
Abstract
Purpose
To investigate the relationship between vaginal microbial community structure and premature ovarian insufficiency (POI).
Methods
Twenty-eight women with POI and 12 healthy women were recruited at Shenzhen Maternity and Child Healthcare Hospital between August and September 2020. Blood samples were collected for glucose tests and detection of sex hormone levels and vaginal secretions were collected for microbial group determination. Vaginal microbial community profiles were analysed by 16S rRNA gene sequencing using the Illumina MiSeq system (Illumina Inc., San Diego, CA, USA).
Results
Compared to the controls, the serum levels of follicle-stimulating hormone, luteinizing hormone, testosterone, and the follicle-stimulating hormone/luteinizing hormone ratio, significantly increased, and oestradiol and anti-Müllerian hormone levels significantly decreased in women with POI. Higher weighted UniFrac values were observed in women with POI than in healthy women. Bacteria in the genera Lactobacillus, Brevundimonas, and Odoribacter were more abundant in the microbiomes of healthy women, while the quantity of bacteria in the genus Streptococcus was significantly increased in the microbiomes of women with POI. Moreover, these differences in microbes in women with POI were closely related to follicle-stimulating hormone, luteinizing hormone, oestradiol, and anti-Müllerian hormone levels and to the follicle-stimulating hormone/luteinizing hormone ratio.
Conclusions
Women with POI had altered vaginal microbial profiles compared to healthy controls. The alterations in their microbiomes were associated with serum hormone levels. These results will improve our understanding of the vaginal microbial community structure in women with POI.
Trial registration
CHICTR, ChiCTR2000029576. Registered 3 August 2020 - Retrospectively registered, https://www.chictr.org.cn/showproj.aspx?proj=48844.
Similar content being viewed by others


Background
Premature ovarian insufficiency (POI) is an ovarian insufficiency syndrome that occurs in women aged < 40 years and affects 1–2% of women. Recently, POI has shown an increasing incidence [1, 2]. Clinically, the condition is characterised by a continuous decline in ovarian function, resulting in earlier cessation of menstruation than normal [2]. Women with POI can also experience comorbidities, including a low chance of natural conception [3], urogenital atrophy [4], decreased bone mineral density [5], increased risk of autoimmune and thyroid disease [6], cognitive dysfunction [7], shortened life expectancy [8], and cardiovascular disease [9].
The cause of POI is currently unknown; however, a number of potential triggers are associated with the development of the disease, including genetic defects, autoimmune dysfunction, enzyme deficiency, surgical intervention, chemotherapy, radiotherapy, and environmental factors [7, 10]. Several studies have indicated that bacterial vaginosis is associated with infertility [11]. Bacterial vaginosis has also been shown to alter the vaginal microbiome [12]. A previous study demonstrated that the vaginal microbiome plays an important role in the pathophysiology of primary ovarian failure, and the relative abundance of bacteria in the genus Lactobacillus was significantly lower in women with primary ovarian failure than in healthy controls [13]. Thus, a relationship may exist between the vaginal microbiome and POI.
In this study, 28 women with POI and 12 healthy women were recruited to study the community profile of the vaginal microbiome. Sequencing of the V3–V4 regions of the 16S rRNA gene in vaginal samples was performed to reveal the differences in the vaginal microbiota between the women with POI and the controls.
Methods
Study cohort
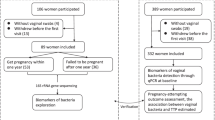
Forty women aged 24 to 40 years were recruited at Shenzhen Maternity and Child Healthcare Hospital between August and September 2020. Twenty-eight women with spontaneous POI and 12 healthy women were included in the study. Spontaneous POI was assessed according to criteria described in a previous report [14]. POI was diagnosed if the patient had primary or secondary amenorrhoea for at least 4 months before the age of 40 years and at least two instances of serum follicle-stimulating hormone (FSH) levels exceeding 40 IU/L with an interval of 4–6 weeks. All control women had normal ovarian function, without a history of menstrual dysfunction and infertility and with regular menstruation and normal FSH levels (< 10 IU/L). Participants were excluded if they had the following conditions: non-46-XX karyotype, family history of POI, pregnancy, tumour, chronic diarrhoea, autoimmune diseases, use of antibiotics/medications within the preceding 3 months, pelvic surgery, gastrointestinal disease, active infections, body mass index (BMI) of < 18.5 or > 23.9 kg/m2, smoking, or were undergoing chemotherapy or radiotherapy treatment. Clinical characteristics were obtained from the participants’ health records.
