
Abstract
Pancreatic ductal adenocarcinoma (PDAC) remains one of the most lethal cancers worldwide, mainly due to its late diagnosis and lack of effective therapies, translating into a low 5-year 12% survival rate, despite extensive clinical efforts to improve outcomes. International cooperative studies have provided informative multiomic landscapes of PDAC, but translation of these discoveries into clinical advances are lagging. Likewise, early diagnosis biomarkers and new therapeutic tools are sorely needed to tackle this cancer. The study of poorly explored molecular processes, such as splicing, can provide new tools in this regard. Alternative splicing of pre-RNA allows the generation of multiple RNA variants from a single gene and thereby contributes to fundamental biological processes by finely tuning gene expression. However, alterations in alternative splicing are linked to many diseases, and particularly to cancer, where it can contribute to tumor initiation, progression, metastasis and drug resistance. Splicing defects are increasingly being associated with PDAC, including both mutations or dysregulation of components of the splicing machinery and associated factors, and altered expression of specific relevant gene variants. Such disruptions can be a key element enhancing pancreatic tumor progression or metastasis, while they can also provide suitable tools to identify potential candidate biomarkers and discover new actionable targets. In this review, we aimed to summarize the current information about dysregulation of splicing-related elements and aberrant splicing isoforms in PDAC, and to describe their relationship with the development, progression and/or aggressiveness of this dismal cancer, as well as their potential as therapeutic tools and targets.
Similar content being viewed by others

Introduction
Pancreatic ductal adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC) is the most common and aggressive type of tumor in the pancreas (80%). PDAC mortality rate is one of the highest of all cancers worldwide, and it is even higher in Europe and North America. It accounts for 4.7% of all cancer-related deaths, almost matching the number of new cases. According to a study involving 28 European countries, it is projected to surpass breast cancer as the third leading cause of cancer death by 2025 [1]. Low survival rates are associated with late diagnosis, the presence of metastasis and the development of drug resistance. PDAC risk factors include age, genetics (in around 10% of cases), tobacco and alcohol use, pancreatitis, and obesity, among others, which generally increase inflammatory pancreatic damage [2].
PDAC develops from pre-invasive lesions. These include cystic lesions such as intraductal papillary mucinous neoplasm (IPMN), mucinous cystic neoplasm (MCN), and intraductal tubulopapillary neoplasm (ITPN). However, the most common lesion is pancreatic intraepithelial neoplasia (PanIN), which is non-cystic. Cystic lesions can be diagnosed using imaging methods, but PanINs cannot be detected early or through methods other than microscopic examination [3]. The progression of PanINs encompasses various grades, advancing towards significant dysplasia. This process is marked by the loss of cell polarity and an enlargement of the nucleus. The transformation of pancreatic tissue has been linked to various mechanisms, including genomic instability and mutations. Multiple studies have demonstrated the presence of some of the most prevalent mutations in PDAC within pre-invasive lesions such as PanINs [4]. For example, it has been shown that the frequency of KRAS mutation increases as the disease advances to a higher grade of dysplasia, but it even appears in the early stages of the lesion. KRAS is the most commonly mutated gene in PDAC (95% of patients) and the main driver of tumorigenesis, with G12D its most prevalent codon mutation [5]. Other frequent mutations have been characterized in PDAC, including mutations in genes encoding tumor suppressor proteins, like CDKN2A, TP53 or SMAD4, or genes involved in essential cell processes, such as chromatin remodeling and DNA damage repair, likely contributing to increased genomic instability [6]. Another cause of DNA damage is telomere shortening, which seems to be an early event in pancreatic lesion. Nevertheless, although the genomic landscape of PDAC is well characterized and most frequent mutations have been proposed as therapeutic targets for PDAC on multiple occasions, current therapies in PDAC do not include these genetic alterations as targets [7, 8]. Along with the described genetic alterations, there are additional factors that reside at different hierarchical levels and contribute to PDAC malignancy. The histological/tissue features comprise a critical level in this tumor type because of the nature, volume, and cellular composition of the stromal compartment, increasing heterogeneity and hindering drug delivery. Likewise, the inflammatory component that accompanies pancreatic damage keeps developing throughout progression to adenocarcinoma. Besides cancer cells, PDAC contains several relevant cell types that comprise an intricate microenvironment, including different classes of fibroblasts, pancreatic stellate cells, cancer stem cells, macrophages, infiltrated lymphocytes, and vascular cells [9]. These cells have been shown to communicate with cancer cells, and their interaction is necessary for tumor progression, promoting tumor growth, angiogenesis, metastasis, and driving drug resistance [10].
Despite the remarkable advances achieved regarding the molecular makeup of PDAC, the number of patients who survive this pathology has only modestly improved in the last years, with a dismal 5year survival rate below 11–12%. Therapeutic approaches are limited, being surgery the only “curative” option, only effective in early diagnosed localized cases (15–20%). In the case of locoregional stage, neoadjuvant treatment may be used to make the tumor removable; while in the case of metastatic disease, surgery is not an option and only chemotherapy is offered. Moreover, chemotherapeutic treatments are limited, and although the latest combinations, including FOLFIRINOX (folinic acid, 5-fluorouracil, irinotecan, and oxaliplatin) or gemcitabine plus nab-paclitaxel, have extended progression-free survival, some treatment regimens are toxic and overall survival remains poor (< 12 months) [11]. For all these reasons, novel research avenues are being explored to develop alternative approaches, more effective treatments and early biomarkers [12]. In this context, the splicing process and its dysregulation has emerged as potential novel molecular tools to combat PDAC.
The spliceosome and the splicing process
Splicing is a complex cellular mechanism by which the immature or precursor RNA is processed, removing the sequences that will not be part of the final RNA, or introns, and binding together the exons that form the mature RNA [13, 14]. However, most of the genes (> 95%) do not undergo this simple cut and paste process, also known as constitutive splicing, but rather they undergo an intricately regulated process, called alternative splicing [13, 15, 16]. This phenomenon allows the generation of different combinations of final sequences through the inclusion and exclusion of concrete groups of exons, which results in a variety of mature RNA transcripts from the same precursor, termed splicing variants or isoforms, that may carry out different or even opposite functions [17]. This is an essential cellular process that ensures an appropriate regulation of gene expression as it enables an increase in the variety of genes and thereby enhances the versatility of the genome [16]. For all these reasons, the accurate regulation of the splicing process is crucial for the correct development and homeostasis of the cell and the organism [18]. The process of splicing and its delicate regulation is carried out by the spliceosome, a ribonucleoproteic complex that recognizes specific RNA sequences to precisely localize the introns and cut them, and subsequently bind the adjacent exons [19]. In mammals, there are two different spliceosomes that act separately: the major spliceosome that processes more than 99% of the introns, and the minor spliceosome that acts over a small and specific set of introns [20]. Accordingly, introns are classified as U2-type (or -dependent, GT-AT) and U12-type (or -dependent, AT-AC), depending on the spliceosome that processes them or the flanking sequences [21]. Both spliceosomes consist of a main core of small nuclear RNAs (snRNAs), known as RNU1, RNU2, RNU4, RNU5 and RNU6 for the major spliceosome; and RNU11, RNU12, RNU4ATAC and RNU6ATAC (RNU5 is present in both), for the minor. These snRNAs are joined to proteins forming small nuclear ribonucleoproteins (snRNP; U1-U6) [19, 20]. In addition, the spliceosomes closely interact with the splicing factors, a diverse set of more than 300 molecules that complete the splicing machinery, helping the snRNPs to select and process the precise sequences, and taking part dynamically in every step of the process, participating in both general tasks as well as very specific events [22, 23].
The splicing process has been classically investigated in simple research models easier to study than human-based systems, like yeast, but the key steps are very well conserved in mammals. Summarizing the explanation by Matera and Wang in 2014 [24] and other studies [21, 25], U1 and U2 recognize and bind to 5’ and 3’ splice sites of the pre-mRNA, respectively. Next, U2 recognizes sequences in the so-called branch point and interacts with U1, forming the pre-spliceosome. In this step, the intron takes the form of a loop, which is called lariat. Then, the preassembled U4-U5-U6 complex is recruited, and several conformational changes take place to form a catalytically active complex, resulting in the U2/U6 structure that catalyzes the splicing reaction. In this step, U1 and U4 are released from the complex. At this point, the first catalytic step is carried out, cutting the binding between the first exon and intron-exon lariat intermediate. Finally, after some further conformational changes, the second catalytic step leads to the separation of intron and second exon and the binding of both exons, leaving the post-spliceosomal complex with the intron lariat free. Finally, U2, U5 and U6 are released. All the described steps are firmly regulated by several spliceosome proteins, which ensure that the cuts and bindings are correct, making possible the sequence recognition and putting together and separating the other components.
