
Abstract
Background
17α-hydroxylase/17,20-lyase deficiency (17-OHD) is a rare form of congenital adrenal hyperplasia caused by CYP17A1 gene variants. Female patients with 17-OHD demonstrate a broad clinical spectrum, including oligomenorrhea or amenorrhea and infertility, often as the sole manifestation. However, no spontaneous pregnancies in affected women have been reported.
Objective
This retrospective cohort study aimed to explore the endocrine characteristics and assisted reproductive technique (ART) performance in women with 17-OHD.
Methods
Five women were referred for primary infertility in a university-affiliated hospital over an eight-year period. The endocrine profiles and cycle characteristics during a total of nine cycles of ovarian stimulation and eight cycles of frozen-thawed embryo transfer (FET) were described in details.
Results
Three cases had homozygous variants and two cases had compound heterozygous variants, including one novel missense variant (p.Leu433Ser) in the CYP17A1 gene. Despite dual-suppression of progesterone (P) production by glucocorticoid and gonadotropin releasing hormone agonist, gradually increased P level, relatively low estradiol concentrations and thin endometrium were observed, negating fresh embryo transfer. During FET cycles, appropriate treatment resulted in low serum P levels and adequate endometrial thickness, leading to four live births.
Conclusions
Our findings demonstrate that continuous elevation of serum P during follicular growth impairs endometrial receptivity, the likely cause of female infertility in 17-OHD. Therefore, female infertility caused by 17-OHD is suggested as an indication for freeze-all strategy, with promising reproductive prognoses following segmented ovarian stimulation and FET treatment.
Similar content being viewed by others

Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by defects in enzymes or proteins involved in cortisol biosynthesis. 17α-hydroxylase/17,20-lyase deficiency (17-OHD) is a rare form of CAH arising from variants in CYP17A1, with an estimated incidence of 1 in 50,000 ~ 100,000 individuals [1, 2]. Worldwide, 17-OHD represents approximately 1% of CAH [3]. However, 17-OHD has been more frequently reported in the Chinese population, and thus considered to be the second most common form of CAH in China [4,5,6,7]. CYP17A1 variants impair cytochrome P450 17α-hydroxylase (P450c17) enzyme expression in the adrenal gland and gonads, resulting in cortisol and sex steroid deficiency, in combination with mineralocorticoid excess [1]. The typical presentations of 17-OHD in 46, XX patients include primary amenorrhea, absent secondary sexual development, low-renin hypertension, and hypokalemia [7,8,9]. It has been reported that some females with 17-OHD have spontaneous menses and pubertal development, but suffer from oligomenorrhea, ovarian cysts and infertility [3, 9]. To the best of our knowledge, no spontaneous pregnancies have been described in affected females due to disordered steroidogenesis. To date, assisted reproductive techniques (ART) seem to be the preferred therapeutic approach to overcome reproductive barriers in such patients [7, 10,11,12,13,14].
Considering infertility impacts quality of life, and 17-OHD is a rare disorder, fertility potential is still poorly understood in this population. Therefore, we conducted a retrospective cohort study to explore the endocrine characteristics and ART performance in females with 17-OHD, in order to expand the management of female infertility.
Materials and methods
In this retrospective cohort study, five unrelated female patients with 17-OHD undergoing ART treatment at the Reproductive Medicine Centre (Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University) between January 2014 and December 2021 were identified. Written informed consent was obtained for ART from each patient. Moreover, this study was approved by the ethics committee of Sun Yat-sen Memorial Hospital, Sun Yat-sen University (SYSKY-2022-143-01).
Clinical and laboratory evaluation
Relevant symptoms, family history and comprehensive fertility examinations of 5 patients were reviewed in detail. Karyotype analysis and anthropometric measurements including height, weight, and blood pressure were conducted. In addition to routine physical examination, the presences of axillary and pubic hair as well as breast and genitalia development were noted. Hormonal and biochemical profiles included follicle stimulating hormone (FSH), luteinizing hormone (LH), prolactin, estradiol (E2), progesterone (P), testosterone (T), anti-Müllerian hormone (AMH), 17-hydroxyprogesterone (17-OHP), dehydroepiandrosterone sulfate (DHEAS), androstenedione, thyroid stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), serum cortisol, serum sodium, and serum potassium.
