
Abstract
Ferroptosis is a novel type of programmed cell death dependent on iron and characterized by the accumulation of lipid peroxides in cells and is closely related to various diseases. Female infertility is a global health concern, which is associated with a variety of factors. The etiology remains unknown in many women with infertility. With further investigation into the pathogenesis of infertility, a growing number of studies have demonstrated the close connections between infertility and ferroptosis. Through a literature review, it is found that ferroptosis is closely involved in endometriosis- and polycystic ovarian syndrome (PCOS)-associated infertility and tubal factor infertility. Iron overload increases the resistance to ferroptosis, and ferroptosis in some cells accelerates endometrial lesion growth. Moreover, iron overload may be hazardous to oocytes. This review may shed some light on the diagnosis and treatment of female infertility.
Similar content being viewed by others
Female infertility refers to the inability of a couple to conceive after at least one year of normal unprotected sexual intercourse, with increasing prevalence in recent years and seriously affects women’s physical and mental health [1].Female infertility is associated with a variety of factors. The etiology still remains unknown in 15–30% of women with infertility [2]. As the pathogenesis of female infertility is being explored by more and more studies, it has been reported that programmed cell death (PCD) plays a key role in human reproduction [3]. PCD is a genetically regulated process of cell suicide that is critical for the development, homeostasis, and integrity of organisms. PCD is divided into several types, including apoptosis, necroptosis, autophagy, ferroptosis and pyroptosis [4, 5]. Ferroptosis is distinct from other forms of programmed cell death. Specifically, the former is dependent on intracellular irons, and nuclei in cells conserve their structural integrity [6]. In recent years, the relationship between ferroptosis and female infertility has aroused academic interest. Identification of novel signaling pathways implicated in female infertility is a prerequisite for satisfying unmet needs. This paper mainly focuses on the relationship between ferroptosis and female infertility based on a literature review and summarizes recent findings on the etiology and pathogenesis of female infertility [7]. Some new directions in female infertility treatment may be identified from the related studies on this topic.
Ferroptosis
Iron is an essential metal for the human body and is vital for maintaining biological homeostasis. Iron oxidation has two states, Fe2+ and Fe3+, which are mainly present intracellularly and extracellularly, respectively. Fe3+ can bind to transferrin (TF) in serum and is subsequently taken up by the TF receptor 1 (TfR1), which is encoded by TFRC on the cell membrane [8].Fe3+ taken up into the cell is reduced to Fe2+ by STEAP3 metalloreductase in the endosome, and then is stored in ferritin and ultimately released again by ferritin phagocytosis-mediated ferritin degradation [9, 10]. Iron metabolism pathway has an irreplaceable role in maintaining the dynamic balance of iron in the body, and the metabolizing molecules related to this pathway include: Divalent metal transporter 1 (DMT1), Transferrinreceptor (TFR), Ferroportin (FPN), Ferritin and so on. When transporter proteins are mutated or absent, iron homeostasis is disrupted, which leads to excessive accumulation of iron, which triggers oxidative cell damage and death. Then cellular oxidative damage and death appear. In addition, it has been shown that the iron-mediated Fenton reaction is involved in the generation of intracellular reactive oxygen species (ROS) [11]. ROS can attack intracellular DNA molecules, lipids, and proteins, among other substances, ultimately leading to intracellular ROS accumulation and depletion of antioxidant capacity [12]. This disrupts the normal cell membrane structure, induces cell death and promotes the development of various disease processes.
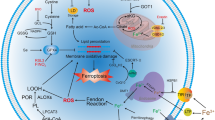
The term “ferroptosis”, defined as a distinct type of iron-dependent non-apoptotic cell death, in 2012 [13]. Ferroptosis is a new type of cell death that is distinct from other cell deaths. Ferroptosis is biochemically, morphologically, and genetically distinct from other types of PCD, such as apoptosis, necrosis, and autophagy. Ferroptosis is featured by iron accumulation, abnormal mitochondrial membrane density, overexpression of relevant biomarkers, lipid peroxidation, and abnormal changes in immune functions [13,14,15,16,17]. Apart from the features above, cells may spread between cells in a wave-like manner after ferroptosis [18]. Although the core molecular mechanism of ferroptosis is closely related to several genes and cell signaling pathways. The role of two types of key mediating pathways has been confirmed: The first is endogenous or enzyme-regulated pathways. For example, glutathione peroxidase 4 (GPX4), as an anti-oxidant defense enzyme against ferroptosis, is usually inhibited. The second is exogenous or transporter-dependent pathways. For example, cysteine or glutamine uptake is obviously reduced, or ferritin uptake is elevated [19]. The mechanism of ferroptosis is illustrated in Fig. 1 [20].