Typically, the introns of mammals are long, and present several decoy splice sites that must not be spliced [15]. As mentioned earlier, alternative splicing is based on the inclusion/exclusion of selected sequences; therefore, a precise regulation is needed to correctly splice each sequence. To this end, cis-regulatory elements are distributed through the RNA, known as splicing regulatory elements, and, depending on their function and location, are classified in exonic/intronic enhancers/silencers (ESE, ISE, ESS and ISS, respectively) [15, 16, 26]. Those sequences recruit trans-regulatory elements, the splicing factors, which will suppress or activate steps of the splicing process (Fig. 1A). However, these events are completely dependent on the context, since the same factor may be a splicing enhancer and a splicing silencer if it binds to an enhancer or silencer element [16, 21, 26].

Splicing process. (A) Summary of the process of splicing, showing how the spliceosome machinery binds RNA in different regulatory sequences (exonic/intronic splicing enhancer [ESE/ISE] and exonic/intronic splicing silencer [ESS/ISS]), cuts the intron out and pastes the flanking exons together. (B) Different possibilities of alternative splicing events
Furthermore, there are additional possibilities for splicing regulation. For instance, the structure of the precursor RNA may alter the accessibility to regulatory domains or even the spliceosome complexes [15]. In addition, the activity of the splicing machinery is finely regulated through modulation of its components, by gene expression regulation via transcription factors, miRNAs, epigenetics, etc [27,28,29], or posttranslational modifications, such as phosphorylation or acetylation [30,31,32] that may affect their location or activity.
This complex regulation allows the correct progression of the splicing process, including the variations that cause alternative splicing. Specifically, there are five different types of alternative splicing: 1) cassette exon skipping, an exon is excluded together with the two flanking introns; 2) mutually exclusive exons, two exons that cannot be included together, one of each is excluded in two different isoforms; 3) intron retention, there is no cutting in the intron resulting in its inclusion in the mature RNA; 4) alternative 3’ splice site and 5) alternative 5’ splice site, the exon is cut in a different site thus it is not fully included in the final RNA [24, 33, 34] (Fig. 1B). Taken together, all this information demonstrates the great complexity of the splicing process and underscores its relevance in controlling the normal functions of the cell.
Splicing is altered in cancer
The precise understanding of the molecular mechanisms underlying the process of alternative splicing has lagged behind that of other fundamental processes, such as transcription or translation. However, evidence was soon found to suggest that alterations in the splicing process were associated with human diseases, and particularly to tumor development and cancer [35]. Actually, there is now ample consensus that dysregulated splicing, originated by mutations or changes in the splicing machinery and/or the abnormal profile of splicing events, is a key phenomenon contributing to all cancer types studied so far, and thus dysregulated splicing participated in all cancer hallmarks, as has been recently reviewed in detail [36, 37]. Nevertheless, while the altering of splicing can be regarded as a common feature in cancer, the specific changes that it involves are specific for each tumor type and, therefore, should be precisely examined in detail in the appropriate samples and representative experimental models. Accordingly, in the present review, we have focused on the dysregulation of splicing in pancreatic cancer.
Splicing dysregulation in PDAC
Despite the impressive growing list of alterations in genes and regulatory mechanisms that have been described to date, these are still insufficient to provide an effective therapeutic strategy to battle PDAC [12]. In this regard, an increasing number of studies indicate that the dysregulation of splicing can play an important role in PDAC progression. Such dysregulation may arise from mutations or alterations in the expression levels of specific components of the splicing machinery. Additionally, alterations in the relative proportions of splice variants, and even the emergence of aberrant variants, might contribute to the intricate landscape of PDAC development (Fig. 2). In the following subsections we will discuss about the alterations in the splicing process that have been described in PDAC.

Splicing alterations in PDAC. Splicing components have been shown to be mutated and their expression dysregulated in PDAC, leading to the disequilibrium in the isoforms or the appearance of aberrant isoforms that cause or promote several cancer features
Dysregulation of splicing machinery components
Alterations in the expression of components of the molecular machinery that operate and control the splicing process have already been described in an extensive list of diseases, including many tumor pathologies [35, 38,39,40]. In PDAC, pioneering studies by Carrigan et al. evaluated expression levels of selected genes in human pancreatic cancer cell lines, discovering a downregulation of 30% of spliceosomal genes, revealing a clear repression of splicing machinery components [41]. More recently, Wang et al. validated some of these changes in PDAC human samples, establishing an expression signature of the spliceosome and splicing regulatory genes that discriminated with high accuracy between tumor and healthy samples [42]. Interactions and implications of alternative splicing in PDAC pathogenesis have been subsequently reviewed [43], emphasizing the promising prospect of dissecting its role in this pathology.
Further work has provided a deeper understanding of the dysregulations in the expression of the splicing machinery in PDAC, revealing that they frequently consist in an altered expression of spliceosome components and/or splicing factors (Table 1), which usually leads to an imbalanced profile of splice variants and/or the appearance of aberrant variants. Considering that the correct functioning of the splicing process regulates the overall balance of RNA variants in the cell, it is not to be unexpected that changes in the expression of splicing-related proteins could dramatically modify cell homeostasis, including key processes in PDAC evolution. This is the case of the overexpression of splicing machinery components that are associated with proliferation and apoptosis, such as SRPK1 [44, 45], CLK1 [46], HNRNPK [47], PTBP3 [48], HNRNPL [49], HNRNPA2B1 [50] and ESRP1 [51]; with metastasis and invasion, such as SF3B1 [52], CLK1 [46], PRPF40A [53], ESRP1 [51], SRSF6 [54] and RBFOX2 [55]; with the acquisition of chemotherapy resistance, such as SRPK1 [44, 45], SRSF1 [56], PTBP1 [57] and SRSF3 [58]; with autophagy, such as SFPQ [59] and PTBP3 [48]; with ubiquitination and degradation of some proteins, such as HRNPU [36]; or with the activation of relevant pathways in PDAC, such as the signaling mediated by KRAS, driven by HNRNPK [47], or the MAP kinases pathway by METTL3 [60], or the pancreatitis and KRASG12D-mediated cancer promoted by SRSF1 [61].
Although current evidence supports that overexpression of splicing machinery components is more frequent in PDAC than initially observed, there are numerous examples where such components are repressed, indicating a putative tumor suppressor role. For example, ESRP1 regulates the expression pattern of FGFR-2 isoforms, attenuating cell growth, migration, invasion, and metastasis in PDAC cell lines [51]. Likewise, SRSF6 hinders pancreatic cancer cells migration and invasion by regulating ECM1 alternative splicing isoforms [54]. Further, RBM10 promotes the appearance of the TERT splicing isoform TERT-S, which in contrast with TERT-FL, is not able to maintain telomeres [62]. SF3B4 inhibits the growth and migration of cancer cells preventing STAT3 phosphorylation [63]. In the same line, downregulation of HNRNPM and RBM5 is associated with an increase in tumor aggressiveness. Specifically, HNRNPM is implicated in the adaptation to a hypovascular environment [64]; while RBM5 expression inversely correlates with KRAS levels and is associated with clinicopathological features and appears to promote tumor progression [65]. Splicing factor CELF2 is downregulated in PDAC and associated to PDAC progression, where its downregulation affects the splicing pattern of CD44, thereby regulating endoplasmic reticulum-associated degradation [66].
Mutations in splicing factors in PDAC
Mutations in genes specifically involved in the splicing process are increasingly recognized as a source of pathological effects in a range of diseases, including cancer [17, 35]. These types of mutations are particularly frequent in pathologies such as acute myeloid leukemia or myelodysplastic syndromes, where they are tightly linked to etiology and offer therapeutic opportunities [67,68,69]. Whole-exome sequencing of PDAC has revealed a number of mutations in key oncogenes and tumor suppressor genes, as KRAS or TP53 [70], which are known to be among the most commonly mutated genes in PDAC [4]. Nonetheless, those studies also identified mutations, although with lower frequency, in genes involved in other essential processes including splicing (Table 2), which conferred a higher tumor heterogeneity in PDAC. Genomic studies have also identified recurrent mutations affecting the early components of the RNA splicing machinery, such as SF3B1 [6]. Mutations in SF3B1 are the most common across multiple tumor types, mainly found in myelodysplastic syndrome [71, 72] and other hematologic malignancies, uveal melanoma and breast cancer [73, 74], where its high mutational frequency altered the capacity of the spliceosome to recognize the pre-RNA pattern [25, 75]. SF3B1 mutations are generally heterozygous, major hotspots identified matched with codon positions K700 and R625, which correspond with deleterious mutations [76,77,78]. However, SF3B1 mutations differ between each pathology, suggesting context-depending functional differences. For example, it has recently been described that SF3B1 K700E mutation increased glycolysis and the Warburg effect in PDAC, by promoting aberrant splicing of PPP2R5A and therefore activating c-Myc signaling, a positive regulator of glycolysis [79]. Thus, while the knowledge on the effects of SF3B1 mutations in PDAC is still limited, it is worth pointing out that several such mutations have been identified in a small but appreciable percentage of patients, such as G740E, N763S, K843R [80], P342T [7], K700E, L773R [6], K700E, Q699_K700delinsHE, N763S, K741K [81].
