
Abstract
Background
Charcot-Marie-Tooth (CMT) disease is one of the most common hereditary peripheral neuropathy. The major clinical features of CMT are progressive muscle weakness of distal extremities and sensory loss. MFN2 encodes a GTPase dynamin-like protein mitofusin 2 and plays an essential role in mitochondrial functions. In previous studies, MFN2 mutations have been linked to neurological disorders including CMT type 2 (CMT2). Here, we report a novel mutation in MFN2 which leads to CMT 2.
Case presentation
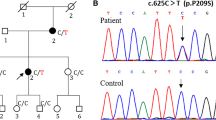
We report a 4-year-old Chinese boy with CMT symptoms including foot-drop gait, running difficulties, frequent falls, slowly progressive atrophy of lower legs with a mildly foot deformity. Nerve conduction velocity study (NCVS) found that no compound motor action potential (CMAP) was elicited in the nervi suralis and tibial nerve. Moreover, the sensory nerve action potential (SNAP) of the nervi suralis was not elicited, which means the peripheral nerves of his lower limbs were damaged. Targeted next-generation sequencing identified a novel heterozygous mutation c.730G > C (p.Val244Leu) in MFN2 in the patient but not in his parents, suggesting that this mutation likely occurred de novo. c.730G > C (p.Val244Leu) in MFN2 is a likely pathogenic mutation for CMT2.
Conclusion
The c.730G > C (p.Val244Leu) mutation in MFN2 is a likely pathogenic mutation for CMT2.
Similar content being viewed by others


Background
Charcot-Marie-Tooth (CMT) disease, also known as hereditary motor sensory neuropathy (HMSN),is one of the most common hereditary peripheral neuropathy with an estimated prevalence of 1 in 2500 [1, 2]. The major clinical features of CMT are progressive muscle weakness of distal extremities and sensory loss [3]. According to the clinical symptoms and electrophysiological characteristics, CMT is categorized into type 1 (CMT1) and type 2 (CMT2) [4]. CMT1 is characterized by hypertrophic demyelination with slow nerve conduction velocities (NCVs) (median nerve conduction velocity <38 m/s), while CMT2 is characterized by normal or mildly reduced NCVs (median nerve conduction velocity >38 m/s) along with axonopathy [1, 4].
MFN2 is located on the short arm of chromosome 1 and encodes mitofusin 2, a GTPase dynamin-like protein of the outer mitochondrial membrane. It plays an essential role in mitochondrial functions including fusion, axonal transport, interoranellar communication and mitophagy [5, 6]. In previous studies, MFN2 mutations have been linked to neurological disorders including CMT2 (named as CMT2A2, OMIM#609260). To date, more than 100 MFN2 mutations have been reported to be associated with CMT2 [6, 7]. Among them, there is just one mutation at the 730th nucleotide in the encoding region (c.730G > A/p.Val244Met) [8].
Here, we reported a 4 year-old-boy with CMT2 symptoms including muscle weakness, difficulty in running and frequent fall. Targeted next-generation sequencing identified a novel heterozygous mutation c.730G > C (p.Val244Leu) in MFN2, which occurred de novo.
Case presentation
The patient was a 4-year-old Chinese boy who couldn’t stand steadily by himself and had suspicious foot-drop gait. Physical examination revealed atrophy of lower extremities (Fig. 1). His left foot showed a mild pescavus deformity and could not lift up by himself. There was no strength in his right hand. Testing of the deep-tendon reflexes showed reduced patellar tendon reflex and ankle reflex. There were no Babinski sign, Chaddock sign, Oppenheim sign and Gordon sign. Muscle strength was reduced with MRC scores of IV on lower extremities. The muscular tension was mildly reduced. NCVS showed damage of peripheral nerves.