Mechanism of ferroptosis. The molecular machinery of ferroptosis involves cellular antioxidant and oxidative systems, and the regulation of ferroptosis includes iron metabolism and lipid peroxidation
Relationship between ferroptosis and infertility
Endometriosis-associated infertility
Endometriosis is a chronic inflammatory disease characterised by the growth of endometrial tissue outside the uterine cavity and occurs in approximately 50% of women with infertility [21]. In normal endometrial tissues, estrogen triggers the rapamycin target protein (mTOR) pathway and inhibits the hypoxia-inducible factor-1/reactive oxygen species/AMP-activated protein kinase (HIF-1/ROS/AMPK) pathway, which results in autophagy of endometrial tissues out of menstruation [22]. Endometrial cells were harvested from patients with endometriosis-associated infertility and examined in some studies. However, it was found that estrogen did not inhibit reactive oxygen species(ROS). Iron overload in endometrial-like tissues outside the uterus finally resulted in a much higher ROS level than in normal in-situ endometrial tissues [23]. Ovarian endometriotic cysts and surrounding ovarian tissues usually exhibit higher iron levels on account of periodically spontaneous bleeding of ectopic lesions [24]. In another study, the increased levels of ferritin and divalent metal transporter 1 (DMT1) were demonstrated in the endometriotic tissues of patients with endometriosis. It was indicated that excess Fe2+ infiltrating into the cytoplasm was stored in ferritin [25]. In that case, if the overall iron utilization decreased, excess Fe2+ would promote sensitivity to ferroptosis. It has been reported that ferroptosis of some endometriotic stromal cells may be elicited after contact with the cyst fluid. Moreover, p38 mitogen-activated protein kinase/signal transducer and activator of transcription 6 (p38 MAPK / STAT6) pathway was involved in ferroptosis-induced vascular endothelial growth factor A (VEGFA) and IL8 upregulation [3]. IL-8 and VEGFA promoted sustained proliferation of endometrial cells, accompanied by obvious angiogenesis and adhesion [26, 27]. The recognition of damage-associated molecular patterns (DAMPs), which are produced or released by damaged and dying cells, promotes sterile inflammation. DAMPs then activate nuclear factor kappa-B (NF-κB) via the advanced glycation end product (AGER) pathway, leading to an immune response [28]. Lipid peroxide 4-hydroxynonenal (4HNE) resulting from ferroptosis can also promote NF-κB activation, thus inducing inflammatory mediator response [29]. Besides, endometriotic stromal cells also contain abundant Fe2+, which may elicit ROS via the Fenton reaction. Then ROS can alter the expression of relevant genes through the transcription factor NF-κB. which contributes to lesion invasion, inflammatory factor production, cell proliferation, and angiogenesis [30, 31].
Prolonged iron overload promotes sustained development of endometriosis, PCOS, and ectopic endometrial lesions, which may directly affect oocyte and embryonic development. The abdominopelvic cavity is the site for oocyte maturation and embryogenesis. The joint action of several factors may lead to microenvironment abnormalities, accelerating impairment of reproductive functions. Iron overload in the peritoneal fluid in infertile patients promotes the downregulation of GPX4 and hence induces lipid peroxidation, directly damaging embryo sac formation. According to another study, heme oxygenase-1 (HMOX1) was significantly upregulated in embryo ferroptosis. HMOX1 promotes ferroptosis, and its overexpression accelerates pro-oxidative reaction, resulting in lipid peroxidation-induced ferroptosis and intracellular iron accumulation [32]. Iron overload may cause embryonic mitochondrial damage, resulting in an elevated ROS level, a decreased ATP level, and a continuous drop in mitochondrial membrane potential [33]. It is evident that HMOX1 may play an important role in mediating embryonic ferroptosis, and its overexpression promotes oxidation and induces ferroptosis through increased iron accumulation and lipid peroxidation [32]. Excessive iron accumulation in cells leads to cytotoxic accumulation, which interferes with oocyte development. The total iron levels, ferritin and TfR1 expression levels in endometrioma-proximal follicles are higher than in endometrioma-distal follicles and healthy ovarian follicles in the ovaries of endometriosis patients. The oocyte retrieval rates in endometrioma-proximal and -distal follicles are lower than those in healthy ovarian follicles [34]. A research found that prolonged iron overload in the follicular fluid interfered with in vitro maturation of mouse oocytes [35]. According to existing studies, iron overload in the peritoneal fluid from infertile patients can cause direct damage to the blastula and oocytes. However, no consensus has been reached as to the pathogenesis of ferroptosis, especially the molecular mechanism of ferroptosis in specific conditions.