The study protocol was approved by the ethics committee of Shenzhen Maternity and Child Healthcare Hospital (Approval number: SFYLS2020–005). Written informed consent was obtained from all participants prior to enrolment. This study was registered at Chinese Clinical Trial Registry (registration No. ChiCTR2000029576).
Sampling
Blood samples were collected for the glucose (GLU) test and sex hormone levels detection on the third day of menstruation. Patients with amenorrhoea provided blood samples whenever they were available. The serum hormone levels were tested using enzyme-linked immunosorbent assay (ELISA) kit (Sino-UK Institute of Biological Technology, Beijing, China) according to the manufacturer’s instructions in the clinical laboratory. Vaginal secretions from the vaginal posterior fornix were collected using empty sterile collection tubes with an inbuilt sterile swab. Samples were stored at − 80 °C until further analysis.
DNA extraction and sequencing
DNA was extracted from vaginal samples. DNA quality was assessed using a NanoDrop™ 2000 ultraviolet spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and electrophoresis on a 1% agarose gel. The 16S rRNA gene was amplified using the 338F forward primer 5′-ACTCCTACGGGAGGCAGCAG-3′ and 806R reverse primer 5′-GGACTACHVGGGTWTCTAAT-3′, targeting the variable (V3–V4) region. All samples were pooled and sequenced using the Illumina MiSeq system (Illumina Inc., San Diego, CA, USA). Raw sequencing data were deposited into the NCBI Sequence Read Archive database (SRA BioProject ID: PRJNA738630).
Sequencing data analysis
The custom Perl script was used to split the sequencing reads from each sample according to the dual index. QIIME 2 (version 2020.08) was used to process the sequence reads [15]. First, we used the command ‘qiime tool import’ to import the sequence data into a QIIME 2 artefact. Second, we used the command ‘qiime data2 denoise-paired’ to exclude chimeric sequences and phiX sequences from the sequence reads. Third, the command ‘qiime2 feature-classifier classify-sklearn’ was used against the Greengenes (13_8 revision) database to assign the taxonomy. The Shannon index was used to represent alpha diversity, which considers both the richness and evenness of microbial communities. The weighted UniFrac distance was used to identify the differences between samples. Both the Shannon index and the UniFrac distance were generated with the command ‘qiime phylogeny align-to-tree-mafft-fasttree’ and ‘qiime diversity core-metrics-phylogenetic’ at a sample depth of 1000. Principal coordinate analysis was then performed based on the weighted UniFrac distance. PICRUSt2.0 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) [16] was used to predict functional composition and abundance.
Statistical analysis
Data analysis was performed using R software (R Foundation for Statistical Computing, Vienna, Austria). Unpaired t-tests were used for comparisons of normally distributed data sets and Wilcoxon rank-sum tests were used for non-normally distributed data. Normally distributed data are expressed as means ± standard deviations and non-normally distributed data are expressed as numbers (percentages). Correlation analysis was performed using Pearson’s correlation coefficient. Statistical significance was set at p < 0.05.
Linear discriminant analysis (LDA) effect size analysis was used to determine the significant differences in microbes between the POI and control groups with an LDA cut-off score of 2.0 [17]. The System-Theoretic Accident Model and Processes (STAMP) software [18] was used to investigate the functional differences between the POI and control groups. A ‘support vector machine’ algorithm with 5-fold cross-validation was used to build a classification model to identify women with POI based on microbial community. Receiver operating characteristic curves were generated, and the area under the curve was calculated to evaluate the model’s performance.
Results
Participant characteristics
Twenty-eight women with POI and 12 healthy controls were recruited for analysis. The clinical characteristics of the two groups are shown in Table 1. The mean ages in the POI and control groups were 34.61 ± 4.37 and 32.5 ± 3.87 years, respectively. The mean BMIs in the POI and control groups were 21.01 ± 1.19 and 20.81 ± 1.60, respectively. Age and BMI did not significantly differ between the two groups. Progesterone (P), prolactin (PRL), and GLU levels were not significantly different between women with and without POI. Women with POI had significantly higher levels of FSH, luteinizing hormone (LH), and testosterone and a higher FSH/LH ratio, but significantly lower oestradiol (E2) and anti-Müllerian hormone (AMH) levels compared to the control women.
