
Abstract
Background
Silver-Russell Syndrome (SRS) is a genetic disorder characterized by intrauterine and postnatal growth restriction and normal head circumference with consequent relative macrocephaly. Addictional findings are protruding forehead in early life, body asymmetry (of upper and lower limbs) and substantial feeding difficulties. Although several genetic mechanisms that cause the syndrome are known, more than 40% of patients with a SRS-like phenotype remain without an etiological diagnosis. In the last few years, different clinical reports have suggested that mutations or deletions of the HMGA2 gene can be responsible for a SRS-like phenotype in patients with negative results of the common diagnostic tests for this syndrome.
Case presentation
We present a 3-year-old male patient with clinical diagnosis of Silver-Russell Syndrome (SRS) associated with a de novo heterozygous deletion of the long arm of the chromosome 12 (12q14.3) encompassing the HMGA2 gene.
Conclusions
Our report confirms the etiological role of HMGA2 as a disease gene in the development of a SRS-like phenotype.
Similar content being viewed by others


Background
Silver-Russell Syndrome (SRS) is a distinct syndromic growth disorder in which prenatal and postnatal growth failure are associated with other characteristic features, including relative macrocephaly at birth, protruding forehead in early life, body asymmetry and substantial feeding difficulties. Almost all children with SRS are born SGA. Postnatal catch-up growth is not seen in the majority of children with SRS [1].
Multiple clinical scoring systems have been proposed for Silver-Russell syndrome (SRS). In their Prospective Study Validating a Clinical Scoring System and Demonstrating Phenotypical-Genotypical Correlations in SRS, Azzi et al. proposed a new scoring system (Netchine-Harbison clinical scoring system), processing a flow-chart for investigation and diagnosis of SRS [2].
SRS can be caused by different genetic mechanisms. The most common abnormalities are related to the epimutation of either the 11p15.5 region or the chromosome 7. In particular, the H19/IGF2:IG-DMR hypomethylation occurs in almost 40% of cases, followed by the maternal uniparental disomy of chromosome 7 (4–10% of cases), the maternal uniparental disomy of 11p15.5 region (less than 1% of cases), and the multilocus hypomethylation with or without ICR2 hypomethylation (less than 1% of cases). Chromosomal rearrangements, such as the duplication of maternal 11p15.5 region and other cryptic chromosomal aberrations, have been reported in less than 1% of cases respectively. However, all these genetic aberrations can be detected in less than 60% of SRS cases, and thus the clinical diagnosis remains without a genetic etiology [1,2,3,4,5].
In the last decade, several studies have suggested the role of HMGA2 as candidate gene in those patients with SRS phenotype and negative result of classical genetic tests for SRS. HMGA2 gene encodes for the HMGA2 protein, member of the “high-mobility group AT-hook” (HMGA) family. These proteins act as architectural transcription factors that regulate the trascriptional activity of several genes. The expression of HMGA proteins is high in early developmental stages in embryos and mesenchymal stem cells, whilst it is almost absent or very low in adult tissues. Additionally, several studies have showed their role in adipocytes differentiation and their overespression in some tumoral tissues [6,7,8].
In 2007, Menten et al. [9] reported SRS patients with the microdeletion of the 12q14.3 region encompassing different genes including HMGA2 and LEMD3 (an OMIM Disease Causing gene associated with Buschke-Ollendorff syndrome and Osteopoikilosis with or without melorheostosis). To our knowledge, other three heterozygous pathogenic variants and two exon deletions involving the HMGA2 gene have been described in patients with clinical diagnosis of SRS [8, 10,11,12].
We report the clinical and genetic characteristics of a patient with SRS phenotype and a de novo 425 Kb microdeletion of 12q14.3 region encompassing the HMGA2 gene but not the LEMD3 gene.
