
Abstract
Background
High-grade serous ovarian cancers (HGSCs) display a high degree of complex genetic alterations. In this study, we identified germline and somatic genetic alterations in HGSC and their association with relapse-free and overall survival. Using a targeted capture of 557 genes involved in DNA damage response and PI3K/AKT/mTOR pathways, we conducted next-generation sequencing of DNA from matched blood and tumor tissue from 71 HGSC participants. In addition, we performed the OncoScan assay on tumor DNA from 61 participants to examine somatic copy number alterations (SCNA).
Results
Approximately one-third of tumors had loss-of-function (LOF) germline (18/71, 25.4%) or somatic (7/71, 9.9%) variants in the DNA homologous recombination repair pathway genes BRCA1, BRCA2, CHEK2, MRE11A, BLM, and PALB2. LOF germline variants also were identified in other Fanconi anemia genes and in MAPK and PI3K/AKT/mTOR pathway genes. Most tumors harbored somatic TP53 variants (65/71, 91.5%). Using the OncoScan assay on tumor DNA from 61 participants, we identified focal homozygous deletions in BRCA1, BRCA2, MAP2K4, PTEN, RB1, SLX4, STK11, CREBBP, and NF1. In total, 38% (27/71) of HGSC patients harbored pathogenic variants in DNA homologous recombination repair genes. For patients with multiple tissues from the primary debulking or from multiple surgeries, the somatic mutations were maintained with few newly acquired point mutations suggesting that tumor evolution was not through somatic mutations. There was a significant association of LOF variants in homologous recombination repair pathway genes and high-amplitude somatic copy number alterations. Using GISTIC analysis, we identified NOTCH3, ZNF536, and PIK3R2 in these regions that were significantly associated with an increase in cancer recurrence and a reduction in overall survival.
Conclusions
From 71 patients with HGCS, we performed targeted germline and tumor sequencing and provided a comprehensive analysis of these 557 genes. We identified germline and somatic genetic alterations including somatic copy number alterations and analyzed their associations with relapse-free and overall survival. This single-site long-term follow-up study provides additional information on genetic alterations related to occurrence and outcome of HGSC. Our findings suggest that targeted treatments based on both variant and SCNA profile potentially could improve relapse-free and overall survival.
Similar content being viewed by others

Background
High-grade serous ovarian cancer (HGSC) is the most common ovarian cancer histologic subtype with the majority of patients diagnosed at a late stage [1]. The standard treatment for HGSC involves both surgical cytoreduction and systemic paclitaxel and platinum chemotherapy with increasing use of targeted treatments of bevacizumab and/or poly ADP-ribose polymerase inhibitors (PARP-i) [2,3,4]. Despite aggressive frontline therapy, many patients with ovarian cancer eventually relapse, resulting in only 20–30% survival after five years [5]. These dismal statistics underscore the need for earlier detection and new treatments following diagnosis.
HGSC originates in the fallopian tube [6, 7] and is characterized by early acquisition of TP53 variants [8, 9]. Other characteristics are extensive somatic copy number alterations (SCNA) [9, 10] with few somatic point variants [11] and inactivation of DNA homologous recombination repair (HRR) primarily through germline and somatic variants of BRCA1 and BRCA2 [9]. Although less frequent, variants in additional Fanconi anemia pathway genes that play key roles in HRR (e.g., BRIP1, PALB2, RAD51C/D), and CHEK2, a DNA damage response gene, have been associated with HGSC [12,13,14,15]. Amplification of regions containing the oncogenes CCNE1, MECOM, and MYC and deletions of regions containing the tumor suppressor genes RB1, NF1, and PTEN are commonly observed [9, 16, 17]. An integrated analysis combining variant data, copy number changes, and changes in gene expression determined that the main pathways altered in HGSC are the HRR, RB1, PI3K/AKT/mTOR, and NOTCH signaling pathways [9].
In this study, we identified and characterized germline and somatic genetic alterations in HGSC and their associations with relapse-free and overall survival. From paired germline and tumor DNA samples from 71 HGSC patients treated at City of Hope (COH), we conducted targeted sequencing of 557 genes in pathways involved in response to DNA damage, DNA repair, cell-cycle regulation, programmed cell death, MAPK, and PI3K/AKT/mTOR signaling including known drivers in HGSC tumorigenesis [18]. In addition, we used the OncoScan assay to determine genome-wide SCNAs and loss of heterozygosity (LOH) in 61 primary ovarian tumors.
Results
Clinical/demographic data
Participants were 71 women with Stage III or IV HGSC with matching germline and tumor samples (Table 1). The median age at diagnosis of cancer was 58 years and median survival was 37.8 months (Table 1). A total of 114 tumors were examined, comprising a mixture of primary debulking, metastatic, and recurrent tumors, often from multiple sites (details in Supplemental Table 1).
Germline and somatic variant identification by pathway
HRR genes
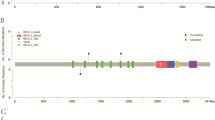
Consistent with previous studies of HGSC [9], we observed germline loss-of-function (LOF) variants in 17 patients (23.9%) in known ovarian cancer HRR predisposition genes including nine (12.7%) in BRCA1, six (8.5%) in BRCA2, two (2.8%) in PALB2, and one in MRE11A (1.4%) (Fig. 1). Patient #56 had germline frame-shift deletions in both BRCA2 and PALB2 with LOH of BRCA2 in the somatic tissue. For patients with germline LOF variants, we checked for LOH in the corresponding tumor. Similar to the HGSC data in The Cancer Genome Atlas (TCGA) [19], we observed a high frequency of LOH with 6/9 germline truncation variants in BRCA1, 5/6 in BRCA2, 1/2 in PALB2, and 1/1 in MRE11A (Supplemental Table 3). We identified somatic LOF BRCA1 variants in five (7.0%) tumors, BRCA2 in three (4.2%) tumors, and BLM in one tumor (1.4%) (Fig. 1). From our OncoScan analysis, we observed two somatic homozygous deletions in BRCA2, one in BRCA1, and two in SLX4 (pink half bar in Fig. 1). Of note, patient #41 carried the H/L germline founder BRCA1 exon 9–12 deletion and had lost the second copy of BRCA1 as seen from Oncoscan analysis. Patient #14 had a germline LOF BRCA2 frame-shift deletion and the OncoScan analysis detected that the tumor had homozygous deletions of BRCA2 and SLX4. In total, 38% (27/71) of HGSC tumors harbored LOF variants in HRR genes. Additionally, a BRIP1 germline missense variant (P47A) in patient #45 is potentially pathogenic (Supplemental Table 3).