Ferroptosis and endometriosis show a bi-directional interaction. On one hand, ferroptosis inducers promote ferroptosis in ectopic endometrial cells and can be used as potential drugs against endometriosis; on the other hand, ferroptotic cells can release a large number of inflammatory cytokines, activating the downstream regulatory network and promoting angiogenesis and proliferation of surrounding tissues. Given the facts above, the downstream reaction triggered by ferroptosis deserves more attention when developing drugs that target ferroptosis in endometriosis.
Polycystic ovarian syndrome-associated infertility
Polycystic ovary syndrome (PCOS) is another common reproductive endocrine disorder related to infertility. Numerous studies have indicated that ferroptosis in the uterus and placenta is closely related to PCOS. One study showed that, iron accumulation increased noticeably in the uterus of pregnant rats treated with 5α-dihydrotestosterone and insulin, while the GPX4 level decreased significantly [36]. Several proteins related to ferroptosis, including acyl-CoA synthetase long-chain family member 4 (Acsl4), transferrin receptor (Tfrc), SLC7A11 (solute carrier family 7 member 1), and GCLC, were abnormally expressed. The above results demonstrate the role of ferroptosis in the occurrence and development of PCOS. Tang et al. [30] carried out an animal experiment to detect iron deposition and lipid peroxide indicators in ovarian tissues in PCOS. They were also concerned with indicators related to cell migration, and it was found that after the inhibition of O-GlcNAc transferase (OGT) in rats with PCOS, the contents of malondialdehyde (MDA) and ROS in ovarian tissues increased rapidly, while the glutathione (GSH) content decreased. These changes were also accompanied by a significant increase in iron deposition. Furthermore, they observed downregulated N-cadherin and vimentin in rats, but abnormally increased E-cadherin, an enzyme with anti-migration activity. According to another study, sustained release of inflammatory factors and rapid increase in the ROS level of ovarian tissues are usually found in PCOS patients. In most cases, PCOS is featured by increased apoptosis in ovarian tissues [37]. However, there is a contradiction in the above report. According to Tang, as the OGT level increased, the ROS level in the letrozole-induced rat model decreased. The findings above remain to be further verified.
Circular RNA (circRHBG) is also involved in the occurrence and development of PCOS by regulating ferroptosis. Zhang D et al. observed granular cells in patients with PCOS, and found that circRHBG was significantly upregulated, which further proved that circRHBG competes with SLC7A11 to bind to miR-515-5 to inhibit ferroptosis in granular cells [37]. They collected peripheral blood samples from PCOS patients. Compared with the normal population, there were significant differential expressions of proteins related to ferroptosis in CD4+T cells in the serum of PCOS patients [37].