In line with the findings on SF3B1, mutations have also been described in other splicing factors like U2AF2, which is involved in pre-RNA branch site (BS) binding; SRSF1, a SR protein that generally promotes exon inclusion [42, 82]; PABPC1, required for poly(A) shortening, the first step in RNA decay [4, 83]; or RBMX, a RNA-binding protein that plays a crucial role in alternative splicing of several pre-RNAs [84].
Collectively, these studies provide compelling evidence that mutations and particularly altered expression in specific components of the splicing machinery are a common feature in PDAC, and that such dysregulations often result in pathological consequences. These observations also point to the aforementioned alterations as potential targets for therapeutic intervention, which is already being exploited via diverse strategies, as discussed below. Nevertheless, the splicing machinery, which integrates all spliceosome components and their associated splicing factors, is extraordinarily complex and there is still much to be learned on its precise expression and regulation in PDAC. A comprehensive understanding of its abnormal functioning will likely help to better comprehend the development and progression of PDAC and will facilitate the discovery of new molecular targets and tools.
Alterations in splicing variants in PDAC
Defects in alternative splicing often result in the appearance of abnormal splicing variants that can play an oncogenic function by conferring advantages to cancer cells. Observations from early studies on alternative splicing in PDAC, employing expression microarray techniques, prompted further analysis that applied more sophisticated bioinformatic approaches to explore the pattern of splicing events and signatures in PDAC cell lines [41] and human tissue [42, 85]. The landscape of alternative splicing in PDAC shows that the most common alterations in the protein-coding genes are skipped exon and alternative first exon, followed by intron retention [42].
In the last two decades, numerous studies have aimed to achieve a deeper understanding of the precise regulation of alternative splicing of individual genes and its mechanistic basis and pathological implications in PDAC (Table 3). A case as paradigmatic as intricate is the study of CD44, a multifunctional cell surface glycoprotein involved in structural and functional roles in cell-cell and cell-matrix interactions. The standard isoform of CD44 (CD44s or CD44h) only contains the five first exons [1,2,3,4,5] and last five exons [16,17,18,19,20,21], while the alternative variants CD44v have variable exons (v1-v10) that are alternatively spliced and incorporated between the exons 5–16, conditioning its final structure and thus its biological role [86]. The CD44 variants CD44v2 and CD44v6, can be detected in human PDAC tissue by immunohistochemistry, where their expression is connected to an increase in mortality rate [87,88,89]. Recently, Zhao et al. delved into the potential role of CD44 isoforms in PDAC cell lines, linking these to an EMT phenotype and higher invasiveness and chemoresistance features [90]. Another study by Zhu et al. described that CD44v3 is associated with poor prognosis in PDAC, with its generation being regulated by splicing factor U2AF1 [91]. A study more focused on metastasis by Xie et al. showed that isoform CD44v6 is essential for liver fibrosis and metastasis from PDAC and could be used as metastasis and prognosis biomarker [92]. Actually, it was demonstrated that peptide inhibitors of this isoform block tumor growth and metastasis in rodent models of PDAC [93]. Thus, although it is still unknown whether and how expression of CD44 variants specifically affect the cellular function of PDAC cells in vivo in patients, alternative splicing of this gene and their variant products comprise likely actionable targets and tools in PDAC.
Splicing variants of relevant receptors have also been described in PDAC. An aberrant variant of secretin receptor (encoded by the SCTR gene) was found, where the third exon is spliced out and therefore residues 44–79 from the NH [2]-terminal tail are eliminated, blocking secretin binding, and thus prompting tumor growth and progression [94, 95]. Moreover, the potential of the secretin receptor variant as an early diagnostic serum biomarker has been proposed [96]. Likewise, an aberrant variant of the cholecystokinin and gastrin receptor, CCKR2, which retains the fourth intron, was identified in PDAC, but was absent in normal pancreas. This variant is constitutively active, may contribute to pathological features in vitro, and it might be associated to a polymorphism of U2AF35, affecting the splicing regulation of this receptor [95]. Furthermore, Ryberg and collaborators reported three novel CCKR2 splice-forms in PDAC, different from the better known CCK2i4svR variant, which might have similar functions [97]. Another example is the prolactin receptor, PRLR, that has been previously linked to carcinogenesis [98]. This gene undergoes alternative splicing in PDAC, allowing for the formation of several splicing isoforms that differ from each other in the intracellular domain and thus they promote the activation of different downstream signaling pathways [99]. The most abundant and best-known isoform is PRLR-LF, by which prolactin mainly transmit its signals. In contrast, the short isoform, PRLR-SF, is not as well understood. A recent study demonstrated that PRLR-SF reduces nucleotide synthesis by inhibiting the pentose phosphate pathway (PPP) through the NEK9-Hippo pathway in PDAC cells and in xenografted tumors in mice, hindering proliferation and tumor growth. PRLR-SF regulates the PPP pathway by reducing the expression of two rate-limiting enzymes G6PD (Glucose-6-phosphate dehydrogenase) and TKT (transketolase). PPP generates both pentose phosphate for nucleic acid synthesis and NADPH for fatty acid synthesis, being a key pathway for cell proliferation which is also upregulated in PDAC cancer stem cells (CSCs) [100]. Therefore, PRLR-SF might play an important role in metabolic reprograming, thus preventing PDAC tumor progression [101].
Fibroblast Growth Factor Receptors comprise a family of tyrosine kinase receptors (FGFR1-4), whose presence, signaling, and therapeutic potential in PDAC has been recently reviewed [102]. Early work demonstrated the existence of different splicing isoforms for FGFR1, 2 and 3 in PDAC [103], where their differential expression was linked to tumor biology. Thus, whereas FGFR1-IIIb and FGFR1-IIIc isoforms are mainly expressed in epithelial and mesenchymal cells, respectively, they are co-expressed in PDAC cells, promoting tumorigenicity by modulating cell proliferation, adhesion, and movement, possibly via activation by FGF5 [104,105,106,107,108].
Also related with epithelial cells are mucins, which protect and lubricate the ducts and are involved in the differentiation and renewal of the epithelium and the modulation of cell adhesion, immune response, and cell signaling. The expression of mucin subtypes and their splice variants are used to classify PDAC in four different subtypes, which were differentially associated to patient survival [109]. Specifically, MUC4 presents several splicing variants, differing in the lack of exons 0–21, being MUC4/Y and MUC4/X [110] the best known isoforms. Interestingly, MUC4 isoforms are mainly expressed in PDAC and correlated with tumor malignancy, while the canonical isoforms are not detectable in normal pancreas [111,112,113].
The BCL2 family is composed by a number of proteins that play critical roles in apoptosis. Alternative splicing of one of its members, BCL2L1, results in the production of two variants with opposite functions, BCL-xL (anti-apoptotic) and BCL-xS (pro-apoptotic), due to the retention/lack of exon 2, respectively. In PDAC, BCL2 dysregulation has been associated with apoptosis resistance, due to BCL2L1 anti-apoptotic isoform overexpression in human tumor tissue [114]. Furthermore, its expression has been related to the progression to high-grade PanINs in mice [115]. The presence of specific BCL2 isoforms is differentially regulated by several splicing factors, where HNRNPF, HNRNPH, KHDRBS1, RBM11, or RBM25 promote the short variant, whereas SRSF1, SRSF9, or SF3B1 promote the longer variant [116]. In PDAC, this regulation has been examined in model cell lines, where the role of SF3B1 in the 5’splice site activation of BCL-xS has been confirmed [52, 117].
Tissue Factor (TF) is a glycoprotein primarily involved in the blood coagulation cascade. Its alternative splicing isoform, known as asTF, excludes the fifth exon and exhibits low prothrombogenic potential [118]. Regulation of asTF has been linked with several splicing factors, specifically with the SR family: SRSF6. In PDAC, asTF has been identified in tumor tissue, correlating with tumor infiltration. Moreover, in PDAC cell lines, asTF promotes tumor vascularization and tumorigenesis by different pathways, such as EGFR and EMT [119]. Likewise, asTF plasmatic levels are found at higher levels in PDAC patients than in healthy subjects, suggesting a potential use as a biomarker [120].
The vascular endothelial growth factor A (VEGFA) gene has 8 exons that can be alternatively spliced in multiple ways. Isoforms generated differ from each other in their affinity for binding sites, tissue localization, and their capacity to be diffusible [121]. VEGFA was previously described as a potential biomarker for benign pancreatic serous cystic neoplasm, which could help differentiate them from other types of lesions that can evolve into PDAC, like intraductal papillary mucinous neoplasms and mucinous cystic neoplasms [121]. Recently, it was observed that VEGFA spliced isoforms show different expression levels in normal pancreas, benign pancreatic serous cystic neoplasms, mucinous cystic neoplasms and intraductal papillary mucinous neoplasms [122].