Oncoplot of LOF germline mutations and non-silent somatic mutations. Only genes with variants in at least 3% of patients are displayed. OS = overall survival. RFS = relapse-free survival
Other Fanconi anemia and DNA damage response genes
In Fanconi anemia pathway genes nominally associated with HGSC, we identified germline LOF variants in FAN1, FANCD2, FANCI, and FANCL (Supplemental Fig. 1). For known ovarian cancer genes involved in cell cycle regulation, as expected, we found a high frequency of somatic TP53 variants (65/71, 91.5%) (Fig. 1, Supplemental Table 4) [8, 9]. We identified a germline CHEK2 frameshift deletion LOF variant in patient #67 with LOH in the tumor and the LOF I157T missense variant in patient #15. From the OncoScan analysis, we identified three somatic homozygous deletions in RB1 (Fig. 1). In DNA damage repair genes nominally or not known to be associated with HGSC, we identified germline LOF variants in cell cycle (MCPH1 and PKMYT1), double-strand break (EME2, POLQ, RECQL5, and SPO11), nucleotide excision repair (ERCC2 and POLK), base excision repair (NEIL3 and POLG), mismatch repair (EXO1), and post-replication repair (RAD18) pathways. Somatic LOF variants involved in cell cycle (RBL2 and TFDP1), mismatch repair (PMS1 and RFC4), nucleotide excision repair (POLK), and base excision repair (POLL) also were identified (Supplemental Fig. 1).
MAPK and PI3K/AKT/mTOR pathway genes
From the combined OncoScan and sequence analyses, we observed somatic variants in MAPK-pathway genes with one in-frame deletion and two homozygous deletions in MAP2K4 and one homozygous deletion in NF1 (Fig. 1 and Supplemental Fig. 1). We observed germline LOF variants in the PI3K/AKT/mTOR genes FKBP11, FKBP7, and IFNA5 along with a somatic LOF variant in PIK3R1 (Supplemental Fig. 1). We also identified three somatic homozygous deletions and one somatic missense variant in PTEN, two somatic homozygous deletions and one somatic frameshift deletion in CREBBP and a somatic hotspot missense variant in PIK3CA (Fig. 1 and Supplemental Fig. 1).
Analysis of multiple tumor samples from the same patient
For the 12 patients with multiple samples from the primary debulking including metastatic sites, the somatic variants were the same except in three patients where one of the multiple tumors from the primary debulking acquired a somatic mutation (Supplemental Table 4). Similarly, for the 16 patients with samples from more than one debulking surgery, no newly acquired somatic variants were observed with a few exceptions (Supplemental Table 4), none of which have been associated with disease progression or response to therapy.
SCNA results
A genome-wide view of the SCNAs in the 61 tumors is shown in Fig. 2. Using Nexus Copy V10, the most frequent broad-arm levels of amplification were chromosomal regions 1q, 3q, 5p, 6p, 7q, 8q, and 20q and the most frequent regions of loss were chromosomal regions 4q, 6q, 8p, 17p, 18q, and 22q. There were 17 regions of amplification on 13 chromosomes and 22 regions of deletion on 16 chromosomes with GISTIC q-value less than 0.05 (Fig. 3). The chromosomal locations, peak limits, and q-values for the 39 regions are listed in Supplemental Table 5. The five most significant regions of amplification were at 19q12 (q = 1.06E-11), 14q32.33 (q = 3.07E-11), 2p11.2 (q = 3.43E-11), 3q26.2 (q = 1.48E-10), and 8q24.21 (q = 2.80E-8). Within these regions are oncogenes CCNE1 in 19q12, MECOM in 3q26.2, MYC in 8q24.21, and MYCL1 in 1p34.2 (q = 7.75E-6). The most significant (q < 0.01) SCNA losses were at 6q26 (q = 1.45E-5; containing MAP3K4 and ARID1B), 7p22.1 (q = 1.12E-3; containing PMS2), 5q21.1 (q = 1.31E-3; containing APC) 10q23.31 (q = 1.61E-3; containing PTEN), 13q14.2 (q = 3.29E-3; containing RB1), and 5q13.1 (q = 4.27E-3; containing PIK3R1).

Overall frequency of SCNAs in 61 primary HGSC tumors. From Nexus Copy Number v. 10, red indicates regions of loss, blue indicates regions of gain, and dark blue indicates regions of high-copy gain

Significantly amplified (panel A) or deleted (panel B) regions of genome. GISTIC analysis was performed to identify significant recurrent focal SCNAs in 61 tumor samples. Genomic positions of SCNA regions are oriented vertically. GISTIC q value and G scores are shown at the bottom and top, respectively. The green line marks GISTIC q value of 0.05. Potential candidate or common cancer genes for each SCNA region are included in the parenthesis next to each SCNA region
Association of LOF in core HRR genes and incidence of high-amplitude SCNAs
In the 61 samples with both sequence and SCNA data from OncoScan, there were LOF variants in the core HRR genes BRCA1, BRCA2, PALB2, in 19 patients (Supplemental Tables 3 and 4). High amplitude SCNAs, in 169 chromosomal regions containing 2369 genes, found in more than 5% samples were included in the test of association between LOF in BRCA1, BRCA2, and PALB2 and incidence of SCNAs in each gene. We found statistically significant associations [P < 0.05 and false discovery rate (FDR) < 0.25] of 262 genes in 10 chromosomal regions (6p25.2, 6p25.3, 8p23.1, 8q23.3, 8q24.13, 8q24.21, 8q24.22, 8q24.23, 8q24.3, 15q26.3) (Supplemental Table 6). All 262 genes had odds ratios (OR) greater than 1.0 (95% confidence interval not including 1.0), indicating that LOF variants in these three genes were associated with increased incidence of high-amplitude SCNAs. The 262 genes were enriched (adjusted P < 0.05) in pathways involved in immune responses and biological functions related to transcriptional regulation (Supplemental Table 7).
