
Abstract
Background
The mechanism by which glioblastoma evades temozolomide (TMZ)-induced cytotoxicity is largely unknown. We hypothesized that mitochondria plays a role in this process.
Methods
RNA transcriptomes were obtained from tumor samples and online databases. Expression of different proteins was manipulated using RNA interference or gene amplification. Autophagic activity and mitochondrial metabolism was assessed in vitro using the respective cellular and molecular assays. In vivo analysis were also carried out in this study.
Results
High SH3GLB1 gene expression was found to be associated with higher disease grading and worse survival profiles. Single-cell transcriptome analysis of clinical samples suggested that SH3GLB1 and the altered gene levels of oxidative phosphorylation (OXPHOS) were related to subsets expressing a tumor-initiating cell signature. The SH3GLB1 protein was regulated by promoter binding with Sp1, a factor associated with TMZ resistance. Downregulation of SH3GLB1 resulted in retention of TMZ susceptibility, upregulated p62, and reduced LC3B-II. Autophagy inhibition by SH3GLB1 deficiency and chloroquine resulted in attenuated OXPHOS expression. Inhibition of SH3GLB1 in resistant cells resulted in alleviation of TMZ-enhanced mitochondrial metabolic function, such as mitochondrial membrane potential, mitochondrial respiration, and ATP production. SH3GLB1 modulation could determine tumor susceptibility to TMZ. Finally, in animal models, resistant tumor cells with SH3GLB1 knockdown became resensitized to the anti-tumor effect of TMZ, including the suppression of TMZ-induced autophagy and OXPHOS.
Conclusions
SH3GLB1 promotes TMZ resistance via autophagy to alter mitochondrial function. Characterizing SH3GLB1 in glioblastoma may help develop new therapeutic strategies against this disease in the future.
Similar content being viewed by others


Background
Glioblastoma (GBM) is the most common primary malignant brain tumor, with an annual age-adjusted incidence rate of 3.19 per 100,000 populations in the US. GBM’s prognosis is disappointing, with a 2-year overall survival rate of only 13.6% [1]. Currently, temozolomide (TMZ) is the most frequently administered alkylating agent for patients with GBM [2]. However, the survival benefit from this chemotherapeutic agent is only modest, as TMZ-taking patients only show a 2.5 months longer of survival compared to those without taking TMZ [3]. One of the common causes leading to treatment failure is the development of resistance. In addition to the mechanism associated with O6-methylguanine-DNA methyltransferase and other DNA-repairing genes [4], our previous study revealed that upregulation of superoxide dismutase 2 (SOD2) enhanced cellular tolerance to reactive oxygen species (ROS), promoting the induction of TMZ resistance [5]. This led to enrichment of specific subsets carrying tumor-initiating cell (TIC) features against the antitumor drug. It also highlighted the ability for tumors to adapt when they encountered stressful environment.
SOD2 functions in mitochondria to modulate intracellular oxidative stress, and thereby regulates cell metabolism [6]. Recent studies have suggested that altered metabolic reprogramming by mitochondria promotes the induction of chemotherapy resistance in cancer cells [7]. In GBM, the altered metabolic reprogramming and the association with the induction of TMZ resistance has likewise been reported [8]. Rabé and colleagues suggested that a transient increase in the oxygen consumption rate (OCR)/extracellular acidification rate (ECAR) ratio might cause cellular resistance to TMZ [9]. However, the promoting factors remain not clear. Autophagy is a regulator of oxidative phosphorylation (OXPHOS) [10]. Autophagy degrades and clears the aggregated proteins and dysfunctional organelles to maintain cellular homeostasis [11]. Upregulation of autophagy can promote either the survival or death of cancer cells, depending on the stimulation received. For example, in a tumor environment with limited nutrients and oxygen, an increase in autophagy allows cancer cells to survive and resume proliferation and initiation [12]. In GBM, certain subsets, particularly those with the capability of self-renewal and differentiation, activate autophagy to survive TMZ treatment and the harsh microenvironment [13].