KRAS mutations are prevalent in more than 95% of pancreatic cancers [123]. The KRAS gene encodes two splicing isoforms, KRAS4A and KRAS4B, products of alternative splicing of the fourth coding exons 4 A and 4B, being mutually exclusive. While KRAS4B is one of the most studied oncogenes, the role of KRAS4A is much less known. In PDAC, both isoforms are detectable, but their specific role has not yet been elucidated [52], although it may parallel that found in colorectal carcinoma, where KRAS4A has been associated to a suppressive and pro-apoptotic activity, while KRAS4B would play an anti-apoptotic effect [124].
Study of EMT-related alternative splice events unveiled that specific splice events from TMC7 and CHECK1 were associated with metastatic PDAC. Additionally, the inclusion of exon 17 of TMC7 was associated to poor prognosis in PDAC, meanwhile its knockdown reduced tumor-related properties [125]. In addition, aberrantly spliced variants have also been described and their role examined in PDAC. For example, while searching genomic variants that could be linked to splicing alterations, an allele was found to promote the generation of a truncated splice variant of the Elongator Acetyltransferase Complex Subunit 2 (ELP2). This aberrant variant acts as tumor suppressor for PDAC by blocking STAT3 oncogenic pathway [126].
As the precision and breadth of RNA sequencing approaches improve and the analysis of splicing receives more attention, the discovery of novel variants and the study of their potential pathogenic role advances further in PDAC will certainly heighten.
Splicing modulation for therapeutic benefit
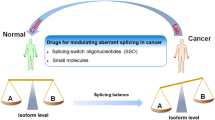
It is now widely accepted that splicing alterations can play important roles in the development and progression of cancer. However, splicing dysregulation can also influence tumor treatment response, as certain aberrant variants modify cellular functions leading to chemotherapy and targeted therapy resistance. Nonetheless, the pathogenic role of altered splicing also provides novel opportunities to tackle cancer, by designing and developing strategies focused on counteracting the effects of splicing errors and employing splicing dysregulation as an actionable therapeutic target. The following are some of the strategies that are being currently applied (Fig. 3).

Splicing components as therapeutic targets. A number of techniques have been developed to chemically regulate splicing, in order to avoid the harmful effect of its dysregulation. Those techniques include chemical inhibitors of the core (top panel), regulators of splicing factors, like kinases inhibitors (middle panel), and oligonucleotides that bind to regulator sequences (bottom panel)
Targeting the splicing core
The widespread alteration of splicing in cancer, among other pathologies, has prompted the development and testing of different types of molecules capable of interacting with specific elements of the spliceosome core and modulating their functioning. Three of these compounds, representative of different chemical natures, are Spliceostatins, Pladienolides and Herboxidienes. Some of these share similar mechanisms aimed at inhibiting SF3B1 and, consequently, interfering with the RNU2 complex, destabilizing it and preventing the transition of the spliceosome complex. SF3B1 is known to physically interact with HIF1α and induce hypoxia, promoting PDAC malignancy [127]. The potential clinical utility of these SF3B1 targeting molecules and their derivates has been demonstrated in several studies. E7107 compound, a Pladienolide B derivative [128], and H3B-8800 [129], a SF3B complex modulator able to kill spliceosome-mutant epithelial and hematologic tumor cells, have been tested in preclinical assays and are currently under clinical evaluation in a phase I study (NCT02841540). In fact, Pladienolide B has been tested in a preclinical PDAC study, demonstrating its capability to decrease multiple cancer features in cells, zebrafish, and mice models by altering relevant signaling pathways and splicing events, and reducing CSC stemness, making CSCs more sensitive to chemotherapy treatment [52]. However, further clinical trial efforts will be required to confirm the toxicology, safety, and potential benefit of compounds targeting the splicing machinery.
Targeting splicing regulatory elements
An alternative approach to manipulate and/or reverse splicing alterations, without blocking the spliceosome machinery core, is based on targeting regulatory proteins that modulate splicing. The use of these splicing regulators could be directed to mutated or altered molecules involved in pathological processes.
Phosphorylation and dephosphorylation of proteins by kinases represent a pivotal regulatory mechanism for multiple biological processes like metabolism, transcription, cell cycle progression, cell movement, apoptosis, and differentiation [130,131,132,133]. Thus, the potential of kinases as therapeutic targets has received considerable attention. Alternative splicing is also regulated by kinases that phosphorylate/dephosphorylate splicing factors, like the SR proteins [134], serving as a signal of nuclear localization and facilitating interaction with other splicing factors. This phosphorylation can be performed by SR protein kinases (SRPKs), topoisomerase 1 (TOP1), protein kinase B (PKB/AKT), NIMA-related kinases (NEK2), PRP4 kinase (PRP4K), dual-specificity tyrosine phosphorylation-regulated kinase 1 A (DYRK1A) [135, 136], cAMP-dependent protein kinase (PKA) [137, 138], or by the family of cdc-like kinases (CLKs) [139]. Thus, over the last decade, an increasing number of studies have revealed that dysregulation of splicing kinases has an important role in tumorigenesis and therapeutic response [140], as it is the case of SM08502, which has been shown to reduce Wnt pathway signalling by inhibiting CLK activity and SR family phosphorylation, leading to the disruption of the spliceosome activity. A Phase 1 clinical trial has been launched to evaluate the safety and pharmacokinetics of orally administered SM08502 in patients with advanced solid tumors [141].
Altogether, these studies emphasize the great potential of modulating splicing regulators for the treatment of cancer. Interestingly, these drugs are already being tested in clinical trials, particularly on oncohematological diseases, also in certain solid tumors. Of particular interest, Bromodomain and Extra-Terminal motif, BET, protein family inhibitors, such as ZEN-3694 are currently evaluated in triple negative breast cancer (clinical trial no. NCT03901469), and metastatic castration-resistant prostate cancer (NCT02705469, NCT02711956) [142]; and OTX015, which was proposed to be used in solid tumors (NCT02259114), and the results on leukemia and lymphoma have been reported [143, 144]. Additionally, there are also core spliceosome inhibitors, such as the case of H3B-8800, a Pladienolide-B derived SF3B1 inhibitor, which specifically targets cancer cells harboring splicing factor mutations [129], which is currently being tested on leukemia and myelodysplastic syndromes, MDS, (NCT02841540) [145]. GSK3368715 is a PRMT1 inhibitor, which regulates numerous nuclear ribonucleoproteins involved in the splicing process [146], and has been used in solid tumors and diffuse large B-cell lymphoma (NCT03666988). Similarly, the PRMT5 inhibitors, JNJ-64619178, GSK3326595 and PF06939999, modify snRNP-associated Sm proteins [147] and are currently being used in solid tumors, non-Hodgkin lymphoma and MDS (NCT03573310, NCT03614728/NCT02783300/NCT04676516 and NCT03854227, respectively). In the same line, the anti-cancer sulfonamide compound E7820, which degrades the splicing factor RBM39 is being assessed in MDS, acute myeloid leukemia (AML), or chronic myelomonocytic leukemia (CMML) (NCT05024994), which have shown acceptable safety [148]. Moreover, the ATR inhibitor Ceralasertib is being tested in MDS and CMML (NCT03770429), although it does not directly affect splicing, those patients harboring splicing factor mutations are more sensitive to this treatment [149]. Despite the promising results of these clinical trials, the use of splicing inhibitors in PDAC has not yet been reported.
Oligonucleotides
Another promising approach to target defects and alterations related to the splicing process is based on the use of short antisense oligonucleotides (ASO), which act by blocking the interaction between proteins and RNAs or between two RNAs. Splice-switching antisense oligonucleotides (SSOs) are nucleotides composed by 15–30 synthetic nucleotides or analogues, chemically modified to avoid enzymatic degradation of the target RNA, which are able to bind specifically to a target complementary sequence and thereby block the binding between splicing factors and pre-RNA [150]. Oligonucleotide therapy has already been approved by the FDA to treat certain diseases, such as Spinal Muscular Atrophy [151]. Their use in cancer is under evaluation [152], after promising preclinical studies showing their potential in various tumor pathologies [153, 154].
Regarding PDAC, pre-clinical in vitro studies showed that OGX-427, an antisense nucleotide complementary to HSP27 (Heat shock protein 27), inhibits proliferation, induces apoptosis, and enhances gemcitabine chemosensitivity in the MIAPaCa-2 PDAC cell line [155]. In fact, a phase II clinical trial in patients with metastatic pancreatic cancer was conducted to prove the efficacy of OGX-427 (Apatorsen; NCT01844817), comparing its effect plus/either gemcitabine/nab-paclitaxel compared to placebo. Despite pre-clinical data showing efficacy in pancreatic cancer cell lines, the addition of Apatorsen to chemotherapy did not improve outcomes in this clinical trial [156]. This and other related studies support the feasibility of using ASO as a tool to modulate splicing-related defects in PDAC, and in fact, there is experimental work underway in this direction.
Conclusion and outlook
PDAC is a highly lethal cancer, often diagnosed at advanced stages. Despite considerable research efforts to find novel therapies, groundbreaking advances remain elusive. The aim of this review was to emphasize the molecular complexity of alternative splicing and the relevance of its alterations in PDAC. Recent studies have revealed significant mutations and alterations in the expression levels of key components of the splicing machinery and splicing variants in PDAC. These alterations are tightly associated with pivotal clinical and molecular features of tumor development or progression.