Overall community structure of the vaginal microbiome of women with POI
To evaluate the community structure of the vaginal microbiota in women with and without POI, sequencing was performed on the V3–V4 regions of the 16S rRNA. In total, 1,903,682 usable reads (47,592 ± 9664 reads per sample) were obtained from all 40 samples, and the mean and median sequence lengths were 423 and 428 base pairs, respectively. The number of reads analysed did not differ between the POI and control samples (47,805 ± 10,475 vs 47,093 ± 7834; p = 0.83), indicating comparable and adequate sequencing coverage.
To explore the dissimilarity in the vaginal microbiota between the two groups, the alpha and beta diversities were represented using the Shannon and weighted UniFrac distance indices, respectively. The POI group exhibited a higher mean Shannon index than the control group; however, this was not statistically significant (1.21 ± 1.08 vs 0.80 ± 0.76; p = 0.19; Fig. 1a). The average weighted UniFrac value was significantly higher in the POI group than in the control group (0.45 ± 0.29 vs 0.26 ± 0.34; p < 0.01; Fig. 1b). Further, the results of principle coordinates analysis performed based on the weighted UniFrac distance showed that the microbial communities were not compositionally different between the two groups (Fig. 1c).

Overall structural differentiation of the vaginal microbiota between the POI and control groups. a Shannon index between the two groups. b Weighted UniFrac value between the two groups. c PCoA plot based on weighted UniFrac value. NG, healthy control group; PCoA, principal coordinates analysis; POI, premature ovarian insufficiency
Characterising the vaginal microbiome in the POI group
Vaginal microbiome communities were dominated by the phyla Actinobacteria, Bacteroidetes, Firmicutes, Fusobacteria, Proteobacteria, and Tenericutes in both the POI and control groups (Fig. 2a). Firmicutes were predominant, accounting for 63.08% ± 40.94 and 84.17% ± 35.40% of the total microbiota on average in the POI and control groups, respectively. The top 10 most abundant genera in both groups were Anaerococcus, Atopobium, Bifidobacterium, Enterococcus, Gardnerella, Lactobacillus, Peptoniphilus, Prevotella, Streptococcus, and Veillonella (Fig. 2b). Compared to the control group, the POI group had increased quantities of Anaerococcus (1.05% vs 0.01%), Atopobium (2.04% vs 1.47%), Enterococcus (2.69% vs 0.00%), Gardnerella (25.94% vs 5.51%), Peptoniphilus (0.84% vs 0.01%), Prevotella (2.32% vs 0.07%), Streptococcus (1.76% vs 0.16%), and Veillonella (0.65% vs 0.01%), but decreased quantities of Bifidobacterium (4.80% vs 8.16%) and Lactobacillus (54.18% vs 83.94%).

Microbial community profiles of the vaginal microbiota in the POI and control groups. a Relative abundances of the dominant phylum. b Relative abundances of the top 10 genera. c Microbes that significantly differed between the POI and control groups. d Pearson correlation between microbes and serum hormones. AMH, anti-Müllerian hormone; E2, oestradiol; FSH, follicle-stimulating hormone; GLU, glucose; LDA, linear discriminant analysis; LH, luteinizing hormone; NG, healthy control group; P, progesterone; POI, premature ovarian insufficiency; PRL prolactin; T, testosterone
The significance of the different genera in the POI and control groups was investigated using LDA effect size analysis. Significantly lower abundances of the genera Lactobacillus (LDA = 5.16, p = 0.02), Odoribacter (LDA = 3.42, p < 0.01), and Brevundimonas (LDA = 3.81, p = 0.0) and a significantly higher abundance of Streptococcus (LDA = 3.90, p = 0.04) were observed in the POI group compared to the control group (Fig. 2c).
Pearson correlation analysis was performed to evaluate the association between the different microbes and serum hormones. The relative proportion of the genus Lactobacillus was significantly correlated with E2 (R = 0.34, p = 0.03) and FSH (R = − 0.31, p = 0.049) levels. The genus Brevundimonas was significantly positively correlated with AMH levels (R = 0.43, p = 0.006). The genus Odoribacter was significantly positively correlated with AMH (R = 0.48, p = 0.002) and E2 (R = 0.38, p = 0.015) levels, and negatively correlated with the FSH/LH ratio (R = − 0.42, p = 0.008), FSH levels (R = − 0.36, p = 0.024), GLU levels (R = − 0.37, p = 0.021), and LH levels (R = − 0.35, p = 0.025). The genus Streptococcus was significantly positively correlated with FSH (R = 0.35, p = 0.025) and LH (R = 0.35, p = 0.029) levels (Fig. 2d).