Case presentation
Our patient is a 3-year-old boy, third child of Caucasian, non-consanguineous and healthy parents. He was born at 39 weeks of gestation via natural delivery, after an unremarkable pregnancy. At birth he was small for gestational age: weight of 2350 g (<3rd centile), length 47 cm (2nd centile), head circumference (HC) 31 cm (<3rd centile). His Apgar scores were 9 at 1 min and 10 at 5 min. His past medical history was significant for a persistent failure to thrive during his first year of life. Several investigations, including a work-up for metabolic and gastrointestinal disorders, multiple urine tests, cranial ultrasound, echocardiogram and abdominal ultrasound, were normal. Endocrinological assessment showed a normal growth hormone (GH) function, but a moderate hypothyroidism, still treated with levothyroxine. At 8 months of age audiological evaluation was normal, while ophtalmologic exam revealed mild pseudostrabismus.
He was referred to our attention at the age of 10 months. His anthropometric parameters were: weight 5900 g (< − 2 SDS), length 64 cm (< − 2 SDS), head circumference (HC) 43.8 cm (9th – 25th centile). In addition, physical examination showed: triangular facies, closed anterior fontanelle, wide forehead, frontal bossing, deeply set eyes, epicanthus inversus, downslanted palpebral fissures, blue sclerae, narrow chin with slight vertical crease, ears posteriorly rotated with prominent anterior crus of antihelix and underdeveloped tragus and antitragus, syndactyly of 2th/3th finger of the foots, clinodactyly of 5th finger of the hands, low muscle mass, asymmetric lower limbs (left almost 1 cm shorter than right). A neuropsychiatric consult was requested, and then a moderate speech delay was detected. The brain MRI was normal.
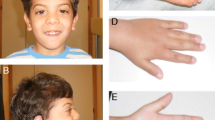
At 2 years, the clinical picture appeared unchanged. His weight was 7500 g (<3rd centile), his lenght was 81 cm at the left side and 80 cm at the right (<3rd centile), his HC was 47,3 cm (10rd centile) and his BMI was 11,7 (<3rd centile). During the last check-up at our Center, at the age of 3 years, no significant changes were revealed regarding the clinical presentation and growth parameters. His weight was 9450 g (<3rd centile), his lenght was 88 cm at the left side and 89 cm at the right (3rd centile), and his HC was 48,2 cm (3rd centile) (Fig. 1).

a, b, c Our patient at 3 years and 4 months of age. Note triangular facies, wide forehead, frontal bossing, deeply set eyes, epicanthus inversus, downslanted palpebral fissures, narrow chin with slight vertical crease, ears posteriorly rotated with prominent anterior crus of anthelix and underdeveloped tragus and antitragus
Applying the Netchine-Harbison CSS (NHCSS) we got a score of 5/6 (the proband met all the criteria for likely SRS except for relative macrocephaly at birth) [2].
Genetic tests
In accordance with the clinical suspicion of SRS assessed through the NHCSS, we proceeded according to the flow-chart proposed by Azzi et al. and performed the classical genetic test for this syndrome. The Methylation-Specific Multiplex Legation-dependent Probe Amplification (MS-MLPA) at the imprinting region 11p15 did not reveal Loss Of Metilation (LOM) and the study of microsatellite markers of chromosome 7 did not detect UniParental Disomy (UPD). Subsequently, consistently with the aforementioned flow-chart, we proceeded with the research of genome-wide microdeletions or microduplications by Array-CGH (Array-Comparative Genome Hybridization). This analysis showed a de novo partial deletion of the long arm of the chromosome 12, 425 Kb-sized, from position 66,358,287, in which there is the most terminal probe delete on the array (cytogenetic locus 12q14.3) to 66,782,791 (cytogenetic locus 12q14.3) (Fig. 2). According with the build UCSC Genome Browser (Hg build 19) several genes lie in this region including the OMIM disease causing genes HMGA2 (OMIM 600698, partially involved), IRAK3 (OMIM 604459) and GRIP1 (OMIM 604597). This microdeletion is not reported in DECIPHER. However, the same database recorded a 1.67 Mb deletion (ID 337828) of unknown significance, encompassing the same genes and maternally inherited, in a female infant with episodic hyperhidrosis, failure to thrive, moderate short stature, facial asymmetry, prominent forehead, proptosis, scoliosis and history of intrauterine growth retardation. A deletion with the same genomic and clinical features (size, genomic coordinates, maternal origin, clinical picture except for the absence of hyperhidrosis) have been previously described by Heldt et al. in a family where it segregated along with Silver-Russell like phenotype. Even if neither Heldt nor the database DECIPHER refer to the case reported by the other, we believe it is reasonably to conclude that the deletions reported in DECIPHER and in Heldt paper are the same [2, 13].