Future studies should aim to achieve a more comprehensive and precise understanding of splicing and its related constellation of molecular components and machineries. To achieve this goal, newly developed technical and methodological strategies should be applied enabling to dissect and modulate splicing components in appropriate models. This approach will leverage this field as an alternative source of omics data to identify new biomarkers and to uncover previously hidden therapeutic targets. Also, the recent use of novel approaches to target splicing (e.g., anti-splicing drugs and oligonucleotides) will likely increase the number and potential extent of therapeutic opportunities.
However, to date, most available studies on this subject have been conducted using established cell lines or resectable tumors. Consequently, they may not fully represent the actual altered spliceosomic landscape in the most vulnerable population of metastatic patients, in whom splicing likely undergoes a more advanced mutational evolution from primary to metastatic tumors. Thus, further studies should focus on this metastatic patient population, through the analysis of metastatic tumor biopsies or with circulating tumor cells (CTC, a useful surrogate model of the metastatic tumor) allowing to examine differences with the primary tumors. These studies may require the use of Patient-Derived Xenografts (PDXs), CTC-derived PDXs, and organoids from metastatic biopsies to address methodological challenges.
Ultimately, the overarching goal is to understand the mechanistic underpinnings of the splicing machinery and dissect its resulting outcomes, the splicing variants. This will allow to obtain a fully detailed spliceosomic landscape in PDAC, and this information may potentiate the development of innovative tools and provide novel avenues to tackle this incurable cancer.
Data Availability
Not applicable.
Abbreviations
- PDAC:
-
Pancreatic ductal adenocarcinoma
- IPMN:
-
Intraductal papillary mucinous neoplasm
- MCN:
-
Mucinous cystic neoplasm
- ITPN:
-
Intraductal tubulopapillary neoplasm
- PanIN:
-
Pancreatic intraepithelial neoplasia
- FOLFIRINOX:
-
Folinic acid, 5-fluorouracil, irinotecan, and oxaliplatin
- snRNAs:
-
Small nuclear RNAs
- snRNP:
-
Small nuclear ribonucleoproteins
- mRNA:
-
Messenger RNA
- ESE:
-
Exonic splicing enhancer
- ISE:
-
Intronic splicing enhancer
- ESS:
-
Exonic splicing silencer
- ISS:
-
Intronic splicing silencer
- miRNA:
-
Micro-RNA
- SR protein:
-
Serine-arginine protein
- CSC:
-
Cancer stem cell
- EMT:
-
Epithelial mesenchymal transition
- ASO:
-
Antisense oligonucleotide
- SSO:
-
Splice-switching antisense oligonucleotide
- FDA:
-
Food and drug administration
- CTC:
-
Circulating tumor cell
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Petersen GM. Familial Pancreatic cancer. Semin Oncol. 2016;43(5):548–53.
Benzel J, Fendrich V. Familial Pancreatic Cancer. Oncol Res Treat. 2018;41(10):611–8.
Murphy SJ, Hart SN, Lima JF, Kipp BR, Klebig M, Winters JL, et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic Tumor. Gastroenterology. 2013;145(5):1098–109. e1.
Lohr M, Kloppel G, Maisonneuve P, Lowenfels AB, Luttges J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic Pancreatitis: a meta-analysis. Neoplasia. 2005;7(1):17–23.
Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405.
Witkiewicz AK, McMillan EA, Balaji U, Baek G, Lin WC, Mansour J, et al. Whole-exome sequencing of Pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744.
Ottenhof NA, de Wilde RF, Maitra A, Hruban RH, Offerhaus GJ. Molecular characteristics of pancreatic ductal adenocarcinoma. Patholog Res Int. 2011;2011:620601.
Neesse A, Algul H, Tuveson DA, Gress TM. Stromal biology and therapy in Pancreatic cancer: a changing paradigm. Gut. 2015;64(9):1476–84.
Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: an essential role in the Tumor microenvironment. Oncol Lett. 2017;14(3):2611–20.
Hester R, Mazur PK, McAllister F. Immunotherapy in pancreatic adenocarcinoma: Beyond Copy/Paste. Clin Cancer Res. 2021;27(23):6287–97.
Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A. Pancreatic cancer: advances and challenges. Cell. 2023;186(8):1729–54.
Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Munoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14(3):153–65.
Baralle FE, Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat Rev Mol Cell Biol. 2017;18(7):437–51.
Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15(2):108–21.
Gallego-Paez LM, Bordone MC, Leote AC, Saraiva-Agostinho N, Ascensao-Ferreira M, Barbosa-Morais NL. Alternative splicing: the pledge, the turn, and the prestige: the key role of alternative splicing in human biological systems. Hum Genet. 2017;136(9):1015–42.
Tyson-Capper A, Gautrey H. Regulation of Mcl-1 alternative splicing by hnRNP F, H1 and K in Breast cancer cells. RNA Biol. 2018;15(12):1448–57.
Rogalska ME, Vivori C, Valcarcel J. Regulation of pre-mRNA splicing: roles in physiology and Disease, and therapeutic prospects. Nat Rev Genet. 2022.
Ritchie DB, Schellenberg MJ, MacMillan AM. Spliceosome structure: piece by piece. Biochim Biophys Acta. 2009;1789(9–10):624–33.
Turunen JJ, Niemela EH, Verma B, Frilander MJ. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA. 2013;4(1):61–76.
Sharp PA, Burge CB. Classification of introns: U2-type or U12-type. Cell. 1997;91(7):875–9.
Anczukow O, Krainer AR. Splicing-factor alterations in cancers. RNA. 2016;22(9):1285–301.
Jurica MS, Moore MJ. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell. 2003;12(1):5–14.
Wang Y, Liu J, Huang BO, Xu YM, Li J, Huang LF, et al. Mechanism of alternative splicing and its regulation. Biomed Rep. 2015;3(2):152–8.
Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer. 2016;16(7):413–30.
Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417(1):15–27.
Griffiths D, Bjoro T, Gautvik K, Haug E. Melatonin reduces the production and secretion of prolactin and growth hormone from rat pituitary cells in culture. Acta Physiol Scand. 1987;131(1):43–9.
Wong JJ, Lau KA, Pinello N, Rasko JE. Epigenetic modifications of splicing factor genes in myelodysplastic syndromes and acute Myeloid Leukemia. Cancer Sci. 2014;105(11):1457–63.
Boutz PL, Chawla G, Stoilov P, Black DL. MicroRNAs regulate the expression of the alternative splicing factor nPTB during muscle development. Genes Dev. 2007;21(1):71–84.
Naro C, Sette C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int J Cell Biol. 2013;2013:151839.
Goncalves V, Jordan P. Posttranscriptional regulation of splicing factor SRSF1 and its role in Cancer Cell Biology. Biomed Res Int. 2015;2015:287048.
Chen Y, Huang Q, Liu W, Zhu Q, Cui CP, Xu L, et al. Mutually exclusive acetylation and ubiquitylation of the splicing factor SRSF5 control Tumor growth. Nat Commun. 2018;9(1):2464.
Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457–63.
Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336.
Bonnal SC, Lopez-Oreja I, Valcarcel J. Roles and mechanisms of alternative splicing in cancer - implications for care. Nat Rev Clin Oncol. 2020;17(8):457–74.
Shen Q, Yu M, Jia JK, Li WX, Tian YW, Xue HZ. Possible molecular markers for the diagnosis of pancreatic ductal adenocarcinoma. Med Sci Monit. 2018;24:2368–76.
Bradley RK, Anczukow O. RNA splicing dysregulation and the hallmarks of cancer. Nat Rev Cancer. 2023;23(3):135–55.
Blazquez-Encinas R, Moreno-Montilla MT, Garcia-Vioque V, Gracia-Navarro F, Alors-Perez E, Pedraza-Arevalo S, et al. The uprise of RNA biology in neuroendocrine Neoplasms: altered splicing and RNA species unveil translational opportunities. Rev Endocr Metab Disord. 2023;24(2):267–82.
Gahete MD, Herman-Sanchez N, Fuentes-Fayos AC, Lopez-Canovas JL, Luque RM. Dysregulation of splicing variants and spliceosome components in Breast cancer. Endocr Relat Cancer. 2022;29(9):R123–R42.
Montero-Hidalgo AJ, Perez-Gomez JM, Martinez-Fuentes AJ, Gomez-Gomez E, Gahete MD, Jimenez-Vacas JM, et al. Alternative splicing in Bladder cancer: potential strategies for cancer diagnosis, prognosis, and treatment. Wiley Interdiscip Rev RNA. 2023;14(3):e1760.
Carrigan PE, Bingham JL, Srinvasan S, Brentnall TA, Miller LJ. Characterization of alternative spliceoforms and the RNA splicing machinery in Pancreatic cancer. Pancreas. 2011;40(2):281–8.