Correlations between the abundant genera and serum hormones were also investigated. The abundance of the genus Anaerococcus was positively correlated with levels of LH (R = 0.40, p = 0.011), PRL (R = 0.42, p = 0.007), and P (R = 0.65, p = 0.0001). The genus Atopobium was positively correlated with P levels (R = 0.33, p = 0.035). The genus Peptoniphilus was positively correlated with levels of FSH (R = 0.35, p = 0.026), LH (R = 0.43, p = 0.005), PRL (R = 0.49, p = 0.001), and P (R = 0.61, p = 0.001). The genus Prevotella was negatively correlated with GLU levels (R = − 0.31, p = 0.048), but positively correlated with P levels (R = 0.31, p = 0.049). The genus Veillonella was significantly positively correlated with FSH (R = 0.33, p = 0.039) and LH (R = 0.33, p = 0.038) levels (Fig. 2d).
A classification model was then constructed based on differentially expressed and abundant genera using the support vector machine algorithm with 5-fold cross-validation. The area under the curve was 0.79, demonstrating that POI could be accurately predicted based on microbial community.
Different metabolic functions between the POI and control groups
Using PICRUSt2.0, the metabolic pathways of the two groups based on the Metacyc database were performed. STAMP analysis identified 16 significantly different pathways between the two groups (Fig. 3), including PWY-7199 (pyrimidine deoxyribonucleosides salvage), GLYCOCAT-PWY (glycogen degradation I), ARGORNPROST-PWY (L-arginine degradation), PWY-7323 (superpathway of GDP-mannose-derived O-antigen building blocks biosynthesis), PWY-6123 (inosine-5′-phosphate biosynthesis I), PWY-7200 (superpathway of pyrimidine deoxyribonucleoside salvage), PWY-6609 (adenine and adenosine salvage III), CALVIN-PWY (Calvin-Benson-Bassham cycle), PWY-6121 (5-aminoimidazole ribonucleotide biosynthesis I), PWY-6122 (5-aminoimidazole ribonucleotide biosynthesis II), PWY-6277 (superpathway of 5-aminoimidazole ribonucleotide biosynthesis), PWY-7196 (superpathway of pyrimidine ribonucleosides salvage), LACTOSECAT-PWY (lactose and galactose degradation I), PWY-5913 (partial TCA cycle [obligate autotrophs]), PWY-7184 (pyrimidine deoxyribonucleotides de novo biosynthesis I), and PWY-6163 (chorismate biosynthesis from 3-dehydroquinate). Excluding the LACTOSECAT-PWY, the remaining 15 pathways were significantly enriched in women with POI.

Metabolic pathways that differed significantly between the POI and control groups. NG, healthy control group; POI, premature ovarian insufficiency; PWY-7199, pyrimidine deoxyribonucleosides salvage; GLYCOCAT-PWY, glycogen degradation I; ARGORNPROST-PWY, L-arginine degradation; PWY-7323, superpathway of GDP-mannose-derived O-antigen building blocks biosynthesis; PWY-6123, inosine-5′-phosphate biosynthesis I; PWY-7200, superpathway of pyrimidine deoxyribonucleoside salvage; PWY-6609, adenine and adenosine salvage III; CALVIN-PWY, Calvin-Benson-Bassham cycle; PWY-6121, 5-aminoimidazole ribonucleotide biosynthesis I; PWY-6122, 5-aminoimidazole ribonucleotide biosynthesis II; PWY-6277, superpathway of 5-aminoimidazole ribonucleotide biosynthesis; PWY-7196, superpathway of pyrimidine ribonucleosides salvage; LACTOSECAT-PWY, lactose and galactose degradation I; PWY-5913, (partial TCA cycle obligate autotrophs); PWY-7184, pyrimidine deoxyribonucleotides de novo biosynthesis I; PWY-6163, chorismate biosynthesis from 3-dehydroquinate
Discussion
Previous reports indicate that the vaginal microbiome contributes significantly to female and neonatal health [19, 20]. In this study, we aimed to determine the overall composition of the vaginal microbiota in women with POI. The vaginal microbiota of both the POI and control groups were composed primarily of the phyla Actinobacteria, Bacteroidetes, and Firmicutes. The genus Lactobacillus was predominant, with low proportions of the genera Anaerococcus, Atopobium, Bifidobacterium, Enterococcus, Gardnerella, Peptoniphilus, Prevotella, Streptococcus, and Veillonella. These results are in accordance with previous observations [20, 21]. Increased patterns of alpha and beta diversity were observed in the POI group, which is consistent with the findings in women with primary ovarian failure [13].