Tubal factor infertility
So far, few reports have been published concerning the relationship between tubal factor infertility and ferroptosis. Ferroptosis is featured by iron-dependent ROS generation and accumulation of lipid peroxides and has close connections with placental dysfunction and trophoblastic damage [38, 39]. Tubal factor infertility is characterized by ectopic pregnancy due to fallopian tube obstruction, which in turn is closely related to iron overload and lipid peroxidation [40]. These are in agreement with the features of ferroptosis. ROS binds to phospholipids and proteins in cell membranes, leading to protein denaturation and aging pigment or lipofuscin. The blood becomes sticky and aggregates. Ferroptosis is mainly achieved via GSH, iron, and polyunsaturated fatty acid (PUFA) biosynthetic pathway. GPX4 uses two GSH molecules as electron donors to inhibit the conversion of lipid peroxides and prevent ferroptosis [41, 42]. Lai et al. used ferroptosis inducer RSL3 to inhibit GPx activity, promoting sustained ROS accumulation [43]. Their study confirmed that the serum containing Huayu Xiaozheng Granule (HYXZ) accelerated the death of RSL3-induced HTR-8/SVneo cells. Intervention with HYXZ resulted in a significant increase in intracellular lipids and ROS, and the LIP content increased as well. Fer-1 helped control lipid peroxide in HYXZ-induced cells, indicating that HYXZ promoted ferroptosis mainly through lipid hydroperoxide production and ROS generated as a by-product. Further, they found that key proteins involved in ferroptosis regulation had abnormal expression in the villous tissue (VT) of TP patients, and that lipid oxidation was activated markedly. In vitro experiments revealed that shikonin promoted RAS-selective lethal 3 (RSL3)-induced ferroptosis and inhibited the invasion and migration abilities of trophoblasts [44]. The above results suggest close connections between tubal pregnancy and ferroptosis, while inhibiting ferroptosis inhibits excessive invasion and migration of trophoblasts. However, given the limited number of studies regarding this topic, the findings above remain to be further confirmed by more studies.
Premature ovarian insufficiency
Premature ovarian insufficiency (POI) is a clinical syndrome in which a woman develops a decline in ovarian function before 40 years old [45]. The clinical manifestations of POI are elevated gonadotropins, decline in estrogen, menstrual disorders and infertility. The pathogenesis of POI is complex and multifactorial and may be caused by defects in primordial follicle formation, follicle recruitment/distribution, follicle growth and development [46, 47]. Recent studies have shown that accelerated follicular atresia is one of the major pathogenic factors [48, 49]. Follicular atresia is mainly caused by programmed death of granulosa cells, such as autophagy, necrosis and apoptosis. It has been shown that iron accumulation occurs in the early stage of follicular atresia, exhibiting abnormal GSH metabolism [50]. In a study of fluoride-induced apoptosis in follicular granulosa cells, results showed that mitochondria in granulosa cells exhibited typical features of ferroptosis, such as reduced mitochondrial volume and reduced mitochondrial crags [51]. This suggests that the reduction of oocytes and granulosa cells in the ovaries of POI mice may be related not only to apoptosis but also to ferroptosis. Furthermore, an ovarian single-cell RNA sequencing reveals an association between perinatal oocyte loss and iron mortality [52]. Zhang’s team identified a new disease-causing mutation, a truncated mutation in the Basonuclin 1 (BNC1) gene, in a large Chinese premature ovarian insufficiency family line [53]. BNC1 is a transcription factor involved in oocyte and follicle production. This research found that BNC1 deficiency in oocytes affects the NF2-YAP-TFRC/ASCL4 signaling axis, upregulates the expression of TFRC and ACSL4, induces oocyte ferroptosis, which ultimately leads to oocyte death and follicular atresia. Inhibition of YAP signaling or ferroptosis significantly rescued BNC1 mutation-induced POI. Chemotherapy exposure has become one of the leading causes of POI. It has been shown that CTX-induced ovarian toxicity is closely related to ferroptosis in granulosa cells [54]. HO-1 mediates oxidative stress, iron release and mitochondrial dysfunction. Knockdown of HO-1 significantly alleviated CTX-induced mitochondrial dysfunction and inhibited iron ferroptosis-induced GPX4 depletion. The treatment of human umbilical cord mesenchymal stem cell-derived exosomes (hUMSC-Exos) reduced the ROS production, free iron ions and lipid peroxidation levels of granulosa cells. The ferroptosis marker proteins Nrf2, xCT and GPX4 also decreased. Nrf2 maintains iron homeostasis by controlling HERC2 (E3 ubiquitin ligase for NCOA4 and FBXL5) and VAMP8 (mediates autophagosome-lysosome fusion) [55]. The Nrf2 inhibitor ML385 significantly attenuated the effects of hUMSC-Exos on granulosa cells [56]. These results suggest that the enhancement of ovarian function in POI mice by hUMSCs-Exos is thought to be related to the inhibition of ferroptosis [57].