Genome-wide array-CGH analysis: 425 Kb deletion of the long arm of the chromosome 12, ranging from 66358287 Mb (12q14.3) to 66782791 Mb (12q14.3)
Materials and methods
DNA of proband and his parents was extracted with QIAamp DNA blood Midi Kit (Qiagen Inc., Valencia, CA) according to the manufacturer’s instructions. The normal control was a commercially available male Promega (Promega Corporation, Madison, WI). Array-CGH analysis was performed using a Human Genome CGH Microarray Kit 8x60K (Agilent) with a resolution of 100–150 kb according to the manufacturer’s instructions. Array slides were analyzed with an Agilent G2505B scanner. Image were analyzed and visualized with CytoGenomics 3.0.6.6 (Agilent Technologies, Santa Clara, CA).
Discussion & conclusions
More than 40% of patients with a SRS-like phenotype remain without an etiological diagnosis [3, 4]. Several genes have been proposed as candidate genes. During the last 10 years, different reports have suggested the role of HMGA2 in the etiology of SRS-like phenotype. To our knowledge, only 28 cases with a SRS-like phenotype due to the haploinsufficiency of HMGA2 have been reported (Table 1). In particular, 23 deletions, detected by a-CGH, have been described. The remaining 5 cases were: a 7 bp deletion [11], a nonsense variant and a frameshift variant [10] and 2 deletions of exon 2 [12] and exons 1–2 [8]. The 23 deletions described so far range between 387 kb and 10.12 Mb, and involve other contiguous genes [9, 12,13,14,15,16,17,18,19,20,21,22,23]. Despite the variable size of these deletions, a critical region of 2.61 Mb has been identified. The OMIM disease causing genes included in this region are: LEMD3 (OMIM 607844), MSRB3 (OMIM 613719), HMGA2 (OMIM 600698), IRAK3 (OMIM 604459), GRIP1 (OMIM 604597).
The HMGA2 gene encodes for the HMGA2 protein, member of the HMGA family. This family of four proteins (HMGA1a, HMGA1b, HMGA1c and HMGA2), encoded by two genes (HMGA1 and HMGA2), can interact with DNA altering its conformation and regulating the transcription of several genes. HMGA2 regulates the transcription of the known fetal growth factor IGF2. Multiple studies on human and animal models have demonstrated a high expression of HMGA2 during fetal period when it may play a crucial role in embryonic development and linear growth. Moreover, HMGA2 has showed to be essential in regulating cell growth, proliferation, differentiation and death, in promoting adipocyte differentiation. Over-expression of HMGA2 has also been found in some tumoral tissues (uterine leiomyoma, gastric cancer, pediatric lipoma) [6,7,8, 12, 24,25,26,27]. These experimental studies, added to the clinical data reported in literature, converge in proving the key role of HMGA2 alterations in pre- and postnatal growth failure and failure to thrive that is otherwise unexplained.
As previously reported, LEMD3 is the causative gene of Buschke-Ollendorff syndrome and Osteopoikilosis with or without melorheostosis (OMIM 166700). All patients with characteristic features of this syndrome result carrying deletions involving also LEMD3, as suggested by data in Table 1 (see superscripts and notes).
