
Abstract
Background
An early diagnosis of CHARGE syndrome is challenging, especially for the primary care physicians who often take care of neonates with multiple congenital anomalies. Here we report eight cases of CHARGE syndrome whose diagnosis was made early in life with the intent to identify the most helpful features allowing a prompt clinical diagnosis.
Methods
Medical records of patients with CHARGE syndrome whose diagnosis was made at the Fondazione IRCCS Ca′ Granda Ospedale Maggiore Policlinico in Milan, Italy were retrospectively reviewed.
Results
Taken together, these patients reflect the considerable phenotypic variability of the syndrome; in one patient, the diagnosis was made immediately after birth because all the major criteria were met. In six patients, presenting with relatively nonspecific defects, a temporal bone computerized tomography scan was essential to achieve the correct diagnosis. In one patient, the diagnosis was made later than the others were. A careful examination revealed the presence of outer, middle, and inner ear anomalies: these elements, in the absence of any additional major criteria, represented for us an important diagnostic clue.
Conclusions
This article suggests that an accurate evaluation of the ear should be made every time CHARGE syndrome is considered as a likely diagnosis even when the standard criteria are not fulfilled.
Similar content being viewed by others

Background
CHARGE syndrome (CS) (OMIM #214800) is an autosomal dominant condition with an occurrence of 1 in 10,000 births [1, 2]. The clinical features of CS were originally described in 1979 by Hall and Hittner [3, 4]. In 1981, Pagon et al. developed the CHARGE acronym (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness). Additional features of this syndrome include cleft lip and palate, hearing loss, tracheoesophageal fistula (TE), and cranial nerve dysfunction such as facial nerve palsy [5]. Some of the congenital abnormalities present in CS can lead to premature death [6].
At present, the clinical criteria elucidated by Blake and Verloes are used together with those of Hall and Hittner. The Blake criteria were slightly adjusted by a consortium and updated in 2009 and include four major and seven minor criteria with the major ones being abnormalities of the ear, coloboma, choanal atresia, cranial nerve dysfunction [5, 7]. Anomalies of the ear could potentially affect the external, internal and middle part with a frequency between 80 and 100% [8]. All four major, or three major and three minor, criteria must be present in order to diagnose CS. In 2005, Verloes proposed a revised set that included semicircular canal defects as a major criterion, anticipated broadening of the phenotypic spectrum, and reduced the number of features necessary for a diagnosis of CS [9]. Blake [5] and Verloes [9] criteria are summarized in Table 1.
CS was previously referred as an association until chromodomain helicase DNA binding protein 7 (CHD7), located on chromosome 8q12.1, was identified as the main gene responsible for the syndrome [10, 11]. Diagnosis now can be confirmed but not excluded by identifying a mutation of this gene found with a detection rate varying between 65 and 90% [8]. The condition is typically sporadic with few familial cases reported [7, 12]. It has a considerable phenotypic variability [2] with no single feature being consistently present and, for this reason, it represents a diagnostic challenge for the primary care physician. Here we report eight different cases of CS whose diagnosis was made early in life.
Methods
We describe a series of eight patients with CS whose diagnosis was made in the Neonatal Intensive Care Unit and neonatal follow up service of our hospital from January 2012 to March 2018. Clinical data, imaging studies and laboratory test results were collected by consulting the infants’ computerized medical charts. All patients underwent a thorough clinical evaluation which included: echocardiography, abdominal ultrasonography, cerebral magnetic resonance imaging, cranial computed tomography (CT) (with the exception of patient 8), audiometry testing, fundoscopy, ear nose throat (ENT), neurological and genetic evaluation. Sequence analysis of the CHD7 gene was performed in Policlinico Sant’Orsola-Malpighi in Bologna, Italy. The other genetic tests (i.e. karyotype and array-comparative genomic hybridization) were performed in our clinic. Informed consent was provided by both parents. The aim of this study was to identify, among all the clinical features, which were the most helpful in reaching the correct diagnosis and differentiating CS from other similar conditions.