Conclusions and outlook
Ferroptosis, a novel type of programmed cell death mediated by iron-dependent lipid peroxidation, has made rapid progress in studies related to disease mechanisms and regulatory pathways. In recent years, the relationship between ferroptosis and infertility and the underlying molecular mechanism have drawn widespread attention. Due to their own specificity, germ cells such as granulosa cells, oocytes and trophoblast cells are highly sensitive to ferroptosis during pathological processes [58]. In recent years, a growing body of research have shown that the molecular mechanisms and signaling pathways of ferroptosis may be closely related to female infertility diseases. It has been generally recognized that iron overload in the peritoneal fluid from infertile patients enhances ferroptosis resistance, and the growth of endometrial lesions can accelerate because of ferroptosis in some cells. The damage caused by iron overload to oocytes in infertile patients may be closely related to the absence of a membrane repair mechanism and unique characteristics of the antioxidant system in immature oocytes. Apart from the findings above, lipid peroxidation and continuous iron accumulation are only intermediate events of ferroptosis, rather than the cause of ferroptosis. Lipid peroxidation plays a role in regulating cell death. Some key factors related to ferroptosis may regulate other forms of cell death, such as GPX4 in the anti-oxidant system that inhibits necrocytosis and apoptosis. However, ferroptosis is regulated by a complex network of genetic and metabolic mechanisms. Current research related to the exact regulatory mechanisms linking cellular ferroptosis and infertility are limited and have inconsistent results. In conclusion, in the field of infertility treatment, cellular ferroptosis and its mechanism in infertility need to be studied systematically and in depth, with a view to providing a reference basis for the treatment of infertility-related diseases.
Methods
An extensive examination of the literature was performed to ensure a comprehensive review on ferroptosis and female infertility. The search was completed using the PubMed database (https://www.ncbi.nlm.nih.gov/pubmed) through May 2024. The search terms included ferroptosis and specific keywords (Endometriosis, Polycystic ovary syndrome, Tubal factor infertility, Premature ovarian insufficiency) based on the authors’ knowledge on the topic. Further, the reference lists of identified articles were manually reviewed.
Data availability
No datasets were generated or analysed during the current study.
References
Phillips K, Olanrewaju R A, Omole F. Infertility: evaluation and management [J]. Am Fam Physician. 2023;107(6):623–30.
Vander Borght M. Fertility and infertility: definition and epidemiology [J]. Clin Biochem. 2018;62:2–10.
Li G, Lin Y, Zhang Y, et al. Endometrial stromal cell ferroptosis promotes angiogenesis in endometriosis [J]. Cell Death Discov. 2022;8(1):29.
Stringer JM, Alesi L R, Winship A L, et al. Beyond apoptosis: evidence of other regulated cell death pathways in the ovary throughout development and life [J]. Hum Reprod Update. 2023;29(4):434–56.
Mowery N, T, Terzian W T H. Nelson A C. Acute lung injury [J]. Curr Probl Surg. 2020;57(5):100777.
Dolma S, Lessnick S L, Hahn W C, et al. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells [J]. Cancer Cell. 2003;3(3):285–96.
Liu M, Wu K, Wu Y. The emerging role of ferroptosis in female reproductive disorders [J]. Biomed Pharmacother. 2023;166:115415.
Andrews N C. Schmidt P J. Iron homeostasis [J]. Annu Rev Physiol. 2007;69:69–85.
Gao M, Monian P, Quadri N, et al. Glutaminolysis and transferrin regulate ferroptosis [J]. Mol Cell. 2015;59(2):298–308.
Kajarabille N, Latunde-Dada GO. Programmed cell-death by Ferroptosis: antioxidants as mitigators [J]. Int J Mol Sci, 2019, 20(19).
Santana-Codina N, Del Rey M Q, Kapner K S, et al. NCOA4-Mediated ferritinophagy is a pancreatic Cancer dependency via maintenance of Iron Bioavailability for Iron-Sulfur Cluster proteins [J]. Cancer Discov. 2022;12(9):2180–97.
Lee S, Hwang N, Seok B G, et al. Autophagy mediates an amplification loop during ferroptosis [J]. Cell Death Dis. 2023;14(7):464.
Dixon SJ, Lemberg K M, Lamprecht M R, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death [J]. Cell. 2012;149(5):1060–72.
Gao M, YI J, Zhu J et al. Role of Mitochondria in ferroptosis [J]. Mol Cell, 2019, 73(2): 354 – 63.e3.
Mao Kaganve, Qu G. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis [J]. Nat Chem Biol. 2017;13(1):81–90.
Matsushita M, Freigang S, Schneider C, et al. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection [J]. J Exp Med. 2015;212(4):555–68.