From analysis of data from 356 HGSC tumor samples in TCGA, 24 had LOF variants in BRCA1 (16 germline, 8 somatic), 25 had LOF variants in BRCA2 (19 germline, 6 somatic), and 2 had somatic LOF variants in PALB2 for a total of 51 patients (14%). Of the 262 SCNA genes significantly associated with variants in one of the three genes, 168 genes had been measured by the Affymetrix SNP 6.0 array in TCGA and had high-amplitude SNCAs in at least 5% of the 356 TCGA samples. For 136 of the 168 genes, the association was validated (P < 0.05 and FDR < 0.25, and consistent direction of OR > 1.0) (Supplemental Table 6); the remaining 36 genes showed a consistent OR (point estimation of OR greater than 1.0) but were not significant at P < 0.05 and FDR < 0.25.
Association of LOF variants in BRCA1, BRCA2, and PALB2 and relapse-free and overall survival
We tested the association of LOF variants in the high-penetrant genes, BRCA1, BRCA2, and PALB2, on overall and relapse-free survival after definitive surgery and adjuvant therapy. For the 61 COH patients with both sequencing and SCNA data, we found 19 LOF variants including 12 germline variants (6 in BRCA1, 5 in BRCA2, 1 in PALB2) and 7 somatic variants (5 in BRCA1, 2 in BRCA2). From TCGA ovarian cancer data, a total of 51 LOF germline or somatic variants (24 in BRCA1, 25 in BRCA2, and 2 in PALB2) were observed in 356 patients. Compared to patients not carrying LOF variants, 19 of 61 COH (panel A) and 51 of 356 TCGA patients (panel C) carrying LOF variants showed significantly (p < 0.05) better relapse-free survival (Supplemental Fig. 2). For overall survival, highly significant overall survival advantage was observed in TCGA patients with LOF variants (panel D), but not in COH patients (panel B), likely due to small sample size (Supplemental Fig. 2).
Association of SCNAs with relapse-free and overall survival
For the 61 primary HGSC tumors, we applied an unsupervised hierarchical clustering algorithm to the 37 of 39 recurrent SCNAs with residual (after removing segments shared with higher peaks) q values less than 0.05 and identified four subgroups of patients in the dendrogram (Fig. 4). In a Kaplan–Meier analysis, the four subgroups had highly significant differences in both relapse-free surivival (log-rank test P = 0.0003) (Fig. 5A) and overall survival (log-rank test p < 0.0001) (Fig. 5B).

Unsupervised hierarchical clustering of 61 primary HGSC samples using Oncoscan data from 37 SCNAs. Changes in copy number for 37 SCNAs with GISTIC residual q value < 0.05 were used to perform unsupervised hierarchical clustering of 61 primary HGSCs samples. Each column represents a tumor sample and rows represent the 37 SCNAs. As shown in the color key, genomic regions of gain are represented in red and regions of loss are represented in blue

Relapse-free (A) and overall survival analysis (B) of four clusters of patients defined by SCNAs. Kaplan–Meier analysis was used to evaluate the difference in survival for COH patients in the four clusters of the unsupervised hierarchical tree in Fig. 4
For high-amplitude SCNAs defined as regions of amplification with ≥ 4 copies or homozygous deletions and where a minimum of 5 of 61 tumors carried a high-amplitude SCNA in the region, we tested the association of SCNA and relapse-free survival and overall survival in 13 of the 17 regions of SCNAs gain and 7 of 22 regions of SCNA loss. High-amplitude SCNA gains in 4 of the 13 regions (3q26.2, 15q26.3, 19p13.12, and 19q12) showed evidence (p < 0.05) of association with a reduction in relapse-free survival (Supplemental Table 8); associations of two (19q12 and 19p13.12) of four regions were validated (p < 0.05) in TCGA data (Table 2). In the 19q12 region, only ZNF536 had a significant association (p = 0.014). In the 19p13.12 region, NOTCH3, ILVBL, SYDE1, and PIK3R2 had p < 0.05. NOTCH3, ILVBL, and SYDE1 shared the same SCNAs distribution across 8 of 61 samples (p value = 0.047); therefore, we selected NOTCH3 as the representative in the following combined analysis of SCNAs in the two validated regions; NOTCH3 has been reported to be a potential oncogene [7]. Compared to patients not carrying high-amplitude gains in ZNF536, PIK3R2, and NOTCH3, patients from both COH (Fig. 6A and B) and TCGA (Fig. 6C and D) carrying high copy gains in at least one of the three genes showed significantly worse relapse-free survival (Fig. 6A and C, p < 0.01) and worse overall survival (Fig. 6B and D, p < 0.05). Homozygous deletions in the seven regions of SCNA loss were not associated with relapse-free or overall survival (Supplemental Table 9).

Relapse-free and overall survival related to high-amplitude SCNAs in NOTCH3, ZNF536, and PIK3R2. Compared to patients not carrying high copy number gains in NOTCH3, ZNF536, and PIK3R2, patients from both COH (panels A and B) and TCGA (panels C and D) carrying high copy gains in at least one of the three genes showed significantly worse relapse-free survival (panel A and C) and overall survival (panel B and D), respectively
We also investigated association of relapse-free and overall survival with the total percentage of genome changes in SCNA. Higher SCNA load was associated with better survival, but the evidence of association was only marginally significant or insignificant in patients from the COH study and from TCGA study, respectively (Supplemental Fig. 3).
Discussion
All germline and somatic genetic variants identified in the 557 sequenced genes are provided in Supplemental Table 10. Consistent with previous studies of HGSC, we observed germline LOF variants in high- and moderate-penetrance HRR pathway genes [9, 12, 15, 19, 20]. In addition to the germline LOF variants in the HRR genes, BRCA1, BRCA2, MRE11A, and PALB2, LOF somatic variants in BRCA1 and BRCA2 were identified including homozygous deletions in BRCA1 and BRCA2. From the combined sequencing and SCNA analysis, we found that approximately one third of the HGSC tumors harbored LOF variants in HRR genes and treatment with PARP inhibitors would be warranted [3].