Among different autophagy regulators, SH3GLB1 (Bax-Interacting Factor 1 or endophilin B1) is indispensable for the initiation of autophagy [14]. This evolutionarily conserved protein is a member of the endophilin protein family, which contains an N-terminal BAR domain and a C-terminal SH3 domain [15]. It recruits beclin1 and activates PI3KC3, which is the crucial step for dimerization and interaction with ultraviolet irradiation resistance–associated gene in forming the early autophagosome [16]. Interestingly, the functional impact of SH3GLB1 is broad and profound, including participating in the processes of mitochondrial dynamics, apoptosis, and endocytosis in membrane reshaping to maintain intracellular homeostasis [14]. Notably, SH3GLB1 is physiologically important for maintaining the function of brain, as it protects neuronal cells from amyloid-β-induced cytotoxicity to induce the progression of Alzheimer’s disease [17].
Given that altered SOD2 and mitochondrial functions promote resistance in GBM, it remains unknown how the related metabolic reaction contributes to the process. Considering that SH3GLB1 is fundamental in mitochondrial function [18], we hypothesized that SH3GLB1 associates with SOD2 to steer the organelle response against TMZ in GBM. Here, we demonstrated that enhanced SH3GLB1 expression was common in recurrent tumors. The protein modulated autophagy and OXPHO to promote TMZ resistance.
Materials and methods
Cell culture
Human GBM cell lines U87MG and A172 (American Type Culture Collection; Manassas, VA, USA), as well as Pt#3 and Pt#5, which both were derived from GBM patients [19], were cultured in 10% fetal bovine serum and antibiotic-supplemented DMEM (Thermo Fisher Scientific). Resistant cells (U87MG-R, A172-R, Pt#3-R, and Pt#5-R) were developed and selected from parental cells by using prolonged TMZ treatment as our previous studies [5, 20]. A primary GBM tumor P1S, which was obtained from a GBM tissue that exhibited therapeutic resistance, was maintained as a patient-derived xenograft in immunodeficient mice before cryopreservation [19].
PCR array
The mRNA of the cells was extracted and processed to cDNA, following by analysis of mitochondria-related genes with RT2 Profiler™ PCR Array Human Mitochondria (#PAHS-087Z, Qiagen, Hilden, Germany).
Analysis of clinical datasets
Online GBM databases from GlioVis for survival [21] and from the Chinese Glioma Genome Atlas (CGGA) [22] for assessment of clinical parameter was utilized to reveal the impact of genes in interest. The data of RSEM (RNA-Seq by Expectation Maximization) values were downloaded for further processing. The expression fold change and significance level (p-value) of mitochondria-related gene expression between primary and resistant GBM RNA-sequencing data were calculated.
Preparation of clinical samples and immunohistochemistry (IHC)
Nine paired primary and recurrent tumor sections were prepared from archived formalin-fixed, paraffin-embedded blocks with their clinical information blind to investigators. All were diagnosed after 2011 and were histologically confirmed for GBM. A DAKO IHC kit (Agilent Technologies, Santa Clara, CA, USA) was used following the manufacturer’s protocol. The staining results of the clinical samples were assessed by a neuropathologist.
Treatment reagents and detecting antibodies
TMZ, mithramycin A, and chloroquine were purchased from Sigma–Aldrich (St. Louis, MO, USA). The detection antibodies were as follows: SH3GLB1 (Proteintech, Rosemont, IL, USA), CD133 (Proteintech), SP1 (Merck Millipore, Burlington, MA, USA), LC3B (Santa Cruz, Dallas, TX, USA), p62 (Cell Signaling, Danvers, MA, USA), caspase 3 (Cell Signaling), actin (Merck Millipore), Atg12 (GeneTex, Irvine, CA, USA), and OXPHOS antibody cocktail (ab110411, Abcam, Cambridge, UK), Vinculin (Thermo Fisher Scientific).
Whole genome RNA sequencing
The 14 paired recurrent and primary frozen samples of high-grade glioma samples were obtained. Total RNA was extracted and quantified following the manufacturer’s protocol (TRIzol; Thermo Fisher Scientific, MA, USA). Strand-Specific RNA Library Kit (Agilent Technologies, Inc., CA, USA) was prepared for mRNA sequencing by Illumina system and AMPure XP magnetic beads (Beckman Coulter, CA, USA) were used for purification. For genetic analysis based on molecular functioning, we applied Ingenuity Pathway Analysis (IPA, Qiagen, Hilden, Germany) on the sequencing data.
