit written communications to patients. J Genet Couns. 2018;27(3):681–8.
Hallowell N, Murton F. The value of written summaries of genetic consultations. Patient Educ Couns. 1998;35(1):27–34.
Sampson JE, Hamman KJ. A current picture of genetic counseling for familial pancreatic cancer. Expert Rev Qual Life Cancer Care. 2016;1(6):425–32.
Young EL, Thompson BA, Neklason DW, Firpo MA, Werner T, Bell R, et al. Pancreatic cancer as a sentinel for hereditary cancer predisposition. BMC Cancer. 2018;18(1):697.
Holter S, Borgida A, Dodd A, Grant R, Semotiuk K, Hedley D, et al. Germline BRCA mutations in a large clinic-based cohort of patients with pancreatic adenocarcinoma. J Clin Oncol. 2015;33(28):3124–9.
Ghiorzo P. Genetic predisposition to pancreatic cancer. World J Gastroenterol. 2014;20(31):10778–89.
Fiederling J, Shams AZ, Haug U. Validity of self-reported family history of cancer: a systematic literature review on selected cancers. Int J Cancer. 2016;139(7):1449–60.
Acknowledgments
We would like to thank the participants for their involvement and the medical practitioners who referred their patients to the screening program. We would like to acknowledge Prof Andrew Biankin and Prof Anthony Gill and Australian Pancreatic Cancer Genome Initiative network for their support and ongoing collaboration: http://pancreaticcancer.net.au/about-collaborators. A special thanks is also given to Dr. Lesley Andrews, A/Prof Judy Kirk and Dr. Marcia Canto for their contribution to the initial screening program development. We also acknowledge Dr. Sam Emmanuel who provided statistical analysis for this study.
We wish to individually acknowledge the NSW and ACT genetics practitioners (clinicians and genetic counsellors) who provided genetic counselling for most screening program participants: Cancer Genetics Unit, St Vincent’s Hospital: Louise Lynagh, Manisha Chauhan, Isabel Danos, Cassandra McDonald; Hereditary Cancer Centre, Prince of Wales Hospital: Dr. Lesley Andrews, Meera Warby, Amanda Willis, April Morrow, Rachel Williams; Hunter Family Cancer Service: Margaret Gleeson, Alexandra Groves, Hereditary Cancer Centre, St George Hospital: Risha Zia; Hereditary Cancer Centre, Wollongong Hospital: Tish Silberbauer, Jessica Duffy, Sian Greening, Emma Healey; Familial Cancer Service, Westmead Hospital: Prof Judy Kirk, Dr. Amanda Seabrook, Emma Edwards, Michelle Bowman, Rebecca Harris, Familial Cancer Service, Royal North Shore Hospital: Dr. Michael Field, Dr. Hilda High, Ashley Crook, Stephanie Burcher, Stephanie White; Familial Cancer Service, Royal Prince Alfred Hospital/Liverpool Hospital: Dr. Annabelle Goodwin, Camron Ebzery, Annabelle Ng, Carol-Ann Verrenkamp, Eliza Courtney, Devika Sathe; Port Macquarie: Kim Frumar, Carol Sorenson; Wagga: Kim Robbins; Taree: Bruce Hopper; ACT: Linda Warwick, Jennifer Hogan, Belinda Dopita, Amanda Engel and Stephanie Heath. Additionally, we thank the other genetics services who also provided genetic counselling for a small number of participants: Sydney Cancer Genetics, Tasmanian Clinical Genetics Service, Austin Hospital, Peter MacCallam Familial Cancer Centre, and Genetic Health Queensland.
Funding
AFPaCC was enabled by a Cancer Institute NSW Translational Health Services Research Grant. The APGI is supported by The Avner Pancreatic Cancer Foundation. The Clinical Research Coordinator positions in both Sydney and Melbourne are currently supported by PanCare Foundation.
Author information
Authors and Affiliations
Contributions
TD assisted in participant recruitment, maintained study data (June 2017 to present), performed some data analysis, prepared revised documents for ethics submission; and drafted the manuscript. SM developed the genetic counselling guidelines, contributed to the research design; assisted in participant recruitment; maintained study data (Sept 2011 – Feb 2017) and revised the final manuscript. AJ contributed to the establishment of AFPaCC, prepared ethics documents and revised the final manuscript. KT contributed to the research design, assisted in participant recruitment, provided genetic counselling and revised the final manuscript. ADS assisted in participant recruitment, provided genetic counselling and revised the final manuscript. DW contributed to the research design; provided revisions to ethics documents, performed screening and revised the final manuscript. AS designed and established the screening program in Australia; prepared documents for ethics submission, recruited patients, performed screening and revised the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee (St Vincent’s Hospital Human Research Ethics Committee Approval number 10/055) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study. This article does not contain any studies with animals performed by any of the authors.
Consent for publication
Participants provided consent for the publication of deidentified data.
Competing interests
The authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Inclusion and exclusion criteria. (DOCX 17 kb)
Additional file 2.
Genetic counselling guidelines. (DOCX 31 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dwarte, T., McKay, S., Johns, A. et al. Genetic counselling and personalised risk assessment in the Australian pancreatic cancer screening program. Hered Cancer Clin Pract 17, 30 (2019). https://doi.org/10.1186/s13053-019-0129-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13053-019-0129-1