Results
In Table 2 we summarize the patients’ clinical features and molecular findings. Among major criteria, choanal atresia was detected only in patient 4. All patients presented with coloboma and hypoplastic or absent semicircular canals with the exception of patient 8 who presented instead with an abnormal right vestibular enlargement. Among minor criteria, rhombencephalic dysfunction, abnormal middle or external ear and psychomotor delay were reported in all patients. Malformation of mediastinal organs (i.e. heart and esophagus), to different degrees, were observed in seven patients.
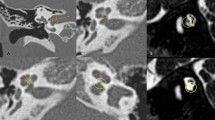
In patients 1–7, the clinical diagnosis of CS was made within the first month of life. On the other hand, the correct diagnosis in patient 8 was made at 18 months of life. Initially, 22q11.2 deletion syndrome was suspected because of the clinical presentation, in particular the marked difficulty in swallowing. Fluorescence in situ hybridization specific for 22q11.2 region along with array-comparative genomic hybridization were performed and both analysis resulted normal. Considering the findings of hypogonadotropic hypogonadism and agenesis of olfactory right bulb, a diagnosis of Kallmann Syndrome was proposed. Molecular analysis with next generation sequencing was also performed but no mutations were found for KAL1, FGFR1, PROKR2, GnRHR, GnRH1, GnRH2, KISSR1, TAC3, TACR3, or HS6ST. The correct diagnosis was finally achieved only by focusing on ear malformations: the patient presented with low set ears with small lobules along with abnormal right vestibular enlargement as seen by cerebral magnetic resonance imaging (MRI) and bilateral aplasia of superior and posterior semicircular canals as well as dysplasia of lateral semicircular canal and vestibulum, dysplasia of the stapes and of the oval window, depicted by CT (Fig. 1). CS was confirmed by molecular analysis of the CHD7 gene which revealed a heterozygous mutation (c.5405-17G > A; IVS25). Patient 4 died at 6 months of age from cardiopulmonary arrest. The other patients are currently alive and all of them present with developmental delay and hearing impairment.

Patient 8’s axial CT image shows the dysplasia of lateral SCC (black arrow) which is only partially present
Discussion
An early diagnosis of CS is important to enable the establishment of a multidisciplinary care team to manage the developmental concerns [12]. This syndrome has a considerable phenotypic variability [7] and many of its features including genital hypoplasia, cleft palate, and heart defect are shared with other syndromes such as 22q.11.2 deletion, Kallmann, Treacher Collins, and VACTERL (vertebral, anorectal, TE, renal and limb defects) [11,12,13]. Moreover, some clinical features may not be fully expressed early in life, some cannot be observed on physical examination, and mental retardation becomes evident only over time. For these reasons, a differential diagnosis can be challenging for the neonatologist who often takes care of newborns with multiple congenital anomalies. In our case series, rhombencephalic dysfunction and ear anomalies were reported in all patients. Multiple cranial nerve involvement produces many ENT concerns including olfactory, facial, glossopharyngeal and vagus nerve involvement. Moreover, choanal stenosis/atresia, cleft lip/palate and TE fistulas may also be present. For this reason, consultation by an ENT physician is essential.
For patient 4 all the major criteria were met and the diagnosis was made immediately after birth. Patients 1–3 and 5–7 presented with relatively nonspecific defects, except for bilateral coloboma and in all these patients, temporal bone CT scan was crucial to obtain the correct diagnosis. In fact, when Verloes proposed revised criteria, semicircular canal defects were included as a major one, as these defects were shown to be a very specific and consistent feature of CS [14, 15]. Patient 8 was the tricky one, and the diagnosis was made much later with respect to the others. He did not express any major criteria but had a significant feeding problem along with renal anomalies, hearing loss, hypogonadotropic hypogonadism, and agenesis of olfactory right bulb. For this reasons, 22.q11.2 deletion syndrome and Kallmann syndrome were initially suspected. In the diagnostic management of this case, focusing on ear anomalies was extremely helpful in pointing to the correct diagnosis. Although an abnormal right vestibular enlargement is not specific for CS, the presence of aplasia of semicircular canals together with the middle and outer ear anomalies was crucial in addressing the proper diagnosis.