Single-cell RNA (scRNA) transcriptomics
Inform consents were given to newly diagnosed GBM patients that were planned for surgical resection to obtain tumor tissues for study. Samples were dissociated using a Brain Tumor Dissociation Kit, papain (Miltenyi Biotec, Bergisch Gladbach, Germany) and ACK RBC Lysing Buffer (Thermo Fisher Scientific). An scRNA library was then constructed by generating gel beads in emulsion (GEM) using a Chromium Next GEM Single Cell 3’GEM, Library & Gel Bead Kit (v3.1; 10X Genomics, Pleasanton, CA, USA). The gel beads were then dissolved, primers were released, and the co-partitioned cells were lysed. cDNA was then synthesized, followed by purification with magnetic bead, barcoding, and amplification by PCR for library construction. RNA sequencing (RNA-seq) was performed using Illumina next-generation sequencing platforms.
For genetic sorting of the scRNA transcriptomics, the raw data were aligned to the GRCh38 reference genome, namely and output into aggregated files that integrated individual cells with their annotated information via CellRanger 4.0 (10X Genomics). Analysis was then performed using Loupe Browser 5.0 (10X Genomics), with the criteria set to exclude low complexity cells (< 1000 genes, < 1800 UMI), high complexity cells (> 6500 genes), and dying cells (> 12% UMI to mitochondrial genes) [23].
For analysis, hierarchical clustering was performed using the Euclidean distance method, and average linkage was determined according to the log2 (fold change) values of the OXPHOS genes [24]. Comparative analysis (similarity matrix) between each cluster were generated using Morpheus (https://software.broadinstitute.org/morpheus). The scores in the similarity matrix were calculated using the Pearson correlation coefficient.
RNA-based gene modulation
Lipofectamine® RNAiMAX reagent (Invitrogen) was used for the transfection of small interfering RNA (siRNA) targeting SH3GLB1 (5′-GGGAAUCAGCAGUACACAUTT-3′ and 3′-AUGUGUACUGCUGAUUCCCTT-5′, GenePharma, Shanghai, China), Sp1 (ON-TARGETplus siRNA; Horizon Discovery, Cambridge, UK), and the control. Long-term modulation was achieved by selecting clones for lentiviral-transfected cells using SH3GLB1 short-hairpin RNA (shRNA; RNAi Core of Academia Sinica, Taipei, Taiwan) or the overexpression vector (GenScript Biotech, NJ, USA).
Western blot analysis
Cell lysates were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked using 5% nonfat milk and incubated overnight with primary antibodies, and labeled with secondary antibodies. A chemiluminescence substrate was used to elicit signals for detection of the intensity.
Cell density assay
For the cell density assay, 5000–20,000 cells were plated in a 24-well plates under treatment until they stabilized. They were stained with 50% ethanol containing 0.5% methylene blue for 90 min and dissolved in 1% N-lauroylsarcosine for measurement with optical density at 570 nm.
Sorting of CD133+ cells
Patient-derived xenograft tumor cells were used as primary cell cultures. They were cultured in serum-free growth factor-supplemented DMEM/F12 (Thermo Fisher Scientific). The cells were collected and disaggregated with repeat pipetting. They were than labeled with fluorochrome-conjugated anti-CD133 antibodies (Miltenyi Biotec) and were sorted by FACSAria™ III (BD Biosciences). The isolated CD133+ cells were then cultured in growth factor-supplemented DMEM/F12. Simultaneously isolated CD133− cells were cultured in serum-supplemented DMEM to maintain the differentiation phenotype.