We also observed LOF variants in Fanconi anemia pathway genes nominally associated with HGSC [9, 19]. These variants included germline LOF variants in FAN1, FANCD2, FANCI, and FANCL. Platinum-based therapies cause interstrand cross links (ICLs) and the Fanconi anemia pathway is critical for resolving ICLs for DNA repair through the HRR pathway [21]. Therefore, variants in Fanconi anemia genes may affect the efficacy of treatment and resultant survival. Interestingly, we observed a somatic nonsense variant in BLM/RECQL3 which codes for a RecQ family DNA helicase, whose deficiency results in Bloom’s syndrome which is characterized by a predisposition to early development of multiple forms of cancer [22]. BLM has been shown to play a critical role in mitosis and multiple steps of HRR-dependent DNA repair [23]. While several germline LOF variants in BLM have been observed in HGSC tumors [19], only one somatic LOF variant was listed in the COSMIC (Catalogue of Somatic Mutations in Cancer) database [24].
Additional DNA repair pathway variants included germline LOF variants in double-strand break repair (EME2, POLQ, RECQL5, and SPO11), nucleotide excision repair (ERCC2 and POLK), base excision repair (NEIL3 and POLG), mismatch repair (EXO1), and post-replication repair (RAD18) pathways. Although uncommon, all these genes have documented germline protein truncating variants from other HGSC studies [19, 25, 26]. Of note, the identical germline splice site variant in EXO1 and the frameshift deletions in ERCC2, POLK, and POLQ were reported in previous HGSC studies. We also observed somatic LOF variants in other DNA repair pathway genes with one each in PMS1 (mismatch repair), POLK (nucleotide excision repair), POLL (base excision repair), and RFC4 (mismatch repair). A protein-truncating variant in RFC4 was listed in COSMIC HGSC tumors whereas only missense variants were listed in PMS1, POLL, and POLK [24].
Alteration of the PI3K/AKT/mTOR pathway is a common occurrence in HGSC [9] and our results support this finding. We observed germline LOF variants in PI3K/AKT/mTOR pathway genes FKBP11, FKBP7, and IFNA5; in previous HGCS genomic studies, germline LOF variants were reported in IFNA5 and FKBP7, but not FKBP11 [19, 25]. Somatic variants in PI3K/AKT/mTOR pathways genes have been reported in HGSCs [9, 19, 27] and we identified LOF variants in CREBBP and PIK3R1. Our OncoScan analysis identified three somatic homozygous deletions in PTEN and two in CREBBP. In addition, we observed one somatic missense variant in PIK3CA, one in CREBBP, and one in PTEN, classified as pathogenic, reinforcing the importance of alteration in the PI3K/AKT/mTOR pathway in HGSC.
We observed variants in several genes involved in cell cycle regulation. As expected, we found a high frequency of somatic TP53 variants [8, 9]. We also observed a germline LOF variant and a potentially pathogenic germline missense variant in the known ovarian cancer gene CHEK2 [15, 19]. Our OncoScan analysis identified three somatic homozygous deletions in RB1 consistent with a previous report that it was a recurrently mutated gene in HGSC [9].
We observed somatic homozygous deletions in the MAPK pathway genes MAP2K4 and NF1. The MAPK signaling pathway regulates many cellular processes, including gene expression, cell cycle, cell survival, cell death, and cell movement [28]. MAP2K4 is listed in COSMIC as potentially having a role in both tumor suppression and as an oncogene [24]. Supporting its role as a tumor suppressor, homozygous focal deletions in MAP2K4 have been identified in ovarian and breast cancers and it is suggested that loss of MAP2K4 could alter function of the JNK (c-Jun N-terminal kinase) pathway [29, 30]. In addition, somatic copy number loss of NF1 is common in HGSC [31, 32]. The NF1 protein has been found to play a role in regulating several intracellular processes and importantly is essential for reducing oncogenic Ras activity, supporting the hypothesis that it acts as a tumor suppressor [33].
For patients where we had multiple tissues from the primary debulking or tissue from multiple surgeries, in the secondary tumor sites, the somatic point mutations were maintained and there were few newly acquired somatic point mutations suggesting that tumor evolution was not through acquisition of somatic mutations in genes in the pathways we studied. This is consistent with prior reports that gene amplifications rather than somatic point mutations are observed in disease progression [34, 35]. We were not able to perform OncoScan analysis for multiple tissues from the same patient.
Along with frequent TP53 variants, a hallmark of HGSC is profound genomic instability [9]. We identified several amplified regions containing driver oncogenes previously observed in HGSC including CCNE1, MECOM, MYC, and MYCL1 and deletion regions containing tumor suppressor genes RB1 and PTEN [9, 16, 17, 34, 36,37,38]. GISTIC identified several regions that, although CNAs, are unlikely to be somatic as the genomic regions they encompass include germline copy number variation (Supplemental Table 5). We only analyzed tumor tissue and so could not distinguish germline from somatic CNAs. These regions include the amplifications regions 14q32.33, 2p11.2, and 2p11.2 and the deletion regions 8p11.22, 22q11.23, and 4q34.3. Regardless of CNA source, they may affect outcome similar to LOF variants regardless of whether germline or somatic.