Previous studies have shown that inflammatory and autoimmune responses are closely related to ovarian function and POI [6, 22]. In this study, compared to the control group, the genera Lactobacillus, Odoribacter, and Brevundimonas were significantly decreased and the genus Streptococcus significantly increased in the POI group. Lactobacilli can promote interleukin (IL)-22 secretion and prevent autoimmune diseases by stimulating the production of antimicrobial peptide [23] and can reinforce the mononuclear phagocytic response [24]. Some species of Odoribacter can induce Th17 cells and protective immunity by promoting IL-1β and IL-6 signalling [25]. Some Streptococcus species produce toxins that activate innate and adaptive host immune responses [26]. Streptococcus agalactiae (also known as group B Streptococcus) is a common bacterial infection during pregnancy, preterm birth, and neonatal infection. Specific Lactobacillus strains could serve as probiotics to prevent vaginal group B Streptococcus colonisation [27]. Moreover, Gardnerella vaginalis can activate NF-κB to promote tumour necrosis factor α secretion [28]; the inflammatory response induced by G. vaginalis can be inhibited by Lactobacillus [13]. Increases in Streptococcus, Gardnerella and a decrease in Lactobacillus species were observed in women with POI. The changes induced by the differing vaginal microbiome in the POI group might affect autoimmunity, which could be related to the development of POI.
Sex hormone levels can affect the defensive ability of the female genital tract and the resident vaginal microbiome during the reproductive years [29, 30]. Thus, changes in sex hormones play an important role in the vaginal microbiome [31]. Oestrogen promotes hyperplasia and increased glycogen production [32]; glycogen can then be converted into lactic acid by lactobacilli, the dominant bacteria in the vagina. This helps maintain the acidic environment of the vagina, inhibiting the growth of pathogens, and strengthening the immune system [33, 34]. In this study, the proportion of Lactobacillus was positively correlated with oestrogen levels, but negatively correlated with FSH levels. Significant decreases in Lactobacillus abundance and oestrogen levels and a significant increase in FSH levels were observed in women with POI. These findings are consistent with the characteristics of women in the initial stages of menopause, who have reduced oestrogen levels, increased FSH levels, and colonisation of a larger number of mixed bacteria due to the change in vaginal pH from acidic to weakly acidic [35, 36]. Moreover, AMH, FSH, LH, PRL, P, and testosterone levels were also related to some microbes, including Streptococcus, Odoribacter, Brevundimonas, Anaerococcus, Atopobium, Peptoniphilus, Prevotella, and Veillonella, increasing the evidence that an altered vaginal microbiota is associated with sex hormones. Accumulating evidence indicates that oestrogen can regulate POI-related symptoms, including GLU and lipid metabolism, bone formation, and inflammatory responses [37, 38], and GLU level was negatively correlated with genera Odoribacter and Prevotella in this study. In addition, PRL can inhibit FSH and gonadotropin-releasing hormone to promote fertility [39], which was positively correlated with Anaerococcus and Peptoniphilus. However, due to the limited data in this study, the mechanism underlying the relationship between these microbes and sex hormones cannot be fully clarified.
Ovarian function is known to be susceptible to damage due to galactose and galactose metabolite accumulation [40]. A previous study found that galactose suppressed the number of ovarian follicles and steroid secretion [41]. Galactose metabolites, including galactose-1-phosphate, galactitol, and uridine diphosphate galactose, play important roles in interfering with ovarian apoptosis and gonadotrophin signalling [40]. The functional analysis results showed that the activity of the LACTOSECAT-PWY pathway decreased significantly in women with POI compared to the control group. This demonstrated that galactose may accumulate in women with POI, damaging ovarian function. The ARGORNPROST-PWY pathway was significantly enriched in women with POI. L-arginine is associated with regulation of ovarian function [42, 43]. This may be unfavourable for ovarian function. In addition, DNA damage results in reproductive dysfunction by activating the mitochondrial apoptosis pathway [44]. Our results showed that many pathways related to ribonucleotide biosynthesis were altered in women with POI. Thus, dysbiosis of the vaginal microbiome in women with POI is closely related to ovarian function.
There are some limitations of this study. The sample size was small, the recruited participants were all from the same hospital, and we could only determine association, not causality. Thus, future studies should include a larger sample size and samples from multiple centres. Metagenome sequencing to study the microbiome at lower level, and transplantation of vaginal microbiome from women with POI to germ-free mice should be considered to investigate the potential causal mechanism.