In regard to the other OMIM disease causing genes included in the critical region, biallelic mutations in MSRB3 gene have been described in cases of deafness autosomal recessive 74 (OMIM 613718), while several polymorphisms of IRAK3 have been associated with increased asthma susceptibility. The GRIP1 gene, known as causative gene of Fraser syndrome transmitted in recessive manner, has been considered as candidate gene for neurodevelopmental disorders in most patients carrying 12q14.3 deletions. This hypothesis arises from the evidence of his high expression in brain tissue in both fetal and postnatal period, and his clear role in synaptic functioning. However, developmental disorders, such as language and/or motor delay, learning difficulties and intellectual disability, have also been described in patients with a 12q14.3 deletion not involving GRIP1 gene [15, 23].
An exclusive role of HMGA2 gene in the pathogenesis of SRS-like cases is supported by the evidence that pre- and postnatal growth failure and underweight, typical features of SRS (the first two items of the Netchine-Harbison CSS), have been described in all patients carrying different aberrations of HMGA2 regardless of their dimension. In fact, there are not significant clinical difference among patients with more or less extensive deletions of the 12q14.3 region (involving multiple genes, as HMGA2), and patients with pathogenic variants or intragenic deletions of HMGA2. Moreover, recently a duplication involving HMG2A in a patient with overgrowth, obesity and tall stature with advanced bone age has been reported [28], confirming the role of HMGA2 gene in growth regulation.
Regarding the other clinical features of the patients described in the literature (see Table 1), relative macrocephaly and a prominent forehead, two further items of the NHCSS, present in our patient, to our knowledge, were described in literature in less than half of the cases. Another item of the NHCSS, the body asymmetry, does not seem to be a typical feature of the 12q14.3 microdeletion syndrome, while it is reported in our proband, who has lower limbs asymmetry. To date, our case represents the only one 12q14.3 microdeletion syndrome with limb asymmetry. Finally, our patient presents a moderate speech delay, in the absence of other neurodevelopmental disorders. The presence of neurodevelopmental disorders (language and/or motor delay, learning difficulties, intellectual disability, behavioural problems), seems to be a distinct and frequent feature of 12q14.3 microdeletion syndrome, as it is reported in about half of patients (see Table 1) [2].
In conclusion, we suggest to investigate HMGA2 gene in all the patients with evocative phenotype for SRS and negative genetic results for MS-MLPA at the imprinting region 11p15 and microsatellite markers of chromosome 7. In order to complete the diagnostic work-up of SRS we recommend array-CGH analysis, and subsequently the sequence analysis and the MLPA of HMGA2 gene.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- SRS:
-
Silver-Russell syndrome
- SD:
-
Standard deviations
- MRI:
-
Magnetic resonance imaging
- MS-MLPA:
-
Metilation-Specific Multiplex Legation-dependent Probe Amplification
- LOM:
-
Lost Of Metilation
- UPD:
-
UniParental Disomy
- a-CGH:
-
Array-Comparative Genome Hybridization
References
Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, Bliek J, et al. Diagnosis and management of silver–Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017;13:105–24.
Azzi S, Salem J, Thibaud N, Chantot-Bastaraud S, Lieber E, Netchine I, Harbison MD. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. 2015;52(7):446–53.
Saal HM. Russell-Silver syndrome. 2002 Nov 2 [Updated 2011 Jun 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle: University of Washington, Seattle; 1993-2019.
Eggermann T, Buiting K, Temple IK. Clinical utility gene card for: Silver–Russell syndrome. Eur J Hum Genet. 2011;19(3). https://doi.org/10.1038/ejhg.2010.202.
Beygo J, Mertel C, Kaya S, Gillessen-Kaesbach G, Eggermann T, Horsthemke B, Buiting K. The origin of imprinting defects in Temple syndrome and comparison with other imprinting disorders. Epigenetics. 2018;13(8):822–8.