Since cancer-driver SCNAs tend to be shorter in length and higher in amplitude than passenger SCNAs [39], we sought to identify genes within high-amplitude SCNAs that were associated with relapse-free and overall survival. We determined that amplifications in NOTCH3, ZNF536, and PIK3R2 were significantly (P < 0.05) associated with shorter relapse-free survival in both our data and TCGA data after adjusting for copy number mutation rate, age at diagnosis, and tumor stage. Debulking status (optimal versus suboptimal) and intra-peritoneal chemotherapy status (yes versus no) were not available in TCGA data. When we included these two variables in our data, the association remained significant for NOTCH3 (P = 0.033) and ZNF536 (P = 0.012), but not for PIK3R2 (P = 0.127). Our finding of the association of NOTCH3 high copy number gain and tumor recurrence fits with many lines of evidence supporting its role as a potent oncogene [9]. The amplification of the region encompassing NOTCH3 (19p13.12) and upregulation of NOTCH3 expression has been detected in a high percentage of HGSCs [40]. The upregulation of the NOTCH3 pathway is associated with tumor progression, drug resistance, and HGSC tumor recurrence [9, 41, 42] suggesting that NOTCH3 inhibitors could be an effective treatment in order to increase sensitivity to platinum-based therapies. There also is growing evidence that PIK3R2, encoding the p85β regulatory subunit of PI3K, is an oncogene. PIK3R2 induces oncogenic signaling in HGSC [43] and high expression is correlated with a significant reduction in overall survival in ovarian cancer patients [44].
The significance of the high copy gain in ZNF536 is more difficult to explain as it has not been associated with cancer. However, it may be due to its location near (within 500 kbp) of CCNE1 at 19q12. The most significantly amplified region from our GISTIC analysis was 19q12. Focal amplification of CCNE1 is a hallmark of HGSC and is associated with chemoresistance [9, 16, 17, 32]. Interestingly, our Firth’s Cox regression analysis of high-amplitude genes significantly associated with relapse-free survival showed that ZNF536 was highly significant (p = 0.014, Table 2) while CCNE1 was not significant (p = 0.22, Table S6). High-level amplification was more frequent in CCNE1 (11 of 61 patients) than in ZNF536 (6 of 61 patients), indicating that CCNE1 amplification may be an earlier amplification event more related to occurrence of HGSC, rather than outcome after therapy.
Knijnenburg and colleagues calculated homologous recombination deficiency (HRD) scores for 33 cancer types from TCGA and found that HGSC had the highest HRD score and that a higher score within HGSCs was associated with better survival [45] likely due to better response to platinum-based chemotherapies. Although we could not calculate HRD scores, one third of our HGSC patients carried germline or somatic LOF variants in core HRR genes. Compared to patients not carrying these LOF variants, patients with these variants showed significantly better relapse-free survival (p < 0.05; Supplemental Fig. 2). In both our tumor samples and TCGA tumor samples, LOF variants in HRR genes were associated with increased incidence of high-amplitude SCNAs at multiple chromosome regions (Supplemental Table 6). This observation suggests that LOF variants in HRR genes caused HRD resulting in greater genomic instability as manifest by increased SCNAs.
HGSC tumors are known for the ability to acquire resistance to the killing effects of various chemotherapeutic agents such as platinum and PARP inhibitors. Platinum resistance can develop from multiple mechanisms including reduced intracellular drug accumulation, intracellular inactivation of the agent, increased DNA repair, or impaired apoptotic signaling pathways [46]. The near ubiquitous somatic mutation of TP53 is likely the key mechanism in which HGSC tumors evade the triggering of apoptosis.
A strength of this study is the single institution experience with detailed treatment and long-term follow-up data on recurrence and survival. Other strengths are the combined data on germline and tumor DNA sequencing, the OncoScan assay for copy number and large rearrangements, and the availability of multiple tissue sites and/or recurrence debulking tissue for a subset of patients. The primary weakness of the study is the small sample size and that not all samples had the SCNA analysis. Because of limited power, we only ran analyses of LOF variants in BRCA1, BRCA2, and PALB2 with relapse-free and overall survival, as well as genes in amplified regions from tumor analysis. A second weakness is that because we used a targeted gene approach, we did not have the data to generate an HRD score. Lastly, the impact of the genomic alterations on the overall prognosis (relapse-free survival and overall survival) did not account for the use of bevacizumab or PARP inhibitors.
Conclusion
In summary, from 71 patients with HGCS, we performed targeted germline and tumor sequencing of genes involved in DNA damage response and PI3K/AKT/mTOR pathways and provided a comprehensive analysis of these 557 genes. We identified germline and somatic genetic alterations including SCNAs and analyzed their associations with relapse-free and overall survival. We validated our results using TCGA data. Amplifications in NOTCH3 and PIK3R2 were significantly associated with shorter relapse-free survival likely because of over-expression where targeted inhibition may be useful. Interestingly, we identified few changes in mutation profiles from primary debulking versus second debulking samples or from the primary site to metastatic sites from HGSC patients with multiple samples. However, we did not investigate SCNAs in multiple tissues from the same individuals. In conclusion, our single-site long-term follow-up study of HGSC patients was consistent with other studies of HGSC. Our findings suggest that targeted treatments based on both variant and SCNA profile potentially could improve relapse-free and overall survival.
Methods
Study subjects
Participants were 71 women with Stage III or IV HGSC who underwent their first debulking surgery between 2002 and 2014 and provided informed consent approved by the City of Hope Institutional Review Board (IRB# 12,358). Subjects were not selected for age or family history. Clinical information was abstracted from medical records and follow-up was through 06/2021. Of the 71 patients, tumor tissue was sequenced from the first debulking surgery for 69 patients; 63 tumors were from the primary site and 6 were from metastatic sites (four tumors in the omentum and two in the pelvis) as no sample was available from the primary site. For two patients, tumor tissue only was available from the second debulking surgery. For 15 patients, we had tumor tissue from both the primary and second debulking surgeries and for 3 patients, we had tumor tissue from 3 surgeries. Twelve patients had multiple tumor sites from the same primary surgery. In total, there were 114 tumor sites sequenced for the 71 patients (details in Supplemental Table 1).
The majority of tumor samples were from banked formalin-fixed-paraffin-embedded (FFPE) sections (n = 67). For 47 tumor samples from 22 patients, we obtained fresh samples at the time of surgical debulking and stored tissues in RNAlater solution (ThermoFisher Scientific, Waltham, MA) at -80 °C. Germline DNA was extracted from peripheral blood mononuclear cells using a phenol/chloroform protocol and tumor DNA was extracted from fresh frozen samples or from FFPE sections using the QIAamp DNA Tissue Kit (Qiagen, Hilden, Germany) (Supplemental Table 1).