In summary, this study demonstrated the dysbiosis of the vaginal microbiome in women with POI and confirmed that Lactobacillus is the predominant genus in the vaginal microbial community of women with POI. The changes in microbial communities are closely related to serum hormones, and the metabolic function of the microbes affects ovarian function. Our results lay a foundation for revealing the interaction between the vaginal microbiota and POI.
Availability of data and materials
The dataset supporting the conclusions of this article is available in the NCBI Sequence Read Archive database (SRA BioProject ID: PRJNA738630).
References
Luisi S, Orlandini C, Regini C, Pizzo A, Vellucci F, Petraglia F. Premature ovarian insufficiency: from pathogenesis to clinical management. J Endocrinol Investig. 2015;38:597–603. https://doi.org/10.1007/s40618-014-0231-1.
Fenton AJ. Premature ovarian insufficiency: pathogenesis and management. J Midlife Health. 2015;6:147–53. https://doi.org/10.4103/0976-7800.172292.
Sassarini J, Lumsden MA, Critchley HOD. Sex hormone replacement in ovarian failure – new treatment concepts. Best Pract Res Clin Endocrinol Metab. 2015;29:105–14. https://doi.org/10.1016/j.beem.2014.09.010.
Portman DJ, MLS G, Vulvovaginal Atrophy Terminology Consensus Conference Panel. Genitourinary syndrome of menopause: new terminology for vulvovaginal atrophy from the International Society for the Study of Women’s Sexual Health and the North American Menopause Society. Menopause. 2014;21:1063–8. https://doi.org/10.1097/GME.0000000000000329.
Bakhsh H, Dei M, Bucciantini S, Balzi D, Bruni V. Premature ovarian insufficiency in young girls: repercussions on uterine volume and bone mineral density. Gynecol Endocrinol. 2015;31:65–9. https://doi.org/10.3109/09513590.2014.958987.
Domniz N, Meirow D. Premature ovarian insufficiency and autoimmune diseases. Best Pract Res Clin Obstet Gynaecol. 2019;60:42–55. https://doi.org/10.1016/j.bpobgyn.2019.07.008.
Gupta A, Tiwari P. Premature ovarian insufficiency: a review. EMJ Repro Health. 2019. https://doi.org/10.33590/emjreprohealth/19-00041.
Mondul AM, Rodriguez C, Jacobs EJ, Calle EE. Age at natural menopause and cause-specific mortality. Am J Epidemiol. 2005;162:1089–97. https://doi.org/10.1093/aje/kwi324.
Goldmeier S, De Angelis K, Rabello Casali K, Vilodre C, Consolim-Colombo F, Belló Klein A, et al. Cardiovascular autonomic dysfunction in primary ovarian insufficiency: clinical and experimental evidence. Am J Transl Res. 2013;6:91–101.
Podfigurna-Stopa A, Czyzyk A, Grymowicz M, Smolarczyk R, Katulski K, Czajkowski K, et al. Premature ovarian insufficiency: the context of long-term effects. J Endocrinol Investig. 2016;39:983–90. https://doi.org/10.1007/s40618-016-0467-z.
Ravel J, Moreno I, Simón C. Bacterial vaginosis and its association with infertility, endometritis, and pelvic inflammatory disease. Am J Obstet Gynecol. 2021;224:251–7. https://doi.org/10.1016/j.ajog.2020.10.019.
Zwittink RD, van den Munckhof EHA, Leverstein-van Hall MA, Boers K, Molijn A, Knetsch CW, et al. The vaginal microbiota in the course of bacterial vaginosis treatment. Eur J Clin Microbiol Infect Dis. 2021;40:651–6. https://doi.org/10.1007/s10096-020-04049-6.
Wang J, Xu J, Han Q, Chu W, Lu G, Chan WY, et al. Changes in the vaginal microbiota associated with primary ovarian failure. BMC Microbiol. 2020;20:230. https://doi.org/10.1186/s12866-020-01918-0.
Guo T, Zheng Y, Li G, Zhao S, Ma J, Qin Y. Novel pathogenic mutations in minichromosome maintenance complex component 9 (MCM9) responsible for premature ovarian insufficiency. Fertil Steril. 2020;113:845–52. https://doi.org/10.1016/j.fertnstert.2019.11.015.
Bolyen E, Rideout JR, Dillon MR, et al. QIIME 2: reproducible, interactive, scalable, and extensible microbiome data science. Peer J. https://doi.org/10.7287/peerj.preprints.27295v2.
Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38:685–8. https://doi.org/10.1038/s41587-020-0548-6.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. https://doi.org/10.1186/gb-2011-12-6-r60.
Parks DH, Beiko RG. STAMP: statistical analysis of metagenomic profiles. In: Nelson KE, editor. Encyclopedia of metagenomics. Boston: Springer; 2015. p. 641–5. https://doi.org/10.1007/978-1-4899-7478-5_780.
Gupta S, Kakkar V, Bhushan I. Crosstalk between vaginal microbiome and female health: a review. Microb Pathog. 2019;136:103696. https://doi.org/10.1016/j.micpath.2019.103696.
Fettweis JM, Serrano MG, Brooks JP, Edwards DJ, Girerd PH, Parikh HI, et al. The vaginal microbiome and preterm birth. Nat Med. 2019;25:1012–21. https://doi.org/10.1038/s41591-019-0450-2.
Serrano MG, Parikh HI, Brooks JP, Edwards DJ, Arodz TJ, Edupuganti L, et al. Racioethnic diversity in the dynamics of the vaginal microbiome during pregnancy. Nat Med. 2019;25:1001–11. https://doi.org/10.1038/s41591-019-0465-8.
Huang Y, Hu C, Ye H, Luo R, Fu X, Li X, et al. Inflamm-aging: a new mechanism affecting premature ovarian insufficiency. J Immunol Res. 2019;2019:8069898. https://doi.org/10.1155/2019/8069898.
Miani M, Le Naour J, Waeckel-Enée E, Verma SC, Straube M, Emond P, et al. Gut microbiota-stimulated innate lymphoid cells support β-defensin 14 expression in pancreatic endocrine cells, preventing autoimmune diabetes. Cell Metab. 2018;28:557–572.e6. https://doi.org/10.1016/j.cmet.2018.06.012.
Ghadimi D, de Vrese M, Heller KJ, Schrezenmeir J. Lactic acid bacteria enhance autophagic ability of mononuclear phagocytes by increasing Th1 autophagy-promoting cytokine (IFN-gamma) and nitric oxide (NO) levels and reducing Th2 autophagy-restraining cytokines (IL-4 and IL-13) in response to mycobacterium tuberculosis antigen. Int Immunopharmacol. 2010;10:694–706. https://doi.org/10.1016/j.intimp.2010.03.014.
Xing C, Wang M, Ajibade AA, Tan P, Fu C, Chen L, et al. Microbiota regulate innate immune signaling and protective immunity against cancer. Cell Host Microbe. 2021. https://doi.org/10.1016/j.chom.2021.03.016.
Ramos-Sevillano E, Ercoli G, Brown JS. Mechanisms of naturally acquired immunity to Streptococcus pneumoniae. Front Immunol. 2019;10:358. https://doi.org/10.3389/fimmu.2019.00358.
Marziali G, Foschi C, Parolin C, Vitali B, Marangoni A. In-vitro effect of vaginal lactobacilli against group B Streptococcus. Microb Pathog. 2019;136:103692. https://doi.org/10.1016/j.micpath.2019.103692.
Vick EJ, Park HS, Huff KA, Brooks KM, Farone AL, Farone MB. Gardnerella vaginalis triggers NLRP3 inflammasome recruitment in THP-1 monocytes. J Reprod Immunol. 2014;106:67–75. https://doi.org/10.1016/j.jri.2014.08.005.
Vitali D, Wessels JM, Kaushic C. Role of sex hormones and the vaginal microbiome in susceptibility and mucosal immunity to HIV-1 in the female genital tract. AIDS Res Ther. 2017;14:39. https://doi.org/10.1186/s12981-017-0169-4.
Wira CR, Rodriguez-Garcia M, Patel MV. The role of sex hormones in immune protection of the female reproductive tract. Nat Rev Immunol. 2015;15:217–30. https://doi.org/10.1038/nri3819.
Xu J, Bian G, Zheng M, Lu G, Chan WY, Li W, et al. Fertility factors affect the vaginal microbiome in women of reproductive age. Am J Reprod Immunol. 2020;83:e13220. https://doi.org/10.1111/aji.13220.
Silva C, Rey R, Elena Nader-Macías M. Effects of estrogen administration on the colonization capability of lactobacilli and Escherichia coli in the urinary tracts of mice. Methods Mol Biol. 2004;268:387–99. https://doi.org/10.1385/1-59259-766-1:387.