Navarra A, Musto A, Gargiulo A, Petrosino G, Pierantoni GM, Fusco A, Russo T, Parisi S. Hmga2 is necessary for Otx2-dependent exit of embryonic stem cells from the pluripotent groundstate. BMC Biol. 2016;14:24.
Xi Y, Shen W, Ma L, Zhao M, Zheng J, Bu S, Hino S, Nakao M. HMGA2 promotes adipogenesis by activating C/EBPβ-mediated expression of PPARγ. Biochem Biophys Res Commun. 2016;472(4):617–23.
Leszinskia GS, Warnckeb K, Hoefelea J, Wagnera M. A case report and review of the literature indicate that HMGA2 should be added as a disease gene for Silver-Russell syndrome. Gene. 2018;663:110–4.
Menten B, Buysse K, Zahir F, Hellemans J, Hamilton SJ, Costa T, Fagerstrom C, Anadiotis G, Kingsbury D, McGillivray BC, Marra MA, Friedman JM, Speleman F, Mortier G. Osteopoikilosis, short stature and mental retardation as key features of a new microdeletionsyndrome on 12q14. J Med Genet. 2007;44(4):264–8.
Abi Habib W, Brioude F, Edouard T, Bennett JT, Lienhardt-Roussie A, Tixier F, Salem J, Yuen T, Azzi S, Le Bouc Y, Harbison MD, Netchine I. Genetic disruption of the oncogenic HMGA2-PLAG1-IGF2 pathway causes fetal growth restriction. Genet Med. 2018;20(2):250–8.
De Crescenzo A, Citro V, Freschi A, Sparago A, Palumbo O, Cubellis MV, Carella M, Castelluccio P, Cavaliere ML, Cerrato F, Riccio A. A splicing mutation of the HMGA2 gene is associated with Silver-Russell syndrome phenotype. J Hum Genet. 2015;60(6):287–93.
Buysse K, Reardon W, Mehta L, Costa T, Fagerstrom C, Kingsbury DJ, Anadiotis G, McGillivray BC, Hellemans J, de Leeuwg N, de Vries BBA, Speleman F, Menten B, Mortier G. The 12q14 microdeletion syndrome: additional patients and further evidence that HMGA2 is an important genetic determinant for human height. Eur J Med Genet. 2009;52(2–3):101–7.
Heldt F, Wallascheka H, Rippergera T, Morlota S, Illiga T, Eggermannb T, Schlegelbergera B, Scholza C, Steinemanna D. 12q14 microdeletion syndrome: a family with short stature and Silver-Russell syndrome (SRS)-like phenotype and review of the literature. Eur J Med Genet. 2018;61(8):421–7.
Mari F, Hermanns P, Giovannucci-Uzielli ML, Galluzzi F, Scott D, Lee B, Renieri A, Unger S, Zabel B, Superti-Furga A. Refinement of the 12q14 microdeletion syndrome: primordial dwarfism and developmental delaywith or without osteopoikilosis. Eur J Hum Genet. 2009;17(9):1141–7.
Spengler S, Schönherr N, Binder G, Wollmann H, Fricke-Otto S, Mühlenberg R, Denecke B, Baudis M, Eggermann T. Submicroscopic chromosomal imbalances in idiopathic Silver-Russell syndrome (SRS): the SRSphenotype overlaps with the 12q14 microdeletion syndrome. J Med Genet. 2010;47(5):356–60.
Lynch SA, Foulds N, Thuresson AC, Collins AL, Annerén G, Hedberg BO, Delaney CA, Iremonger J, Murray CM, Crolla JA, Costigan C, Lam W, Fitzpatrick DR, Regan R, Ennis S, Sharkey F. The 12q14 microdeletion syndrome: six new cases confirming the role of HMGA2 in growth. Eur J Hum Genet. 2011;19(5):534–9.
Bibb AL, Rosenfeld JA, Weaver DD. Report of a mother and daughter with the 12q14 microdeletion syndrome. Am J Med Genet A. 2012;158A:417–22.