Wang J, Dumartin L, Mafficini A, Ulug P, Sangaralingam A, Alamiry NA, et al. Splice variants as novel targets in pancreatic ductal adenocarcinoma. Sci Rep. 2017;7(1):2980.
Kawalerski RR, Leach SD, Escobar-Hoyos LF. Pancreatic cancer driver mutations are targetable through distant alternative RNA splicing dependencies. Oncotarget. 2021;12(6):525–33.
Hayes GM, Carrigan PE, Beck AM, Miller LJ. Targeting the RNA splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res. 2006;66(7):3819–27.
Hayes GM, Carrigan PE, Miller LJ. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res. 2007;67(5):2072–80.
Chen S, Yang C, Wang ZW, Hu JF, Pan JJ, Liao CY, et al. CLK1/SRSF5 pathway induces aberrant exon skipping of METTL14 and cyclin L2 and promotes growth and Metastasis of Pancreatic cancer. J Hematol Oncol. 2021;14(1):60.
Escobar-Hoyos LF, Penson A, Kannan R, Cho H, Pan CH, Singh RK, et al. Altered RNA splicing by mutant p53 activates oncogenic RAS Signaling in Pancreatic Cancer. Cancer Cell. 2020;38(2):198–211. e8.
Ma J, Weng L, Jia Y, Liu B, Wu S, Xue L, et al. PTBP3 promotes malignancy and hypoxia-induced chemoresistance in Pancreatic cancer cells by ATG12 up-regulation. J Cell Mol Med. 2020;24(5):2917–30.
Qiao L, Xie N, Bai Y, Li Y, Shi Y, Wang J, et al. Identification of upregulated HNRNPs Associated with Poor Prognosis in Pancreatic Cancer. Biomed Res Int. 2019;2019:5134050.
Chen ZY, Cai L, Zhu J, Chen M, Chen J, Li ZH, et al. Fyn requires HnRNPA2B1 and Sam68 to synergistically regulate apoptosis in Pancreatic cancer. Carcinogenesis. 2011;32(10):1419–26.
Ueda J, Matsuda Y, Yamahatsu K, Uchida E, Naito Z, Korc M, et al. Epithelial splicing regulatory protein 1 is a favorable prognostic factor in Pancreatic cancer that attenuates pancreatic metastases. Oncogene. 2014;33(36):4485–95.
Alors-Pérez E, Blázquez-Encinas R, Alcalá S, Viyuela-García C, Pedraza-Arevalo S, Herrero-Aguayo V, et al. Dysregulated splicing factor SF3B1 unveils a dual therapeutic vulnerability to target Pancreatic cancer cells and cancer stem cells with an anti-splicing drug. J Exp Clin Cancer Res. 2021;40(1):382.
Huo Z, Zhai S, Weng Y, Qian H, Tang X, Shi Y, et al. PRPF40A as a potential diagnostic and prognostic marker is upregulated in Pancreatic cancer tissues and cell lines: an integrated bioinformatics data analysis. Onco Targets Ther. 2019;12:5037–51.
Li M, Wu P, Yang Z, Deng S, Ni L, Zhang Y, et al. miR-193a-5p promotes Pancreatic cancer cell Metastasis through SRSF6-mediated alternative splicing of OGDHL and ECM1. Am J Cancer Res. 2020;10(1):38–59.
Jbara A, Lin KT, Stossel C, Siegfried Z, Shqerat H, Amar-Schwartz A et al. RBFOX2 modulates a metastatic signature of alternative splicing in Pancreatic cancer. Nature. 2023.
Adesso L, Calabretta S, Barbagallo F, Capurso G, Pilozzi E, Geremia R, et al. Gemcitabine triggers a pro-survival response in Pancreatic cancer cells through activation of the MNK2/eIF4E pathway. Oncogene. 2013;32(23):2848–57.
Calabretta S, Bielli P, Passacantilli I, Pilozzi E, Fendrich V, Capurso G, et al. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in Pancreatic cancer cells. Oncogene. 2016;35(16):2031–9.
Wang ZW, Pan JJ, Hu JF, Zhang JQ, Huang L, Huang Y, et al. SRSF3-mediated regulation of N6-methyladenosine modification-related lncRNA ANRIL splicing promotes resistance of Pancreatic cancer to gemcitabine. Cell Rep. 2022;39(6):110813.
Tsukahara T, Haniu H, Matsuda Y. The PTB-Associated Splicing Factor/Peroxisome proliferator-activated receptor Gamma Axis regulates autophagosome formation in human Pancreatic Cancer cells. Biores Open Access. 2015;4(1):319–25.
Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in Pancreatic cancer cells. Int J Oncol. 2018;52(2):621–9.
Wan L, Lin KT, Rahman MA, Ishigami Y, Wang Z, Jensen MA et al. Splicing factor SRSF1 promotes Pancreatitis and KRASG12D-Mediated Pancreatic Cancer. Cancer Discov. 2023.
Xiao W, Chen X, Li X, Deng K, Liu H, Ma J, et al. RBM10 regulates human TERT gene splicing and inhibits Pancreatic cancer progression. Am J Cancer Res. 2021;11(1):157–70.
Zhou W, Ma N, Jiang H, Rong Y, Deng Y, Feng Y, et al. SF3B4 is decreased in Pancreatic cancer and inhibits the growth and migration of cancer cells. Tumour Biol. 2017;39(3):1010428317695913.
Takino JI, Sato T, Hiraishi I, Nagamine K, Hori T. Alterations in glucose metabolism due to decreased expression of Heterogeneous Nuclear Ribonucleoprotein M in Pancreatic Ductal Adenocarcinoma. Biology (Basel). 2021;10(1).
Peng J, Valeshabad AK, Li Q, Wang Y. Differential expression of RBM5 and KRAS in pancreatic ductal adenocarcinoma and their association with clinicopathological features. Oncol Lett. 2013;5(3):1000–4.
Lai S, Wang Y, Li T, Dong Y, Lin Y, Wang L, et al. N6-methyladenosine-mediated CELF2 regulates CD44 alternative splicing affecting tumorigenesis via ERAD pathway in Pancreatic cancer. Cell Biosci. 2022;12(1):125.
Brierley CK, Steensma DP. Targeting splicing in the treatment of myelodysplastic syndromes and other myeloid Neoplasms. Curr Hematol Malig Rep. 2016;11(6):408–15.
Larsson CA, Cote G, Quintas-Cardama A. The changing mutational landscape of acute Myeloid Leukemia and Myelodysplastic Syndrome. Mol Cancer Res. 2013;11(8):815–27.
Chen S, Benbarche S, Abdel-Wahab O. Splicing factor mutations in hematologic malignancies. Blood. 2021;138(8):599–612.
Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6.
Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365(15):1384–95.
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–9.
Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal Melanoma. Nat Genet. 2013;45(2):133–5.
Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, et al. SF3B1 and other novel cancer genes in chronic lymphocytic Leukemia. N Engl J Med. 2011;365(26):2497–506.
Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7:10615.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Read A, Natrajan R. Splicing dysregulation as a driver of Breast cancer. Endocr Relat Cancer. 2018;25(9):R467–R78.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
Yang JY, Huo YM, Yang MW, Shen Y, Liu DJ, Fu XL, et al. SF3B1 mutation in Pancreatic cancer contributes to aerobic glycolysis and Tumor growth through a PP2A-c-Myc axis. Mol Oncol. 2021;15(11):3076–90.
Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32(2):185–203. e13.
Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of Pancreatic cancer. Nature. 2016;531(7592):47–52.
Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22(9):976–86.
Behm-Ansmant I, Gatfield D, Rehwinkel J, Hilgers V, Izaurralde E. A conserved role for cytoplasmic poly(A)-binding protein 1 (PABPC1) in nonsense-mediated mRNA decay. EMBO J. 2007;26(6):1591–601.
Heinrich B, Zhang Z, Raitskin O, Hiller M, Benderska N, Hartmann AM, et al. Heterogeneous nuclear ribonucleoprotein G regulates splice site selection by binding to CC(A/C)-rich regions in pre-mRNA. J Biol Chem. 2009;284(21):14303–15.
Yang C, Wu Q, Huang K, Wang X, Yu T, Liao X, et al. Genome-wide profiling reveals the landscape of prognostic alternative splicing signatures in pancreatic ductal adenocarcinoma. Front Oncol. 2019;9:511.
Screaton GR, Bell MV, Jackson DG, Cornelis FB, Gerth U, Bell JI. Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons. Proc Natl Acad Sci U S A. 1992;89(24):12160–4.
Rall CJ, Rustgi AK. CD44 isoform expression in primary and metastatic pancreatic adenocarcinoma. Cancer Res. 1995;55(9):1831–5.
Gansauge F, Gansauge S, Zobywalski A, Scharnweber C, Link KH, Nussler AK, et al. Differential expression of CD44 splice variants in human pancreatic adenocarcinoma and in normal pancreas. Cancer Res. 1995;55(23):5499–503.