CYP17A1 gene analysis
Genomic DNA was extracted from peripheral blood sample using a commercial kit (Qiagen, Germany). The reference sequence of CYP17A1 was retrieved from the UCSC Genome Browser on Human Feb. 2009 Assembly (hg19) (http://genome.ucsc.edu). All coding regions and flanking introns were amplified by genomic DNA polymerase chain reaction (PCR) using specific primers designed by Oligo 6.0 (http://www.oligo.net/downloads.html). The 2015 American College of Medical Genetics and Genomics (2015 ACMG) standards and guidelines were used to classify the variations [15]. Variant annotation and pathogenicity prediction were performed using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and MutationTaster (http://www.mutationtaster.org/).The Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/) and 1000 Genomes (1000G) browser (https://www.internationalgenome.org/) were used to determine allele frequencies of variants.
Fertility treatment
All patients underwent conventional in vitro fertilization (IVF) treatments depending on normal profiles of partners. Variable controlled ovarian stimulations (COS) were performed in our clinic based on features of ovarian reserve, such as gonadotropin releasing hormone agonist (GnRH-a) long protocol, GnRH antagonist protocol, luteal phase stimulation and progestin-primed ovarian stimulation (PPOS). However, two patients had attempted IVF treatments using ultra-long GnRH-a protocol and short-acting GnRH-a long protocol in other hospitals before referral to our clinic. All available embryos were cryopreserved by vitrification. In frozen-thawed embryo transfer (FET) cycles, patients were given oral glucocorticoids and GnRH-a injections to suppress progesterone excess from both adrenal and gonadal sources. When progesterone value was well controlled in the range of follicular phase, artificial endometrial preparation was started with oral estradiol valerate at a dosage of 4-6 mg/d (Progynova; Bayer Schering Pharma, France). When endometrial thickness exceeded 7 mm, endometrial transformation was applied with progesterone administration until pregnancy testing. A maximum of three frozen-thawed embryos were transferred. At the time of transfer, the use of oral glucocorticoids was stopped. If pregnancy was achieved, luteal support was maintained until 12 weeks gestation.
Results
Clinical manifestations
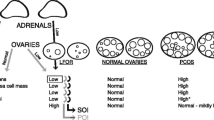
As shown in Table 1, barring Patient 4 (P4), all patients had spontaneous menarche, three whom complained of prolonged menstrual bleeding. Because of primary amenorrhea, P4 received glucocorticoid therapy to establish menstruation after the diagnosis of 17-OHD, but her menses onset was characterized by oligomenorrhea. All cases were referred for primary infertility, and comprehensive infertility evaluations revealed little except bilateral tubal obstruction in Patient 3 (P3). Three patients (P1, P2, P5) presented with recurrent ovarian cysts. All patients developed normal secondary sexual characteristics, however, pubic and axillary hair were scarce or absent. Unlike individuals with classic 17-OHD, they exhibited normal blood pressure.
Hormonal profiles
Patient 1 (P1) was diagnosed with 17-OHD after ovarian stimulation in our clinic, whereas the remaining patients had earlier diagnoses and had a history of glucocorticoid treatment prior to referral. As shown in Table 2, several hormone features were common amongst all patients. Pituitary hormone profiles including serum FSH, LH, prolactin and TSH were normal in most cases, while mild LH elevation was observed in P2 and P5. Serum E2, T, DHEA and androstenedione levels were in the low-normal range. Follicular phase progesterone was high in all patients except P4, who was on glucocorticoid replacement therapy. Basal 17-OHP levels were within the reference range of follicular phase. In accordance with the clinical features of each patient, ACTH, cortisol and electrolytes were within normal ranges.