Alyaqoub F, Pyatt RE, Bailes A, Brock A, Deeg C, McKinney A, Astbury C, Reshmi S, Shane KP, Lamb Thrush D, Sommer A, Gastier-Foster JM. 12q14 microdeletion associated with HMGA2 gene disruption and growth restriction. Am J Med Genet A. 2012;158A:2925–30.
Takenouchi T, Enomoto K, Nishida T, Torii C, Okazaki T, Takahashi T, Kosaki K. 12q14 microdeletion syndrome and short stature with or without relative macrocephaly. Am J Med Genet A. 2012;158A:2542–4.
Nso-Roca AP, Marco FC, Ricote JM, Ruizl MJ. Endocrinological anomalies in a patient with 12q14 microdeletion syndrome. Completing phenotype of this exceptional short stature condition. J Pediatr Endocrinol Metab. 2014;27(5–6):539–43.
Mc Cormack A, Sharpe C, Gregersen N, Smith W, Hayes I, George AM, Love DR. 12q14 microdeletions: additional case series with confirmation of a macrocephaly region. Case Rep Genet. 2015;2015:192071.
Raymond L, Francou B, Petit F, Tosca L, Briand-Suleau A, Metay C, Martinovic J, Cordier AJ, Benachi A, Pineau D, Guiochon-Mantel A, Goossens M, Tachdjian G, Brisset S. Complex translocation t(1;12;14)(q42;q14;q32) and HMGA2 deletion in a fetus presenting growth delay and bilateral cataracts. Eur J Med Genet. 2015;58(11):591–6.
Fischetto R, Palumbo O, Ortolani F, Palumbo P, Leone MP, Causio FA, Pesce S, Digilio MC, Carella M, Papadia F. Clinical and molecular characterization of a second family with the 12q14 microdeletionsyndrome and review of the literature. Am J Med Genet A. 2017;173(7):1922–30.
Cleynen I, Van de Ven WJM. The HMGA proteins: a myriad of functions (review). Int J Oncol. 2008;32(2):289–305.
Dadone B, Refae S, Lemarie-Delaunay C, Bianchini L, Pedeutour F. Molecular cytogenetics of pediatric adipocytic tumors. Cancer Genet. 2015;208:469–81.
Lagana AS, Vergara D, Favilli A, La Rosa VL, Tinelli A, Gerli S, Noventa M, Vitagliano A, Triolo O, Rapisarda AMC, Vitale SG. Epigenetic and genetic landscape of uterine leiomyomas: a current view over a common gynecological disease. Arch Gynecol Obstet. 2017;296:855–67.
Zhu J, Wang H, Xu S, Hao Y. Clinicopathological and prognostic significance of HMGA2 overexpression in gastric cancer: a meta-analysis. Oncotarget. 2017;8:100478–89.
Dória S, Alves D, Pinho MJ, Pinto J, Leão M. 12q14 microduplication: a new clinical entity reciprocal to the microdeletion syndrome? BMC Med Genet. 2020;13(1):2.
Acknowledgements
We would like to thank the patient family, and all the participants of this study.
Funding
The authors declare no funding for this work.
Author information
Authors and Affiliations
Contributions
FM, MB and MP made substantial contributions to conception and design, acquisition of data and analysis and interpretation of data. MM performed array-CGH analysis. FM and MB wrote the manuscript. TF and MP reviewed and revised the manuscript. FM, MB, ES, DP and MP were involved in the genetic diagnosis and care. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The parents of the patient described in this case report signed written informed consent for take part in the study.
Consent for publication
Written informed consent for publication of medical data, genetic testing results and identifying images was obtained from the patient’s parents.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mercadante, F., Busè, M., Salzano, E. et al. 12q14.3 microdeletion involving HMGA2 gene cause a Silver-Russell syndrome-like phenotype: a case report and review of the literature. Ital J Pediatr 46, 108 (2020). https://doi.org/10.1186/s13052-020-00866-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-020-00866-9