Gotoda T, Matsumura Y, Kondo H, Saitoh D, Shimada Y, Kosuge T, et al. Expression of CD44 variants and its association with survival in Pancreatic cancer. Jpn J Cancer Res. 1998;89(10):1033–40.
Zhao S, Chen C, Chang K, Karnad A, Jagirdar J, Kumar AP, et al. CD44 expression level and isoform contributes to Pancreatic Cancer cell plasticity, invasiveness, and response to Therapy. Clin Cancer Res. 2016;22(22):5592–604.
Zhu H, Zhou W, Wan Y, Lu J, Ge K, Jia C. CD44V3, an alternatively spliced form of CD44, promotes Pancreatic Cancer Progression. Int J Mol Sci. 2022;23(20).
Xie Z, Gao Y, Ho C, Li L, Jin C, Wang X, et al. Exosome-delivered CD44v6/C1QBP complex drives Pancreatic cancer liver Metastasis by promoting fibrotic liver microenvironment. Gut. 2022;71(3):568–79.
Matzke-Ogi A, Jannasch K, Shatirishvili M, Fuchs B, Chiblak S, Morton J, et al. Inhibition of Tumor Growth and Metastasis in Pancreatic Cancer models by Interference with CD44v6 Signaling. Gastroenterology. 2016;150(2):513–25. e10.
Korner M, Hayes GM, Rehmann R, Zimmermann A, Friess H, Miller LJ, et al. Secretin receptors in normal and diseased human pancreas: marked reduction of receptor binding in ductal neoplasia. Am J Pathol. 2005;167(4):959–68.
Ding WQ, Cheng ZJ, McElhiney J, Kuntz SM, Miller LJ. Silencing of secretin receptor function by dimerization with a misspliced variant secretin receptor in ductal pancreatic adenocarcinoma. Cancer Res. 2002;62(18):5223–9.
Hayes GM, Carrigan PE, Dong M, Reubi JC, Miller LJ. A novel secretin receptor splice variant potentially useful for early diagnosis of pancreatic carcinoma. Gastroenterology. 2007;133(3):853–61.
Ryberg A, Borch K, Monstein HJ. Expression of multiple forms of 3’-end variant CCK2 receptor mRNAs in human pancreatic adenocarcinomas. BMC Res Notes. 2011;4:131.
Bernard V, Young J, Binart N. Prolactin - a pleiotropic factor in health and Disease. Nat Rev Endocrinol. 2019;15(6):356–65.
Hu ZZ, Meng J, Dufau ML. Isolation and characterization of two novel forms of the human prolactin receptor generated by alternative splicing of a newly identified exon 11. J Biol Chem. 2001;276(44):41086–94.
Brandi J, Dando I, Pozza ED, Biondani G, Jenkins R, Elliott V, et al. Proteomic analysis of Pancreatic cancer stem cells: functional role of fatty acid synthesis and mevalonate pathways. J Proteom. 2017;150:310–22.
Nie H, Huang PQ, Jiang SH, Yang Q, Hu LP, Yang XM, et al. The short isoform of PRLR suppresses the pentose phosphate pathway and nucleotide synthesis through the NEK9-Hippo axis in Pancreatic cancer. Theranostics. 2021;11(8):3898–915.
Kang X, Lin Z, Xu M, Pan J, Wang ZW. Deciphering role of FGFR signalling pathway in Pancreatic cancer. Cell Prolif. 2019;52(3):e12605.
Kornmann M, Beger HG, Korc M. Role of fibroblast growth factors and their receptors in Pancreatic cancer and chronic Pancreatitis. Pancreas. 1998;17(2):169–75.
Kornmann M, Lopez ME, Beger HG, Korc M. Expression of the IIIc variant of FGF receptor-1 confers mitogenic responsiveness to heparin and FGF-5 in TAKA-1 pancreatic ductal cells. Int J Pancreatol. 2001;29(2):85–92.
Liu Z, Ishiwata T, Zhou S, Maier S, Henne-Bruns D, Korc M, et al. Human fibroblast growth factor receptor 1-IIIb is a functional fibroblast growth factor receptor expressed in the pancreas and involved in proliferation and movement of pancreatic ductal cells. Pancreas. 2007;35(2):147–57.
Cras-Meneur C, Scharfmann R. FGFR1-IIIb is a putative marker of pancreatic progenitor cells. Mech Dev. 2002;116(1–2):205–8.
Liu Z, Neiss N, Zhou S, Henne-Bruns D, Korc M, Bachem M, et al. Identification of a fibroblast growth factor receptor 1 splice variant that inhibits Pancreatic cancer cell growth. Cancer Res. 2007;67(6):2712–9.
Kornmann M, Ishiwata T, Matsuda K, Lopez ME, Fukahi K, Asano G, et al. IIIc isoform of fibroblast growth factor receptor 1 is overexpressed in human Pancreatic cancer and enhances tumorigenicity of hamster ductal cells. Gastroenterology. 2002;123(1):301–13.
Thompson CM, Cannon A, West S, Ghersi D, Atri P, Bhatia R, et al. Mucin expression and Splicing Determine Novel subtypes and Patient Mortality in Pancreatic Ductal Adenocarcinoma. Clin Cancer Res. 2021;27(24):6787–99.
Chaturvedi P, Singh AP, Batra SK. Structure, evolution, and biology of the MUC4 mucin. FASEB J. 2008;22(4):966–81.
Choudhury A, Moniaux N, Winpenny JP, Hollingsworth MA, Aubert JP, Batra SK. Human MUC4 mucin cDNA and its variants in pancreatic carcinoma. J Biochem. 2000;128(2):233–43.
Xie K, Zhi X, Tang J, Zhu Y, Zhang J, Li Z, et al. Upregulation of the splice variant MUC4/Y in the Pancreatic cancer cell line MIA PaCa-2 potentiates proliferation and suppresses apoptosis: new insight into the presence of the transcript variant of MUC4. Oncol Rep. 2014;31(5):2187–94.
Choudhury A, Moniaux N, Ringel J, King J, Moore E, Aubert JP, et al. Alternate splicing at the 3’-end of the human pancreatic tumor-associated mucin MUC4 cDNA. Teratog Carcinog Mutagen. 2001;21(1):83–96.
Friess H, Lu Z, Andren-Sandberg A, Berberat P, Zimmermann A, Adler G, et al. Moderate activation of the apoptosis inhibitor bcl-xL worsens the prognosis in Pancreatic cancer. Ann Surg. 1998;228(6):780–7.
Ikezawa K, Hikita H, Shigekawa M, Iwahashi K, Eguchi H, Sakamori R, et al. Increased Bcl-xL expression in pancreatic neoplasia promotes carcinogenesis by inhibiting senescence and apoptosis. Cell Mol Gastroenterol Hepatol. 2017;4(1):185–200e1.
Antonopoulou E, Ladomery M. Targeting splicing in Prostate Cancer. Int J Mol Sci. 2018;19(5).
Massiello A, Roesser JR, Chalfant CE. SAP155 binds to ceramide-responsive RNA cis-element 1 and regulates the alternative 5’ splice site selection of Bcl-x pre-mRNA. FASEB J. 2006;20(10):1680–2.
Leppert U, Eisenreich A. The role of tissue factor isoforms in cancer biology. Int J Cancer. 2015;137(3):497–503.
Unruh D, Turner K, Srinivasan R, Kocaturk B, Qi X, Chu Z, et al. Alternatively spliced tissue factor contributes to Tumor spread and activation of coagulation in pancreatic ductal adenocarcinoma. Int J Cancer. 2014;134(1):9–20.
Unruh D, Sagin F, Adam M, Van Dreden P, Woodhams BJ, Hart K, et al. Levels of alternatively spliced tissue factor in the plasma of patients with Pancreatic Cancer may help Predict aggressive Tumor phenotype. Ann Surg Oncol. 2015;22(Suppl 3):1206–11.
Woolard J, Bevan HS, Harper SJ, Bates DO. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation. 2009;16(7):572–92.
Yip-Schneider MT, Wu H, Schmidt CM. Novel expression of vascular endothelial growth factor isoforms in the pancreas and pancreatic cystic lesions. Biochimie. 2021;181:234–9.
Plowman SJ, Williamson DJ, O’Sullivan MJ, Doig J, Ritchie AM, Harrison DJ, et al. While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable. Mol Cell Biol. 2003;23(24):9245–50.
Luo F, Ye H, Hamoudi R, Dong G, Zhang W, Patek CE, et al. K-ras exon 4A has a tumour suppressor effect on carcinogen-induced murine colonic adenoma formation. J Pathol. 2010;220(5):542–50.
Tian J, Chen C, Rao M, Zhang M, Lu Z, Cai Y, et al. Aberrant RNA splicing is a primary link between genetic variation and Pancreatic Cancer risk. Cancer Res. 2022;82(11):2084–96.
Weng Y, Qian H, Hong L, Zhao S, Deng X, Shen B. Identification of EMT-related alternative splicing event of TMC7 to promote invasion and migration of Pancreatic cancer. Front Immunol. 2022;13:1089008.