Variant detection in CYP17A1
Sanger sequencing revealed six variants in CYP17A1 (Table 3). Three individuals had homozygous variants and two were compound heterozygotes. With the exception of P1 (Fig. 1), the remaining patients’ genetic reports came from other hospitals. In P1, the novel variant c.1298T > C was unreported in dbSNP, and 1000 genomes database. The minor allele frequency (MAF) in gnomAD was very low (0.000007). The c.1298T > C missense variant, predicted to result in loss of both 17α-hydroxylase and 17,20-lyase was designated damaging by in silico predictive tools, PolyPhen-2 and Mutation Taster. In P3, the variant c.985_987delinsAA results in a frameshift, potentially impacting splicing. The variant c.1346G > A was reported in dbSNP (rs752164207) and gnomAD at a low frequency (0.00000-0.000008). In silico analysis of the 1346G > A variant by PolyPhen-2 and Mutation Taster, deemed it to be probably damaging and disease causing. The same position variation c.1346 was included in HGMD and showed pathogenic (HGMD Accession Number: HM0669).

The sequencing chromatogram of Patient 1
Cycle characteristics of ovarian stimulation
Over a span of eight years, 5 patients underwent 9 cycles of COS in total (Table 4). Besides poor ovarian response in P4, the majority of patients showed normal ovarian response using GnRH-a or GnRH antagonist protocol. With exogenous gonadotropin stimulation, multiple follicles matured, but an abnormal sex hormone pattern was noted throughout the cycle, characterized by gradually increased follicular phase P level and relatively low E2 concentration. Late follicular-phase E2 was disproportionate compared to the number of dominant follicles. Overall, thin endometria were observed during ovarian stimulation. P3 had adequate endometrial thickness as E2 level on human chorionic gonadotropin (HCG) trigger day reached over 500ng/L. In all patients, ovarian stimulation resulted in available oocytes (ranging from 1 to 16) and viable embryos (ranging from 0 to 7). High P levels and inappropriate endometrial thickness negated fresh embryo transfer and all available embryos were cryopreserved.
Cycle characteristics and pregnancy outcomes of frozen embryo transfer
In total, 8 FET cycles were conducted (Table 5). With dual-suppression by oral glucocorticoids and GnRH-a administration, serum P levels on transformation day were usually very low or undetectable. For P5, dexamethasone was administrated only during FET cycle, since there was no dominant follicle growth and her P concentration was kept at a stable-low level. Adequate endometrial thickness was found after exogenous oestrogen supplementation, ranging from 7.8 to 17.0 mm. Two embryos were transferred in six FET cycles, four FET cycles developed into live births. FET failed in P2 mainly due to poor control of serum P level in FET cycle. Despite having three additional frozen embryos in our centre, she has not returned for further embryo transfer.
Discussion
This study describes our centre’s experience in managing 5 cases of infertility in women with 17-OHD (46, XX). Our study focused on ART performance in 17-OHD patients including fresh and frozen embryo transfer. Results demonstrated that the fertility potential of females with 17-OHD was promising, and segmented ovarian stimulation and FET significantly improved chances of successful live births.
Although 17-OHD was first described in 1966 [16] and despite global cases of 17-OHD, little is known about fertility in these patients. CYP17A1 encodes the key enzyme P450c17, which mediates both 17α-hydroxylase and 17,20-lyase activities. Combined enzyme deficiency is the most common form of 17-OHD [2, 9]. To date, there is no specific definition for complete or partial 17-OHD, and the clinical manifestation is mainly based on residual enzyme activity. Sometimes the diagnosis is quite challenging as phenotype variability occurs. The five patients described herein were defined as partial 17-OHD, given that they presented with androgen and oestrogen deficiency without mineralocorticoid excess and developed normal female secondary sex characteristics without hypertension or hydro-electrolyte imbalance.