Bayir H, Anthonymuthu T S, Tyurina Y Y, et al. Achieving life through death: Redox Biology of Lipid Peroxidation in ferroptosis [J]. Cell Chem Biol. 2020;27(4):387–408.
Riegman M, Sagie L, Galed C, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture [J]. Nat Cell Biol. 2020;22(9):1042–8.
Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications [J]. Cell Res. 2021;31(2):107–25.
Li Y, He Y, Cheng W, et al. Double-edged roles of ferroptosis in endometriosis and endometriosis-related infertility [J]. Cell Death Discov. 2023;9(1):306.
Ni Z, Li Y, Song D, et al. Iron-overloaded follicular fluid increases the risk of endometriosis-related infertility by triggering granulosa cell ferroptosis and oocyte dysmaturity [J]. Cell Death Dis. 2022;13(7):579.
Shen H H, Zhang T, Yang H L, et al. Ovarian hormones-autophagy-immunity axis in menstruation and endometriosis [J]. Theranostics. 2021;11(7):3512–26.
Cacciottola L, Donnez J, Dolmans MM. Can endometriosis-related oxidative stress pave the way for New Treatment targets? [J]. Int J Mol Sci, 2021, 22(13).
Li Y, Zeng X. Erastin induces ferroptosis via ferroportin-mediated iron accumulation in endometriosis [J]. Hum Reprod. 2021;36(4):951–64.
Woo JH, Choi Y S, Choi JH. Iron-storage protein ferritin is upregulated in endometriosis and Iron overload contributes to a migratory phenotype [J]. Biomedicines, 2020, 8(11).
Sikora J, Smycz-Kubanska M, Mielczarek-Palacz A et al. Abnormal peritoneal regulation of chemokine activation-the role of IL-8 in pathogenesis of endometriosis [J]. Am J Reprod Immunol, 2017, 77(4).
Hsu C Y, Hsieh T H, Tsai C F, et al. miRNA-199a-5p regulates VEGFA in endometrial mesenchymal stem cells and contributes to the pathogenesis of endometriosis [J]. J Pathol. 2014;232(3):330–43.
Gong T, Liu L, Jiang W, et al. DAMP-sensing receptors in sterile inflammation and inflammatory diseases [J]. Nat Rev Immunol. 2020;20(2):95–112.
Jang E J, Kim D H, Lee B, et al. Activation of proinflammatory signaling by 4-hydroxynonenal-src adducts in aged kidneys [J]. Oncotarget. 2016;7(32):50864–74.
Defrère S, González-Ramos R, Lousse JC, et al. Insights into iron and nuclear factor-kappa B (NF-kappaB) involvement in chronic inflammatory processes in peritoneal endometriosis [J]. Histol Histopathol. 2011;26(8):1083–92.
González-Ramos R, Van Langendonckt A, Defrère S, et al. Involvement of the nuclear factor-κB pathway in the pathogenesis of endometriosis [J]. Fertil Steril. 2010;94(6):1985–94.
Fernández-Mendívil C, Luengo E, Trigo-Alonso P, et al. Protective role of microglial HO-1 blockade in aging: implication of iron metabolism [J]. Redox Biol. 2021;38:101789.
Chen X, Zhou Y, Wu D, et al. Iron overload compromises preimplantation mouse embryo development [J]. Reprod Toxicol. 2021;105:156–65.
Sanchez A M, Papaleo E, Corti L, et al. Iron availability is increased in individual human ovarian follicles in close proximity to an endometrioma compared with distal ones [J]. Hum Reprod. 2014;29(3):577–83.
Li A, Ni Z, Zhang J, et al. Transferrin Insufficiency and Iron overload in Follicular Fluid Contribute to Oocyte Dysmaturity in Infertile Women with Advanced endometriosis [J]. Front Endocrinol (Lausanne). 2020;11:391.
Teo C F, Wollaston-Hayden E E Wellsl. Hexosamine flux, the O-GlcNAc modification, and the development of insulin resistance in adipocytes [J]. Mol Cell Endocrinol. 2010;318(1–2):44–53.
Shah JS, Sabouni R, Cayton Vaught K C, et al. Biomechanics and mechanical signaling in the ovary: a systematic review [J]. J Assist Reprod Genet. 2018;35(7):1135–48.