The lower extremities of the patient
The patient is the third child in his family. His two sisters who are four and two years older were completely healthy. No family history of similar problems was presented. The patient was one of the two twins when his mother was in pregnancy. Unfortunately his twin brother died in the uterus. Since his mother’s amniotic fluid broke early, he was born by caesarean with 5.73 pounds in weight. There was no asphyxia at birth. During his growing progress, he could walk at the age of 1 year and 7 to 8 months albeit not steadily with suspicious foot-drop gait. He was easy to fall when running with progressive aggravation. He had normal intelligence. NCVS showed normal motor nerve conduction velocity (MCV) of upper limbs but showed slower sensory nerve conduction velocity (SCV). In addition, neither compound motor action potential (CMAP) or sensory nerve action potential (SNAP) of lower limbs was elicited, indicating that the peripheral nerves of his lower limbs were damaged (Tables 1 and 2). When it comes to brain and spinal MRI, the brain MRI image indicates the possibility of periventricular leukomalacia (PVL) while the spinal MRI image shows no apparent abnormality (Fig. 2). As for lumbar puncture, the cerebrospinal fluid examination results were normal. The leucocyte count was zero, the protein and sugar quantifications were in normal control range and the immunoglobulin quantifications were as well.

The brain MRI image of the patient
Since the clinical features of the patient was suspicious of CMT, we sequenced 133 candidate genes for hereditary motor sensory neuropathy (HMSN) and identified a novel heterozygous mutation c.730G > C (p.Val244Leu) in MFN2. This mutation was not present in either of his parents.
Discussion
MFN2 is located on the short arm of chromosome 1. It encodes mitofusin 2, a GTPase dynamin-like protein of the outer mitochondria membrane. MFN2 has an essential role in mitochondrial functions including fusion, axonal transport, interorganellar communication and mitophagy [5, 6]. Because mitochondria are crucial organelles present in almost all cells of human body (except erythrocyte) to provide ATP for metabolic processes and oxidative phosphorylation [9], mutations of MFN2 may cause mitochondria dysfunction and affect high energetic demand tissues. Since the first heterozygous mutation in the MFN2 has been found by Züchner et al in 2004, more than 100 MFN2 mutations have been reported to be associated with CMT [6, 7, 10]. Based on the existing reports, these mutations can be either gain or loss of function in mitochondria. In some cases, certain MFN2 mutations are associated with specific clinical features [11]. c.730G > A (p.Val244Met), a mutation occurring at the same position as the one reported in this study, has been reported to cause CMT2 by K.Kijima et al in 2005. The Japanese kid with the c.730G > A mutation showed CMT2 phenotype with no sensory impairments. The age of onset was 10 months and the symptom included walking difficulties [8]. In our study, the patient’s symptom at the onset was abnormal gait when he started to walk at the age of 1 year and 7 to 8 months. Then he showed running difficulties, frequent falls and slowly progressive atrophy of lower legs with a mild deformity at his left foot. On the other side, this boy did have sensory loss at his lower limbs. All of these symptoms agree with CMT phenotypes. Because of his median nerve conduction velocity is 50.0 m/s (>38 m/s), we classified it as CMT2.
Based on previously reports and the clinical symptoms of the patient, we believe that the novel MFN2 mutation identified in this study has a deleterious nature and is the likely cause of CMT2 for the following reasons:. First, the c.730G > C mutation changes an amino acid from valine to leucine and may affect protein features. Second, p.Val244Leu in MFN2 was predicted to be disease-causing by all three computational tools (Mutation Taster, PROVEAN and SIFT). Third, this mutation changed an evolutionarily conserved amino acid (Phylop score is 5.51) (Table 3). Four, according to previous reports, there is one CMT2A disease causing mutation at the same position (c.730G > A/p.Val244Met). In summary, the de novo pattern is consistent with the previously reported autosomal dominant inheritance of CMT2A2.
Conclusion
The c.730G > C (p.Val244Leu) mutation in MFN2 is a likely pathogenic mutation for CMT2.
Ethics approval and consent to participate
Written informed consent was obtained from the parents of the patient for participate with ethics approval.