Besides the aforementioned hormonal abnormalities, ovarian cyst and cyst rupture provided clinical clues as to the diagnosis of 17-OHD in some female patients [14, 17,18,19,20]. Ovarian cyst formation was ascribed to high P level or elevated gonadotropins without an oestrogen-triggered LH surge [9, 21]. The gynecological complaints of three patients (P1, P2, P5) were consistent with previous studies, and fortunately, none required ovarian surgery. Furthermore, P5 had a misdiagnosis of polycystic ovarian syndrome (PCOS) and underwent repeated hysteroscopy due to refractory endometrial cavity fluid. She had primary infertility and no history of endometrial tuberculosis, or other underlying causes of endometrial cavity fluid. 17-OHD should be distinguished from PCOS, especially in cases with PCO-like ovaries, elevated LH, and good ovarian reserve.
CYP17A1 is located on chromosome 10q24.32 and consists of eight exons, encoding a 508 amino acid protein. Until now, variants in CYP17A1 (OMIM * 609,300) exceeding more than 150, have included point variants, indel variants, splice site alterations, and large deletions (9). In our study, genetic analyses revealed one novel missense variant c.1298T > C (p.Leu433Ser), which was recently reported in Chinese hypospadias patients with karyotype 46, XY [22]. All variants in this study were predicted deleterious or likely to be pathogenic. Therefore, establishing a genetic diagnosis aided clinical management and therapy.
Spontaneous pregnancy has not been documented in women with 17-OHD and primary infertility. In previous studies, ovarian histology confirmed numerous primary and secondary ovarian follicles [23, 24], but follicular maturation was arrested in some affected women [25]. Mild forms of 17-OHD such as seen in P1 and P3, were associated with regular menstruation and normal genitalia, but primary infertility still supervened. The impairment of reproductive capacity in 17-OHD is presumed to be multifactorial including impaired folliculogenesis, anovulation, thin endometria and cervical dysmucorrhea.
What’s more, in our study even with dual-suppression of P production by GnRH-a administration and ongoing glucocorticoid therapy during ovarian stimulation, serum P level increased during the follicular phase. The distinctive response to ovarian stimulation in 17-OHD proved to be quite a useful tool. Firstly, as follicles grow, endogenous progesterone biosynthesis is activated resulting in P overproduction, hence natural conception is scarce and failed ovulation induction without IVF has only ever been reported in the literature [26, 27]. Secondly, since fresh embryo transfer was out of the question in 17-OHD patients, the choice of ovarian stimulation regimen was tailored to account for ovarian reserve and menstrual cycle regularity. Nearly half the COS cycles in this study were GnRH-a long protocol, thus ART procedures were prolonged. In this respect, we would like to propose 17-OHD as an indication for a freeze-all strategy in IVF and suggest flexible COS options in this population, in order to shorten the time to pregnancy. Thirdly, the traditional timing of triggering final oocyte maturation is generally related to follicle size, growing follicle cohort, hormonal data, experience of previous cycles, etc. [28]. In 17-OHD, the criteria for triggering was mainly based on the size of leading follicles, not taking into account E2 or P levels.
We searched the literature and found that the reproductive prognosis of 17-OHD was acceptable, and ART overrode the steroid biosynthetic defect state and helped affected females conceive with their own gametes [7, 10,11,12,13,14]. The successful therapeutic achievements in our study were a direct result of segmented ovarian stimulation and FET. It was reported that hypoplastic uteri was found in some cases [20, 29, 30], and uterine dysfunction might be deemed to impair fertility. As for our results, all 5 cases showed adequate endometrial thickness in FET cycles after exogenous oestrogen supplement. In our experiences, fertility potential in most cases of partial 17-OHD will not be compromised if oestrogen treatment improves uterine volume and endometrial thickness.
In summary, our findings demonstrate that continuous elevation of serum P during follicle growth impairs endometrial receptivity, the main cause of female infertility in partial 17-OHD. We suggest that primary infertility caused by 17-OHD should be an indication for a freeze-all strategy, aided by segmented ovarian stimulation and FET treatment.
Data Availability
The data in the current study can be found in the electronic medical record system of Reproductive Medicine Centre, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University.
References
El-Maouche D, Arlt W, Merke DP. Congenital Adrenal Hyperplasia Lancet. 2017;390:2194–210.