High-throughput sequencing and variant calling
We used KAPA Hyper Prep Kits (Kapa Biosystems, Wilmington, MA) to create barcoded paired-end libraries with 300-bp inserts and hybridized the bar-coded samples to a custom NimbleGen SeqCap (Roche, Basel, Switzerland) or SureSelectXT (Agilent, Santa Clara, CA) targeted-gene capture kit. We used custom bait designs which included 557 candidate cancer susceptibility genes involved in the response to DNA damage, DNA repair, cell cycle regulation, programmed cell death, and PI3K/AKT/mTOR pathways (Supplemental Table 2). Following capture, samples were sequenced with 2 × 100 bp paired end reads on a GAIIx or HiSeq 2500 (Illumina, San Diego, CA) in the COH Integrative Genomics Core (IGC) with germline samples sequenced to an average coverage of 84.1-fold and tumor samples sequenced to an average coverage of 84.5-fold. Paired-end reads from each sample were aligned to human reference genome (GRCh37/hg19) using the Burrows-Wheeler Alignment Tool (BWA, v0.7.5a-r405) under default settings [47], and the aligned binary alignment map (BAM) sequence files were sorted and indexed using SAMtools (v0.1.19) [48]. The sorted and indexed BAM files were processed by Picard MarkDuplicates (v1.105, http://picard.sourceforge.net/) to remove duplicate sequencing reads. A pileup file was then created via mpileup in SAMtools [48] and germline and somatic variants were called using Varscan2 (v.2.2.8) [49]. These variants were subsequently annotated using ANNOVAR [50].
Germline variant filtering
We evaluated variants using Ingenuity Variant Analysis (IVA) Qiagen Clinical Insight version 1.2 (Qiagen Inc, Alameda, CA). IVA used the following content versions: Ingenuity Knowledge Base (X-release), gnomAD (v2.1.1) [51], PhyloP [52], Sorting Intolerant from Tolerant (SIFT) [53], the Human Gene Mutation Database (HGMD, 2019.3), Catalogue of Somatic Mutations in Cancer (COSMIC, v89) [24], and Clinvar (2019–11-06) [54]. Variants with a frequency greater than 2% in the gnomAD database were removed. All remaining variants were visually checked using the Integrative Genomics Viewer [55] to confirm that there were no sequencing errors.
We applied American College of Medical Genetics and Genomics (ACMGG) guidelines to the variants using the ACMGG calling algorithm in IVA [56]. IVA categorizes variants based on standard ACMGG variant calling recommendations (i.e., PVS1, BS2, etc.) by searching available databases and literature for known information for each variant in addition to running in silico models as described above. All ACMGG-called pathogenic (P) or likely pathogenic (LP) variants, as well as protein-truncating variants of unknown significance (PTVUS) were individually evaluated using the available literature and ClinVar to make a final call [54]. PTVUS include frameshift variants, stop codon changes, or variants that disrupt a splice site up to two bases into the intron. For simplicity, we categorized all P, LP, and PTVUS variants as loss-of-function (LOF) variants.
Assay to test for Hispanic/Latina (H/L) germline founder BRCA1 exon 9–12 deletion
The 11 H/L patients in our study were tested for the germline founder BRCA1 exon 9–12 deletion [57]. The deletion was detected by PCR amplification of the mutant and wild type alleles, using specific primers based on the Weitzel et al. method [57]. The PCR products were resolved on a 1.5% agarose gel to identify the amplification of the truncated and wild-type alleles.
OncoScan assay
OncoScan was performed in the Cytogenetics Core on DNA from 46 FFPE and 15 fresh-frozen tissues from 61 of the 71 patients (Supplemental Table 1). Sufficient DNA samples were not available for tumors from 10 patients for the assay. For the assay, 75 ng of DNA was used for the Molecular Inversion Probe (MIP) amplification, labeling, and hybridization steps (Thermo Fisher Scientific/Affymetrix, Santa Clara, CA). The CEL files (raw data) were then input into Chromosome Analysis Suite (ChAS) software (Thermo Fisher Scientific/Affymetrix, Santa Clara, CA) for processing and normalization with reference files specific for FFPE or fresh tissue. The generated OSCHP files were then loaded to the Nexus Express software (BioDiscovery, El Segundo, CA) to generate per-sample segmentation using the TuScan (Tumor Scan) algorithm developed by Affymetrix.
Determination of loss of heterozygosity (LOH)
We used the output from the OncoScan assay [58] to assess LOH on DNA from the 61 tumors (Supplemental Table 1). For the 10 patients in which we did not have OncoScan data, we used the variant allele frequencies from tumor sequence to determine LOH (> 20% increase of variant allele frequency over normal was used for defining LOH). For regions demonstrating LOH where there was targeted sequencing data, we confirmed that those demonstrating LOH from OncoScan data also demonstrated LOH in the sequencing data.
Identification of significant SCNAs
The segmentation files served as input files for the GISTIC2.0 program [39] on the gene pattern server (https://genepattern.broadinstitute.org/gp) to identify significant SCNAs using a q-value cutoff < 0.05. The GISTIC “All Lesion File,” which includes changes in copy number for each SCNA (row) and sample (column), was used as an input matrix for the hclust function in R [59] to perform unsupervised hierarchical clustering analysis with Pearson correlation coefficient as a distance metric and a complete linkage method to measure closeness between two clusters.
Association analysis between LOF variants in core HRR pathway genes and incidence of high-amplitude SCNAs
We tested the association between LOF variants in BRCA1, BRCA2, and PALB2 and incidence of high-amplitude SCNAs measured by the OncoScan assay using Firth’s logistic regression model. In the model, high-amplitude SCNAs (yes or no in a tumor sample with a GISTIC2-threshold score of 2 or -2 for a specific gene) was the outcome variable and LOF variant (yes or no in a tumor sample with a germline or somatic LOF variant in at least one core HRR pathway gene) was the predictor variable. This association test was performed only for genes with high-amplitude SCNAs in at least 5% of tumor samples (reducing the effect of rare SCNA events).