Simmler P, Cortijo C, Koch LM, Galliker P, Angori S, Bolck HA, et al. SF3B1 facilitates HIF1-signaling and promotes malignancy in Pancreatic cancer. Cell Rep. 2022;40(8):111266.
Eskens FA, Ramos FJ, Burger H, O’Brien JP, Piera A, de Jonge MJ, et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res. 2013;19(22):6296–304.
Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. 2018;24(4):497–504.
Cohen P. Protein kinases–the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1(4):309–15.
Ardito F, Giuliani M, Perrone D, Troiano G, Lo Muzio L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (review). Int J Mol Med. 2017;40(2):271–80.
Fleuren ED, Zhang L, Wu J, Daly RJ. The kinome ‘at large’ in cancer. Nat Rev Cancer. 2016;16(2):83–98.
Deng YN, Bellanti JA, Zheng SG. Essential kinases and transcriptional regulators and their roles in autoimmunity. Biomolecules. 2019;9(4).
Martin Moyano P, Nemec V, Paruch K. Cdc-Like Kinases (CLKs): Biology, Chemical Probes, and therapeutic potential. Int J Mol Sci. 2020;21(20).
Ding S, Shi J, Qian W, Iqbal K, Grundke-Iqbal I, Gong CX, et al. Regulation of alternative splicing of tau exon 10 by 9G8 and Dyrk1A. Neurobiol Aging. 2012;33(7):1389–99.
Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD, et al. Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun. 2019;10(1):131.
Shi J, Qian W, Yin X, Iqbal K, Grundke-Iqbal I, Gu X, et al. Cyclic AMP-dependent protein kinase regulates the alternative splicing of tau exon 10: a mechanism involved in tau pathology of Alzheimer Disease. J Biol Chem. 2011;286(16):14639–48.
Kvissel AK, Orstavik S, Eikvar S, Brede G, Jahnsen T, Collas P, et al. Involvement of the catalytic subunit of protein kinase A and of HA95 in pre-mRNA splicing. Exp Cell Res. 2007;313(13):2795–809.
Aubol BE, Wu G, Keshwani MM, Movassat M, Fattet L, Hertel KJ, et al. Release of SR proteins from CLK1 by SRPK1: a symbiotic kinase system for Phosphorylation Control of Pre-mRNA Splicing. Mol Cell. 2016;63(2):218–28.
Fackenthal JD, Godley LA. Aberrant RNA splicing and its functional consequences in cancer cells. Dis Model Mech. 2008;1(1):37–42.
Tam BY, Chiu K, Chung H, Bossard C, Nguyen JD, Creger E, et al. The CLK inhibitor SM08502 induces anti-tumor activity and reduces wnt pathway gene expression in gastrointestinal cancer models. Cancer Lett. 2020;473:186–97.
Aggarwal RR, Schweizer MT, Nanus DM, Pantuck AJ, Heath EI, Campeau E, et al. A phase Ib/IIa study of the Pan-BET inhibitor ZEN-3694 in combination with Enzalutamide in patients with metastatic castration-resistant Prostate Cancer. Clin Cancer Res. 2020;26(20):5338–47.
Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, et al. Bromodomain inhibitor OTX015 in patients with Lymphoma or Multiple Myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016;3(4):e196–204.
Berthon C, Raffoux E, Thomas X, Vey N, Gomez-Roca C, Yee K, et al. Bromodomain inhibitor OTX015 in patients with acute Leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3(4):e186–95.
Steensma DP, Wermke M, Klimek VM, Greenberg PL, Font P, Komrokji RS et al. Phase I first-in-human dose escalation study of the oral SF3B1 modulator H3B-8800 in myeloid Neoplasms. Leukemia. 2021.
Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, Adams ND, et al. Anti-tumor activity of the type I PRMT inhibitor, GSK3368715, synergizes with PRMT5 inhibition through MTAP loss. Cancer Cell. 2019;36(1):100–14. e25.
Gonsalvez GB, Tian L, Ospina JK, Boisvert FM, Lamond AI, Matera AG. Two distinct arginine methyltransferases are required for biogenesis of Sm-class ribonucleoproteins. J Cell Biol. 2007;178(5):733–40.
Bewersdorf JP, Stahl MF, Taylor J, Chandhok NS, Watts J, Derkach A, et al. A phase II clinical trial of E7820 for patients with Relapsed/Refractory myeloid malignancies with mutations in splicing factor genes. Blood. 2022;140(Supplement 1):9065–7.
Brunner AM, Liu Y, Mendez LM, Garcia JS, Amrein PC, Neuberg DS, et al. Inhibition of ATR with AZD6738 (Ceralasertib) for the treatment of Progressive or Relapsed Myelodysplastic syndromes and Chronic Myelomonocytic Leukemia: Safety and Preliminary Activity from a phase Ib/II study. Blood. 2021;138(Supplement 1):1521.
Havens MA, Hastings ML. Splice-switching antisense oligonucleotides as therapeutic Drugs. Nucleic Acids Res. 2016;44(14):6549–63.
Stein CA, Castanotto D. FDA-Approved Oligonucleotide therapies in 2017. Mol Ther. 2017;25(5):1069–75.
Corey DR. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat Neurosci. 2017;20(4):497–9.
Shahbazi R, Ozpolat B, Ulubayram K. Oligonucleotide-based theranostic nanoparticles in cancer therapy. Nanomed (Lond). 2016;11(10):1287–308.
Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nat Rev Drug Discov. 2020;19(10):673–94.
Baylot V, Andrieu C, Katsogiannou M, Taieb D, Garcia S, Giusiano S, et al. OGX-427 inhibits Tumor progression and enhances gemcitabine chemotherapy in Pancreatic cancer. Cell Death Dis. 2011;2:e221.
Ko AH, Murphy PB, Peyton JD, Shipley DL, Al-Hazzouri A, Rodriguez FA, et al. A Randomized, Double-Blinded, phase II trial of Gemcitabine and Nab-Paclitaxel Plus Apatorsen or Placebo in patients with metastatic Pancreatic Cancer: the RAINIER Trial. Oncologist. 2017;22(12):1427–e129.
Dal Molin M, Zhang M, de Wilde RF, Ottenhof NA, Rezaee N, Wolfgang CL, et al. Very Long-term Survival Following Resection for Pancreatic Cancer Is Not Explained by Commonly Mutated Genes: Results of Whole-Exome Sequencing Analysis. Clinical Cancer Research. 2015; 21(8) 1944-1950.
Noë M, Niknafs N, Fischer CG, Hackeng WM, Guthrie VB, Hosoda W, et al. Genomic characterization of malignant progression in neoplastic pancreatic cysts. Nature Communications. 2020; 11(1):4085
Lowder CY, Dhir T, Goetz AB, Thomsett HL, Bender J, Tatarian T, et al. A step towards personalizing next line therapy for resected pancreatic and related cancer patients: A single institution’s experience. Surgical Oncology. 2020; 33118-125.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10000 patients. Nature Medicine. 2017; 23(6) 703-713
Notta F, Chan-Seng-Yue M, Lemire M, Li Y, Wilson GW, Connor AA, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature. 2016; 538(7625) 378-382
Acknowledgements
Not applicable.
Funding
This work has been supported by Spanish Ministry of Economy [MINECO; BFU2016-80360-R (to JPC)] and Ministry of Science and Innovation [MICINN; PID2019-105201RB-I00 (to JPC), PID2019-105564RB-I00 (to RML)]. Instituto de Salud Carlos III, co-funded by European Union (ERDF/ESF, “Investing in your future”) [FIS DTS Grant DTS20/00050 (to RML)); Postdoctoral Grant Sara Borrell CD19/00255 (to AIC); Predoctoral contract FI17/00282 (to EAP)]. Spanish Ministry of Universities [FPU20/03958 (to VGV); FPU18/02275 (to RBE); postdoctoral contract under the program María Zambrano funded by the European Union Next Generation-EU (to SPA)]. Asociación Cáncer de Páncreas and AESPANC 2022 (to JPC); Boehringer Ingelheim Fonds travel grant (EAP). Junta de Andalucía (BIO-0139). GETNE2016 and GETNE2019 Research grants (to JPC); and CIBERobn. Part of this work was supported by COST (European Cooperation in Science and Technology – www.cost.eu) through the COST Action TRANSPAN (CA21116).
Author information
Authors and Affiliations
Contributions
Conceptualization, EAP, SPA, AIC, JPC; writing—original draft preparation, EAP, SPA, RBE, MTMM, VGV, IB, RML, BS, AIC, JPC; writing—review and editing, EAP, SPA, RBE, MTMM, VGV, IB, RML, BS, AIC, JPC. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Alors-Pérez, E., Pedraza-Arevalo, S., Blázquez-Encinas, R. et al. Splicing alterations in pancreatic ductal adenocarcinoma: a new molecular landscape with translational potential. J Exp Clin Cancer Res 42, 282 (2023). https://doi.org/10.1186/s13046-023-02858-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-023-02858-z