Consent to publish
Written informed consent was obtained from the parents of the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Availability of data and materials
The main data that supported the conclusion of the manuscript are presented in the paper and additional supporting files.
Endnotes
The endnotes we used were reference and included in the reference section.
Abbreviations
- CMT:
-
is the abbreviation for Charcot-Marie-Tooth disease
- CMT2:
-
is the abbreviation for Charcot-Marie-Tooth disease type 2
- NCVS:
-
is the abbreviation for nerve conduction velocity study
- CMAP:
-
is the abbreviation for compound motor action potential
- SNAP:
-
is the abbreviation for sensory nerve action potential
- HMSN:
-
is the abbreviation for hereditary motor sensory neuropathy
- MCV:
-
is the abbreviation for motor nerve conduction velocity
- SCV:
-
is the abbreviation for sensory nerve conduction velocity
- PVL:
-
is the abbreviation for leukomalacia
References
Geir JB, Jette CS, Ana L, Helle H, Michael BR. MFN2 point mutations occur in 3.4 % of Charcot-Marie-Tooth families. An investigation of 232 Norwegian CMT families. BMC Med Genet. 2010; doi: 10.1186/1471-2350-11-48
Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin Genet. 1974;6:98–118.
Gemignani F, Melli G, Alfieri S, Inglese C, Marbini A. Sensory manifestations in Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2004;9(1):7-14.
Jani-Acsadi A, Ounpuu S, Pierz K, Acsadi G. Pediatric Charcot-Marie-Tooth disease. Pediatr Clin North Am. 2015; doi: 10.1016/j.pcl.2015.03.012.
Van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014; doi: 10.1016/j.bbamcr.2014.03.009.
Stuppia G, Rizzo F, Riboldi G, Del Bo R, Nizzardo M, Simone C, et al. MFN2-related neuropathies: Clinical features, molecular pathogenesis and therapeutic perspectives. J Neurol Sci. 2015; doi: 10.1016/j.jns.2015.05.033.
Bergamin G1, Dalla Torre C, Cacciavillani M, Lucchetta M, Boaretto F, Campagnolo M, et al. Novel mutation of the mitofusin 2 gene in a family with Charcot–Marie–Tooth disease type 2. Muscle Nerve. 2014; doi: 10.1002/mus.23985
Kijima K, Numakura C, Izumino H, Umetsu K, Nezu A, Shiiki T, et al. Mitochondrial GTPase mitofusin 2 mutation in Charcot–Marie–Tooth neuropathy type 2A. Hum Genet. 2005;116(1-2):23–7.
Ranieri M1, Brajkovic S, Riboldi G, Ronchi D, Rizzo F, Bresolin N, et al. Mitochondrial fusion proteins and human diseases. Neurol Res Int. 2013; doi: 10.1155/2013/293893.
Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot–Marie–Tooth neuropathy type 2A. Nat Genet. 2004;36(5):449–51.
Verhoeven K, Claeys KG, Züchner S, Schröder JM, Weis J, Ceuterick C, et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot–Marie–Tooth type 2. Brain. 2006;129(Pt 8):2093–102.
Acknowledgments
We would like to thank the support from the National Natural Science Foundation of China (8127137&81571180).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LL diagnosed the patient and made substantial contributions to acquire the data, participated in the design of the study and carried out the molecular genetic studies. YY participated in the sequence alignment and drafted the manuscript and accomplished the work of revising. Both authors read and approved the final manuscript.
Authors’ information
LL is director, Department of Pediatric Neurology, Xinhua Hospital Affiliated to Shanghai Jiaotong University School of Medicine.
YY is a medical student of Soochow University.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, Y., Li, L. A novel p.Val244Leu mutation in MFN2 leads to Charcot-Marie-Tooth disease type 2. Ital J Pediatr 42, 28 (2016). https://doi.org/10.1186/s13052-016-0237-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-016-0237-8