Pathway analysis of genes with significant association between LOF variants in core HRR pathway genes and incidence of high-amplitude SCNAs. The list of genes with statistically significant associations (P < 0.05 and FDR < 0.25) from the Firth’s logistic regression served as input for functional interpretation of the gene list using the g:Profile webserver (http://biit.cs.ut.ee/gprofiler/) and pathway enrichment analysis using Qiagen Ingenuity Pathway analysis (https://digitalinsights.qiagen.com/).
Analyses of effect of LOF variants in HRR core genes on relapse-free and overall survival
We conducted Kaplan–Meier analyses and log-rank tests to assess differences in relapse-free and overall survival between women carrying LOF variants (germline or somatic) in BRCA1, BRCA2, and PALB2 and those without variants in these genes. Relapse-free survival was defined as time from date of last chemotherapy treatment to cancer recurrence. For relapse-free survival, time was censored at death if the cause of death was not from ovarian cancer or at last contact if the patient was still alive at last contact date.
Analyses of effect of high-amplitude SCNAs on relapse-free and overall survival
Because cancer driver SCNAs tend to be shorter in length and higher in amplitude than passenger SCNAs [39], we only analyzed high-amplitude SCNAs defined as GISTIC-threshold scores of 2 (high-level amplification of > 4 copies) and -2 (homozygous deletion) [11]. A multivariate Cox regression model was used to assess the association between high-amplitude SCNAs and relapse-free and overall survival, adjusting for age at diagnosis, tumor stage, surgery (optimal vs. suboptimal debulking), type of adjuvant chemotherapy (intra-venous vs. intra-peritoneal), and SCNA rate (the number of SCNAs per sample generated by the Nexus Express software).
TCGA SCNA, variant, and clinical data for late-stage (3 or 4) HGSC patients (n = 356) [9] were downloaded from the cBioPortal website (http://www.cbioportal.org) [60]. Because the COH HGSC patients were all stage III and IV tumors, we only included data from TCGA of HGSC with stage III or IV tumors (356/489). These data were used for validation testing of results.
Availability of data and materials
The data sets supporting the results of this article are available and can be accessed in NCBI (National Center for Biotechnology Information) dbGAP (database of Genotypes and Phenotypes) with accession number phs003198.v1.p1.
Abbreviations
- ACMGG:
-
American College of Medical Genetics and Genomics
- BAM:
-
Binary alignment map
- BWA:
-
Burrows-Wheeler Alignment
- ChAS:
-
Chromosome Analysis Suite
- COH:
-
City of Hope
- FDR:
-
False-discovery rate
- FFPE:
-
Formalin-fixed-paraffin-embedded
- HGSC:
-
High-grade serous ovarian cancer
- H/L:
-
Hispanic/Latina
- HRR:
-
Homologous recombination repair
- ICL:
-
Interstrand cross links
- IGC:
-
Integrative Genomics Core
- JNK:
-
C-Jun N-terminal kinase
- LOF:
-
Loss-of-function
- LOH:
-
Loss of heterozygosity
- LP:
-
Likely pathogenic
- P:
-
Pathogenic
- PTVUS:
-
Protein-truncating variants of unknown significance
- SCNA:
-
Somatic copy number alterations
- TCGA:
-
The Cancer Genome Atlas
- TuScan:
-
Tumor Scan
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
Laine A, Sims TT, Le Saux O, Ray-Coquard I, Coleman RL. Treatment Perspectives for Ovarian Cancer in Europe and the United States: Initial Therapy and Platinum-Sensitive Recurrence after PARP Inhibitors or Bevacizumab Therapy. Curr Oncol Rep. 2021;23(12):148.
Kurnit KC, Fleming GF, Lengyel E. Updates and New Options in Advanced Epithelial Ovarian Cancer Treatment. Obstet Gynecol. 2021;137(1):108–21.
Vaughan S, Coward JI, Bast RC Jr, Berchuck A, Berek JS, Brenton JD, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11(10):719–25.
Matulonis UA, Sood AK, Fallowfield L, Howitt BE, Sehouli J, Karlan BY. Ovarian cancer. Nat Rev Dis Primers. 2016;2:16061.
Ducie J, Dao F, Considine M, Olvera N, Shaw PA, Kurman RJ, et al. Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat Commun. 2017;8(1):990.
Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, et al. High grade serous ovarian carcinomas originate in the fallopian tube. Nat Commun. 2017;8(1):1093.
Ahmed AA, Etemadmoghadam D, Temple J, Lynch AG, Riad M, Sharma R, et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J Pathol. 2010;221(1):49–56.
Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609-15.
Gorringe KL, Jacobs S, Thompson ER, Sridhar A, Qiu W, Choong DY, et al. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin Cancer Res. 2007;13(16):4731–9.
Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45(10):1127–33.
Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016;2(4):482–90.
Loveday C, Turnbull C, Ramsay E, Hughes D, Ruark E, Frankum JR, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. 2011;43(9):879–U90.
Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42(5):410–U65.
Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108(44):18032–7.
Etemadmoghadam D, deFazio A, Beroukhim R, Mermel C, George J, Getz G, et al. Integrated genome-wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clin Cancer Res. 2009;15(4):1417–27.
Engler DA, Gupta S, Growdon WB, Drapkin RI, Nitta M, Sergent PA, et al. Genome wide DNA copy number analysis of serous type ovarian carcinomas identifies genetic markers predictive of clinical outcome. PLoS ONE. 2012;7(2):e30996.
Mabuchi S, Kuroda H, Takahashi R, Sasano T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol Oncol. 2015;137(1):173–9.
Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MDM, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun. 2014;5:3156.
Pennington KP, Walsh T, Harrell MI, Lee MK, Pennil CC, Rendi MH, et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin Cancer Res. 2014;20(3):764–75.
Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016;35(9):909–23.
German J, Sanz MM, Ciocci S, Ye TZ, Ellis NA. Syndrome-causing mutations of the BLM gene in persons in the Bloom’s Syndrome Registry. Hum Mutat. 2007;28(8):743–53.
Larsen NB, Hickson ID. RecQ Helicases: Conserved Guardians of Genomic Integrity. Adv Exp Med Biol. 2013;767:161–84.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47(D1):D941–7.