Li J, McCormick J, Bocking A, Reid G. Importance of vaginal microbes in reproductive health. Reprod Sci. 2012;19:235–42. https://doi.org/10.1177/1933719111418379.
Reid G, Dols J, Miller W. Targeting the vaginal microbiota with probiotics as a means to counteract infections. Curr Opin Clin Nutr Metab Care. 2009;12:583–7. https://doi.org/10.1097/MCO.0b013e328331b611.
Brotman RM, Shardell MD, Gajer P, Fadrosh D, Chang K, Silver MI, et al. Association between the vaginal microbiota, menopause status, and signs of vulvovaginal atrophy. Menopause. 2018;25:1321–30. https://doi.org/10.1097/GME.0000000000001236.
Mirmonsef P, Modur S, Burgad D, Gilbert D, Golub ET, French AL, et al. Exploratory comparison of vaginal glycogen and Lactobacillus levels in premenopausal and postmenopausal women. Menopause. 2015;22:702–9. https://doi.org/10.1097/GME.0000000000000397.
Baker L, Meldrum KK, Wang M, Sankula R, Vanam R, Raiesdana A, et al. The role of estrogen in cardiovascular disease. J Surg Res. 2003;115:325–44. https://doi.org/10.1016/S0022-4804(03)00215-4.
Li JY, Chassaing B, Tyagi AM, Vaccaro C, Luo T, Adams J, et al. Sex steroid deficiency–associated bone loss is microbiota dependent and prevented by probiotics. J Clin Invest. 2016;126:2049–63. https://doi.org/10.1172/JCI86062.
Donato J Jr, Frazão R. Interactions between prolactin and kisspeptin to control reproduction. Arch Endocrinol Metab. 2016;60:587–95. https://doi.org/10.1590/2359-3997000000230.
Liu G, Hale GE, Hughes CL. Galactose metabolism and ovarian toxicity. Reprod Toxicol. 2000;14:377–84. https://doi.org/10.1016/S0890-6238(00)00096-4.
Campbell BK, Kendall NR, Onions V, Scaramuzzi RJ. The effect of systemic and ovarian infusion of glucose, galactose and fructose on ovarian function in sheep. Reproduction. 2010;140:721–32. https://doi.org/10.1530/REP-10-0185.
Battaglia C, Salvatori M, Maxia N, Petraglia F, Facchinetti F, Volpe A. Adjuvant L-arginine treatment for in-vitro fertilization in poor responder patients. Hum Reprod. 1999;14:1690–7. https://doi.org/10.1093/humrep/14.7.1690.
Battaglia C, Regnani G, Marsella T, Facchinetti F, Volpe A, Venturoli S, et al. Adjuvant L-arginine treatment in controlled ovarian hyperstimulation: a double-blind, randomized study. Hum Reprod. 2002;17:659–65. https://doi.org/10.1093/humrep/17.3.659.
Liu J, Yang M, Jing L, Ren L, Wei J, Zhang J, et al. Silica nanoparticle exposure inducing granulosa cell apoptosis and follicular atresia in female Balb/c mice. Environ Sci Pollut Res Int. 2018;25:3423–34. https://doi.org/10.1007/s11356-017-0724-5.
Code availability
Not applicable.
Funding
This work was supported by the Sanming Project of Medicine in Shenzhen (No. SZSM201612046), the Guangdong Provincial Administration of Traditional Chinese Medicine (No. 20201294), and the Internal Research Project of Shenzhen Maternity and Child Healthcare Hospital (No. FYA2018006). The funders had no role in the study design; data collection, analysis, and interpretation; decision to publish; or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
J Wu: Protocol/Project development, Manuscript writing. Y Ning: Protocol/Project development. L Tan: Data collection. Y Chen: Data collection. X Huang: Designed computational framework, Data analysis. Y Zhuo: Designed computational framework, Data analysis, Manuscript writing. J Wu and Y Ning have contributed equally to this paper, Y Zhuo is the corresponding author. All authors have discussed the results and contributed to the final manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was performed in line with the principles of the Declaration of Helsinki and was approved by the Ethics Committee of Shenzhen Maternity and Child Healthcare Hospital (SFYLS2020–005), Shenzhen, China.
Written informed consent was obtained from all the participants prior to enrolment.
Consent for publication
The authors affirm that patients signed informed consent regarding publishing their data.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, J., Ning, Y., Tan, L. et al. Characteristics of the vaginal microbiome in women with premature ovarian insufficiency. J Ovarian Res 14, 172 (2021). https://doi.org/10.1186/s13048-021-00923-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13048-021-00923-9