Beharier O, Kajiwara K. Ferroptosis, trophoblast lipotoxic damage, and adverse pregnancy outcome [J]. Placenta. 2021;108:32–8.
Ng S W, Norwitz S G, Norwitz E R. The impact of Iron overload and ferroptosis on Reproductive disorders in humans: implications for preeclampsia [J]. Int J Mol Sci, 2019, 20(13).
Xie Y, Hou W, Song X, et al. Ferroptosis: process and function [J]. Cell Death Differ. 2016;23(3):369–79.
Ko P J Magtanongl, Dixon SJ. Emerging roles for lipids in non-apoptotic cell death [J]. Cell Death Differ. 2016;23(7):1099–109.
Deng L, He S, Guo N, et al. Molecular mechanisms of ferroptosis and relevance to inflammation [J]. Inflamm Res. 2023;72(2):281–99.
Lai YL, Zeng FL, Feng M, Chen ZY, Gao J, Deng GP. A study on the mechanism of Huayu Xiaozheng Granule in regulating the sensitivity of trophocytes to ferroptosis to treat tubal pregnancy [J]. China J Traditional Chin Med Pharm. 2022;37(08):4366–70.
Lai Y, Zeng F, Chen Z, et al. Shikonin could be used to treat Tubal pregnancy via enhancing ferroptosis sensitivity [J]. Drug Des Devel Ther. 2022;16:2083–99.
Nash Z Daviesm. Premature ovarian insufficiency [J]. BMJ. 2024;384:e077469.
Rossetti R, Ferrari I. Genetics of primary ovarian insufficiency [J]. Clin Genet. 2017;91(2):183–98.
Goswami D, Conway G S. Premature ovarian failure [J]. Hum Reprod Update. 2005;11(4):391–410.
Sullivan SD, Castrillon D H. Insights into primary ovarian insufficiency through genetically engineered mouse models [J]. Semin Reprod Med. 2011;29(4):283–98.
Jin M, Yu Y, Huang H. An update on primary ovarian insufficiency [J]. Sci China Life Sci. 2012;55(8):677–86.
Zhang J, Liu Y, Yao W, et al. Initiation of follicular atresia: gene networks during early atresia in pig ovaries [J]. Reproduction. 2018;156(1):23–33.
Zhao W P, Wang H W, Liu J, et al. Mitochondrial respiratory chain complex abnormal expressions and fusion disorder are involved in fluoride-induced mitochondrial dysfunction in ovarian granulosa cells [J]. Chemosphere. 2019;215:619–25.
Wang JJ, Ge W, Zhai Q Y, et al. Single-cell transcriptome landscape of ovarian cells during primordial follicle assembly in mice [J]. PLoS Biol. 2020;18(12):e3001025.
Wang F, Liu Y, Ni F, et al. BNC1 deficiency-triggered ferroptosis through the NF2-YAP pathway induces primary ovarian insufficiency [J]. Nat Commun. 2022;13(1):5871.
Chen H, Nie P, Li J, et al. Cyclophosphamide induces ovarian granulosa cell ferroptosis via a mechanism associated with HO-1 and ROS-mediated mitochondrial dysfunction [J]. J Ovarian Res. 2024;17(1):107.
Anandhan A, Dodson M. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8 [J]. Sci Adv. 2023;9(5):eade9585.
Zhou Y, Huang J. Human mesenchymal stem cells derived exosomes improve ovarian function in chemotherapy-induced premature ovarian insufficiency mice by inhibiting ferroptosis through Nrf2/GPX4 pathway [J]. J Ovarian Res. 2024;17(1):80.
Geng Z, Nie X et al. Ling L,. Electroacupuncture May Inhibit Oxidative Stress of Premature Ovarian Failure Mice by Regulating Intestinal Microbiota [J]. Oxid Med Cell Longev, 2022, 2022: 4362317.
Zhang J, Su T, Fan Y, et al. Spotlight on iron overload and ferroptosis: Research progress in female infertility [J]. Life Sci. 2024;340:122370.
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
Fan Peiyin, Wang Yuxian and Zhang Jiali wrote the main manuscript text. Xu Jian reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
peiyin, F., yuxian, W., jiali, Z. et al. Research progress of ferroptosis in female infertility. J Ovarian Res 17, 183 (2024). https://doi.org/10.1186/s13048-024-01508-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-024-01508-y