Wolthers OD, Rumsby G, Techatraisak K, Honour JW, Hindmarsh PC. 17-Hydroxylase/17,20 lyase Deficiency diagnosed during Childhood. Horm Res. 2002;57:133–6.
Marsh CA, Auchus RJ. Fertility in patients with genetic deficiencies of cytochrome P450c17 (CYP17A1): combined 17-hydroxylase/17,20-lyase Deficiency and isolated 17,20-lyase Deficiency. Fertil Steril. 2014;101:317–22.
Tian Q, Yao F, Zhang Y, Tseng H, Lang J. Molecular study of five chinese patients with 46XX partial 17a-hydroxylase/17,20-lyase Deficiency. Gynecol Endocrinol. 2012;28:234–8.
Han B, Xue L, Fan M, Zhao S, Liu W, Zhu H et al. Clinical and molecular Manifestation of Fifteen 17OHD patients: a Novel mutation and a founder effect. Endocrine. 2016;53:784–90.
Chen H, Yuan K, Zhang B, Jia Z, Chen C, Zhu Y, et al. A novel compound heterozygous CYP17A1 variant causes 17α-Hydroxylase/17, 20-Lyase Deficiency. Front Genet. 2019;10:996.
Xu Y, Jiang S, Yan Z, Niu Y, Du W, Liu B, et al. Phenotypic heterogeneity and fertility potential of patients with 17-Hydroxylase/17,20-lyase Deficiency. J Clin Endocrinol Metab. 2022;107:e2610–8.
Sun M, Mueller JW, Gilligan LC, Taylor AE, Shaheen F, Noczyńska A, et al. The broad phenotypic spectrum of 17α-hydroxylase/17,20-lyase (CYP17A1) Deficiency: a Case Series. Eur J Endocrinol. 2021;185:729–41.
Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol. 2017;165:71–8.
Levran D, Ben-Shlomo I, Pariente C, Dor J, Mashiach S, Weissman A. Familial partial 17,20-desmolase and 17alpha-hydroxylase deficiency presenting as infertility. J Assist Reprod Genet. 2003;20:21–8.
Bianchi PH, Gouveia GR, Costa EM, Domenice S, Martin RM, de Carvalho LC, et al. Successful live birth in a woman with 17α-Hydroxylase Deficiency through IVF Frozen-Thawed embryo transfer. J Clin Endocrinol Metab. 2016;101:345–8.
Kitajima M, Miura K, Inoue T, Murakami Y, Kitajima Y, Murakami N, et al. Two consecutive successful live birth in woman with 17α hydroxylase Deficiency by frozen-thaw embryo transfer under hormone replacement Endometrium Preparation. Gynecol Endocrinol. 2018;34:381–4.
Blumenfeld Z, Koren I. Successful delivery in 17,20-Lyase Deficiency. J Clin Endocrinol Metab. 2021;106:1882–6.
Xia Y, Shi P, Xia J, Zhang H, Xu L, Kong X. Novel mutations of the CYP17A1 gene in four chinese 46,XX cases with partial 17a-hydroxylase/17,20-lyase deficiency. Steroids. 2021;173:108873.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Biglieri EG, Herron MA, Brust N. 17-hydroxylation deficiency in man. J Clin Invest. 1966;45:1946–54.
Singhellakis PN, Panidis D, Papadimas J, Demertzi H, Tsourdis A, Sotsiou F, et al. Spontaneous sexual development and menarche in a female with 17 alpha-hydroxylase deficiency. J Endocrinol Invest. 1986;9:177–83.
ten Kate-Booij MJ, Cobbaert C, Koper JW, de Jong FH. Deficiency of 17,20-lyase causing giant ovarian cysts in a girl and a female phenotype in her 46,XY sister: case report. Hum Reprod. 2004;19:456–9.
Tian QJ, Zhang YW, Lu ZL, Sha GH, Gao H. Incomplete P450 17 alpha enzyme deficiency: report of six cases. Chin J Obstet Gxnecol. 2007;42:670–4.