In CS, ear abnormalities are extremely frequent being found in > 90% of patients. Although semicircular canal anomalies are highly penetrant features in this syndrome, all the three segments of the ear can be affected and, in fact, ear anomalies are included both in major and in minor criteria [8, 16]. External malformations usually involve an abnormal shape and position of the pinnae, a cup shape wide helix, frequently small or absent lobules (Fig. 2). Middle ear involvement includes ossicular malformations, in particular the aplasia or dysplasia of the incus, of the stapes and oval and round windows, and chronic serus otitis which contributes to conductive hypoacusia [8, 16]. Inner ear abnormalities include cochlear and vestibular anomalies such as dysplasia of the vestibulum and varying degrees of cochlea hypoplasia and malformations, as well as aplasia or dysplasia of semicircular canals which is quite characteristic of CS [8, 16]. Patient 8 presented with low set ears with small lobules, a right mild conductive hearing loss, and an abnormal right vestibular enlargement detected by cerebral MRI as well as bilateral aplasia of semicircular canals. These elements reinforced the hypothesis of CS, which was confirmed on molecular analysis. It is interesting to note that the mutation detected in this case (c.5405-17G > A; IVS25) was previously reported as associated with a mild phenotype, especially in a familial case supporting genotype – phenotype correlation [17].

External typical aspect of ears in our patients
Conclusion
An early pediatric clinical diagnosis of CS remains a complicated task [18]. Which anomaly or combination of anomalies carries the greatest diagnostic weight is not entirely clear. When CS is considered as a likely diagnosis but the criteria are not fulfilled, our experience suggests that a careful observation of the ear could be helpful. Moreover, a CT scan and a MRI of the temporal bone should be obtained to look for the suggestive middle and inner ear defects. Although it is known that with appropriate imaging, abnormalities of the semicircular canals are found in as many as 95% of CHARGE individuals [14, 19,20,21,22,23], this study underlines the importance of a prompt recognition of these signs in the neonatal age to make early diagnosis and timely care.
Availability of data and materials
No datasets were generated or analysed during the current study.
Abbreviations
- Array –CGH:
-
array comparative genome hybridization
- CS:
-
CHARGE syndrome
- TE:
-
Tracheoesophageal fistula
- CT:
-
Cranial computed tomography
- ENT:
-
Ear nose throat
- MRI:
-
Magnetic resonance imaging
- NGS:
-
Next generation sequencing
- ASD:
-
Atrial septal defect
- CHD7:
-
Chromodomain helicase DNA binding protein
- CNS:
-
Central nervous system
- PDA:
-
Patent ductus arteriosus
- SCC:
-
Semicircular canal
- VSD:
-
Ventricular septal defect
References
Sanlaville D, Verloes A. CHARGE syndrome: An update. Eur J Hum Genet. 2007;15:389–99.
Van Ravenswaaij-Arts CMA, Blake K, Hoefsloot L, Verloes A. Clinical utility gene card for: CHARGE syndrome - update 2015. Eur J Hum Genet Nature Publishing Group. 2015;23:1–4. https://doi.org/10.1038/ejhg.2015.15.
Hall BD. Choanal atresia and associated multiple anomalies. J Pediatr. 1979;95:395–8.
Hittner HM, Hirsch NJ, Kreh GM, Rudolph AJ. Colobomatous microphthalmia, heart disease, hearing loss, and mental retardation. A syndrome. J Pediatr Ophthalmol Strabismus. 1979;16:122–8.
Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis. 2006;1:1–8.
Bergman JEH, Blake KD, Bakker MK, du Marchie Sarvaas GJ, Free RH, van Ravenswaaij-Arts CMA. Death in CHARGE syndrome after the neonatal period. Clin Genet. 2010;77:232–40.
Bergman JEH, Janssen N, Hoefsloot LH, Jongmans MCJ, Hofstra RMW, van Ravenswaaij-Arts CMA. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2011;48:334–42.
Lalani SR, Hefner MA, Belmont JW, Davenport SL. CHARGE syndrome summary clinical characteristics diagnosis clinical diagnosis. Gene Rev. 2006; Available from: https://www.ncbi.nlm.nih.gov/books/NBK1117/pdf/Bookshelf_NBK1117.pdf.
Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet. 2005;133(A):306–8.
Janssen N, Bergman JEH, Swertz MA, Tranebjaerg L, Lodahl M, Schoots J, et al. Mutation update on the CHD7 gene involved in CHARGE syndrome. Hum Mutat. 2012;33:1149–60.
Zentner GE, Layman WS, Martin DM, Scacheri PC. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. 2010;152:674–86.
Hughes SS, Welsh HI, Safina NP, Bejaoui K, Ardinger HH. Family history and clefting as major criteria for CHARGE syndrome. Am J Med Genet A. 2014;164:48–53.
Jyonouchi S, McDonald-McGinn DM, Bale S, Zackai EH, Sullivan KE. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11.2 deletion syndrome: A comparison of immunologic and nonimmunologic phenotypic features. Pediatrics. 2009;123:e871–7.
Lemmerling M, Dhooge I, Mollet P, Mortier G, Van Cauwenberge PKM. CT of the temporal bone in the CHARGE association. Neuroradiology. 1998;40:462–5.
Amiel J, Attieé-Bitach T, Marianowski R, Cormier-Daire V, Abadie V, Bonnet D, et al. Temporal bone anomaly proposed as a major criteria for diagnosis of CHARGE syndrome. Am J Med Genet. 2001;99:124–7.
Kaplan J, Hudgins L. Neonatal presentations of charge syndrome and VATER/VACTERL association. Neoreviews. 2008;9:e299–304.
Bilan F, Legendre M, Charraud V, Manière B, Couet D, Gilbert-Dussardier B, et al. Complete screening of 50 patients with CHARGE syndrome for anomalies in the CHD7 gene using a denaturing high-performance liquid chromatography-based protocol: New guidelines and a proposal for routine diagnosis. J Mol Diagnostics. 2012;14:46–55. Elsevier Inc. https://doi.org/10.1016/j.jmoldx.2011.08.003.
Allen T. CHARGE syndrome: diagnosis and clinical management in the NICU. Adv Neonatal Care. 2012;12:336–42.
Tellier AL, Cormier-Daire V, Abadie V, Amiel J, Sigaudy S, Bonnet D, et al. CHARGE syndrome: report of 47 cases and review. Am J Med Genet. 1998;76:402–9.
Wiener-Vacher SR, Amanou L, Denise P, Narcy P, Manach Y. Vestibular function in children with the CHARGE association. Arch Otolaryngol Head Neck Surg. 1999;125:342–7.
Abadie V, Wiener-Vacher S, Morisseau-Durand MP, Porée C, Amiel J, Amanou L, et al. Vestibular anomalies in CHARGE syndrome: investigations on and consequences for postural development. Eur J Pediatr. 2000;159:569–74.
Morimoto AK, Wiggins RH, Hudgins PA, Hedlund GL, Hamilton B, Mukherji SK, et al. Absent semicircular canals in CHARGE syndrome: radiologic spectrum of findings. Am J Neuroradiol. 2006;27:1663–71.
Bauer PW, Wippold FJ II, Goldin JRPL. Cochlear implantation in children with CHARGE syndrome. Arch Otolaryngol Head Neck Surg. 2002;128:1013–7.
Acknowledgements
We thank the patients’ parents to participate to this study. This work has been generated within the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN-ITHACA).
Funding
No funding to declare.
Author information
Authors and Affiliations
Contributions
MFB and BLC conceived the research, collected, interpreted data, drafted the manuscript; LC, RV, VF contributed to interpret data, reviewed and revised the manuscript; MC and CR performed genetic tests and molecular analysis; SG, OP, CC, PFA, ES, and FT collected clinical data, reviewed and revised the manuscript; FM conceived the research, reviewed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The parents of patients give informed consent for publication of this article.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bedeschi, M.F., Crippa, B.L., Colombo, L. et al. A case series of CHARGE syndrome: identification of key features for a neonatal diagnosis. Ital J Pediatr 46, 53 (2020). https://doi.org/10.1186/s13052-020-0806-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-020-0806-8