Dicks E, Song H, Ramus SJ, Oudenhove EV, Tyrer JP, Intermaggio MP, et al. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget. 2017;8(31):50930–40.
Subramanian DN, Zethoven M, McInerny S, Morgan JA, Rowley SM, Lee JEA, et al. Exome sequencing of familial high-grade serous ovarian carcinoma reveals heterogeneity for rare candidate susceptibility genes. Nat Commun. 2020;11(1):1640.
Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell. 2018;33(4):690–705 e9.
Burotto M, Chiou VL, Lee JM, Kohn EC. The MAPK pathway across different malignancies: a new perspective. Cancer. 2014;120(22):3446–56.
Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70.
Davis SJ, Choong DY, Ramakrishna M, Ryland GL, Campbell IG, Gorringe KL. Analysis of the mitogen-activated protein kinase kinase 4 (MAP2K4) tumor suppressor gene in ovarian cancer. BMC Cancer. 2011;11:173.
Lee S, Zhao L, Rojas C, Bateman NW, Yao H, Lara OD, et al. Molecular Analysis of Clinically Defined Subsets of High-Grade Serous Ovarian Cancer. Cell Rep. 2020;31(2):107502.
Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521(7553):489–94.
Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356(6371):713–5.
Li C, Bonazzoli E, Bellone S, Choi J, Dong W, Menderes G, et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc Natl Acad Sci U S A. 2019;116(2):619–24.
Malek JA, Mery E, Mahmoud YA, Al-Azwani EK, Roger L, Huang R, et al. Copy number variation analysis of matched ovarian primary tumors and peritoneal metastasis. PLoS ONE. 2011;6(12):e28561.
George SH, Milea A, Sowamber R, Chehade R, Tone A, Shaw PA. Loss of LKB1 and p53 synergizes to alter fallopian tube epithelial phenotype and high-grade serous tumorigenesis. Oncogene. 2016;35(1):59–68.
Wang YK, Bashashati A, Anglesio MS, Cochrane DR, Grewal DS, Ha G, et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat Genet. 2017;49(6):856–65.
Kaveh F, Baumbusch LO, Nebdal D, Borresen-Dale AL, Lingjaerde OC, Edvardsen H, et al. A systematic comparison of copy number alterations in four types of female cancer. BMC Cancer. 2016;16(1):913.
Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12(4):R41.
Park JT, Li M, Nakayama K, Mao TL, Davidson B, Zhang Z, et al. Notch3 gene amplification in ovarian cancer. Cancer Res. 2006;66(12):6312–8.
Park JT, Chen X, Trope CG, Davidson B, Shih Ie M, Wang TL. Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am J Pathol. 2010;177(3):1087–94.
Jung SG, Kwon YD, Song JA, Back MJ, Lee SY, Lee C, et al. Prognostic significance of Notch 3 gene expression in ovarian serous carcinoma. Cancer Sci. 2010;101(9):1977–83.
Rao L, Mak VCY, Zhou Y, Zhang D, Li X, Fung CCY, et al. p85beta regulates autophagic degradation of AXL to activate oncogenic signaling. Nat Commun. 2020;11(1):2291.
Eckstein N, Servan K, Hildebrandt B, Politz A, von Jonquieres G, Wolf-Kummeth S, et al. Hyperactivation of the insulin-like growth factor receptor I signaling pathway is an essential event for cisplatin resistance of ovarian cancer cells. Cancer Res. 2009;69(7):2996–3003.
Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018;23(1):239–54 e6.
Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31(15):1869–83.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Koboldt DC, Zhang QY, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–76.
Wang K, Li MY, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38(16).
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15(8):1034–50.
Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40(Web Server issue):W452-7.
Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(D1):D980–5.
Robinson JT, Thorvaldsdottir H, Wenger AM, Zehir A, Mesirov JP. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017;77(21):e31–4.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Weitzel JN, Lagos VI, Herzog JS, Judkins T, Hendrickson B, Ho JS, et al. Evidence for common ancestral origin of a recurring BRCA1 genomic rearrangement identified in high-risk Hispanic families. Cancer Epidemiol Biomarkers Prev. 2007;16(8):1615–20.
Foster JM, Oumie A, Togneri FS, Vasques FR, Hau D, Taylor M, et al. Cross-laboratory validation of the OncoScan(R) FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med Genomics. 2015;8:5.
R Core Team (2013). R: A language and environment for statistical computing. R Foundation for Statistical Computing V, Austria. URL http://www.R-project.org/.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Acknowledgements
Research reported in this publication included work performed in the Integrative Genomics Core and the Research Pathology Shared Resource supported by the National Cancer Institute of the National Institutes of Health under grant number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. SLN is partially supported by the Morris and Horowitz Families Endowed Professorship.
Funding
This work was funded by pilot funding from the City of Hope and work performed in the cores was funded by the National Institutes of Health (NIH P30CA033572).
Author information
Authors and Affiliations
Contributions
Conception and design: SLN and MCC. Administrative support: LS. Provision of study materials or patients: LAL, RM, MTW, ESH, THD, PSL, AAH, SW, MCC, and SLN. Collection and assembly of data: AWA, YCD, LS, SW, and SLN. Data analysis and interpretation: AWA, YCD, CDW, ST, VB, and SLN. Manuscript writing: AWA, YCD, and SLN. Final approval of manuscript: All authors. Accountable for all aspects of the work: All authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All individuals provided written informed consent and were enrolled into the study through a City of Hope Institutional Review Board approved protocol (IRB#12358). All methods were carried out in accordance with relevant guidelines and regulations and experimental protocols were approved through a City of Hope Institutional Review Board approved protocol (IRB#12358).
Consent for publication
Not applicable.
Competing interests
Both Drs. Wakabayashi and Cristea now work at Regeneron; they worked at City of Hope when the data were generated. Dr. Han receives research funding from Vergent Biosciences. None of the other authors report conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Adamson, A.W., Ding, Y.C., Steele, L. et al. Genomic analyses of germline and somatic variation in high-grade serous ovarian cancer. J Ovarian Res 16, 141 (2023). https://doi.org/10.1186/s13048-023-01234-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-023-01234-x