Carvalho LC, Brito VN, Martin RM, Zamboni AM, Gomes LG, Inácio M, et al. Clinical, hormonal, ovarian, and genetic aspects of 46,XX patients with congenital adrenal hyperplasia due to CYP17A1 defects. Fertil Steril. 2016;105:1612–9.
Gomes LG, Bachega TASS, Mendonca BB. Classic congenital adrenal hyperplasia and its Impact on Reproduction. Fertil Steril. 2019;111:7–12.
Shaomei W, Yongbin P, Daiyue Y, Zhaorong H, Huirong Y, Nan L, et al. Whole exome sequencing applied to 42 Han Chinese patients with posterior hypospadias. Steroids. 2022;184:109041.
Neuwinger Araki S, Chikazawa K, Sekiguchi I, Yamauchi H, Motoyama M, Tamada T. Arrest of follicular development in a patient with 17 alpha-hydroxylase deficiency: folliculogenesis in association with a lack of estrogen synthesis in the ovaries. Fertil Steril. 1987;47:169–72.
Neuwinger J, Licht P, Munzer B, Sir-Petermann T, Siebzehnrübl E, Wildt L. Substitution with testosterone as aromatizable substrate for induction of follicular maturation, estradiol production and ovulation in a patient with 17 alpha-hydroxylase deficiency. Exp Clin Endocrinol Diabetes. 1996;104:400–8.
Miura K, Yasuda K, Yanase T, Yamakita N, Sasano H, Nawata H, et al. Mutation of cytochrome P-45017 alpha gene (CYP17) in a japanese patient previously reported as having glucocorticoid-responsive hyperaldosteronism: with a review of japanese patients with mutations of CYP17. J Clin Endocrinol Metab. 1996;81:3797–801.
Meirow D, Schenker JG, Rosler A. Ovarian hyperstimulation syndrome with low oestradiol in non-classical 17 alpha-hydroxylase, 17,20-lyase deficiency: what is the role of oestrogens? Hum Reprod. 1996;11:2119–21.
Matsuzaki S, Yanase T, Murakami T, Uehara S, Nawata H, Yajima A. Induction of endometrial cycles and ovulation in a woman with combined 17alpha-hydroxylase/17,20-lyase deficiency due to compound heterozygous mutations on the p45017alpha gene. Fertil Steril. 2000;73:1183–6.
Ovarian Stimulation TEGGO, Bosch E, Broer S, et al. ESHRE guideline: ovarian stimulation for IVF/ICSI. Hum Reprod Open. 2020;2020:hoaa009.
Ben-Nun I, Siegal A, Shulman A, Ghetler Y, Kaneti H, Lunenfeld B, et al. Induction of artificial endometrial cycles with oestradiol implants and injectable progesterone: establishment of a viable pregnancy in a woman with 17-alpha-hydroxylase deficiency. Hum Reprod. 1995;10:2456–8.
Taniyama M, Tanabe M, Saito H, Ban Y, Nawata H, Yanase T. Subtle 17alpha-hydroxylase/17,20-lyase deficiency with homozygous Y201N mutation in an infertile woman. J Clin Endocrinol Metab. 2005;90:2508–11.
Funding
This study is performed without any supporting fund.
Author information
Authors and Affiliations
Contributions
PP and LZ conceived the study and wrote the first draft of the manuscript. JH, XC, RN, QZ, DY and YL contributed to the patients’ treatments and revised the manuscript critically. This study was supervised by DY and YL. All authors contributed to the article and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors have nothing to declare.
Ethics approval and consent to participate
The study was approved by the institutional ethics committee of Sun Yat-sen Memorial hospital, Sun Yat-sen University (No. SYSKY-2022-143-01).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pan, P., Zheng, L., Huang, J. et al. Endocrine profiles and cycle characteristics of infertile 17α-hydroxylase/17,20-lyase Deficiency Patients undergoing assisted Reproduction Treatment: a retrospective cohort study. J Ovarian Res 16, 111 (2023). https://doi.org/10.1186/s13048-023-01190-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-023-01190-6