Fluorescent labeling of autophagy
Cells were transfected with a control vector or LC3B-EGFP plasmid (courtesy of Chun Hei Antonio Cheung, National Cheng Kung University, Tainan, Taiwan) using Lipofectamine LTX reagent (Invitrogen) to label autophagosomes. Autolysosomes were stained with acridine orange (Sigma–Aldrich), which is a cell-permeable green fluorophore that shifts to red fluorescence in acidic vesicular organelles. Detection was performed through fluorescence microscopy and flow cytometry.
Measurement of the mitochondrial function
For the live cell metabolic assay, an XF24 Analyzer (Seahorse Bioscience; Agilent Technologies, Santa Clara CA, USA) was utilized. Cells were seeded at a density of 2 × 104 cells/well into their specialized 24-well microplates (Seahorse XFe24 FluxPak, Part #102340–001, Agilent Technologies). The cartridge sensor was soaked using XF Calibrant in a CO2-free incubator at 37 °C overnight. The culture medium was replaced by NaHCO3-free DMEM, followed by calibration for respiratory rate. Basal OCR levels were first measured followed by measurements of the other parameters after stimulation with oligomycin (10 μM) for ATP production and proton leak; carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (2 μM) for maximal respiration and spare respiratory capacity; a combination of rotenone and antimycin A (5 μM) for nonmitochondrial respiration. Three measurements were recorded in each well.
To study membrane potential (ΔΨm), a ΔΨm-dependent dye, JC-1 (Invitrogen, Waltham, MA, USA) was used. This allowed cells to generate signals of red and green fluorescence from the aggregated form at high ΔΨm and the monomeric form at low ΔΨm, respectively. Detection was performed using flow cytometry.
Chromatin immunoprecipitation (ChIP)
The study was performed with an EZ-Magna Chip A/G kit (Merck Millipore). Proteins were cross-linked to DNA with the addition of 1% formaldehyde for 10 min and then quenched by 125 mM glycine. Cells were treated with protease inhibitors and lysed in the lysis buffer. The samples were then incubated with the antibody and magnetic beads at 4 °C overnight. After magnetic separator, they were incubated at 62 °C with proteinase K. DNA was released by reverse cross-linking and was purified. Subsequent qPCR was performed using SH3GLB1 forward and reverse primers as follows: 5′-CAAGCATACAGAGGCGCCGAG-3′ and 5′-CAAGAATGGGTCAGTCGGCTC-3′, respectively.
Tumor xenograft mouse model
The studies were approved and processed with advice from the animal ethics board. U87MG-R-luciferase-EGFP cells with or without shSH3GLB1 were used to create a xenograft model in 6–7-week-old male NOD-SCID mice. Cells were injected into the subcutaneous flank area or brain for inoculation at numbers of 2 × 106 or 1 × 105, respectively. Ten days later, the animals were randomly assigned for treatment. The Luciferase activity was measured by using an IVIS imaging system (Xenogen, Alameda, CA, USA) after intraperitoneal injection of firefly substrate (80 mg/kg; Promega, Madison, WI, USA). Mice were euthanized if severe neurological or intolerable physical signs appeared. The tumor tissue was extracted for subsequent experiments. The intracranial experiments were conducted according to previous studies [5] to do survival analysis.
Statistics
Data were statistically analyzed using Prism (version 7.02, GraphPad, La Jolla, CA, USA). Differences in continuous variables were calculated using unpaired, two-tailed Student’s t-test. The linear correlation between two variables was measured by using Person’s correlation coefficient (r). Statistical significance was set at p < 0.05.
Results
Recurrent GBM shows enhanced SH3GLB1 and OXPHOS gene signatures
SOD2-knockdown A172-R cells were used to find the potential mediating genes that were associated with SOD2 for metabolic alternation (Supplementary Fig. S1A). The result was cross-examined with profiles obtained from the CGGA database that included 140 primary and 109 recurrent GBM tumors (Fig. 1A). Eight genes that could be significant in resistance-related mitochondrial functions and tumor recurrence were analyzed. Among them, SH3GLB1 exhibited a higher correlation with SOD2 (Pearson’s r = 0.524, p < 0.001, Fig. 1B left panel). The two genes also showed significant correlation in the TCGA database and in our samples (Supplementary Fig. S1B).

SH3GLB1 and OXPHOS-related genes are associated with recurrent GBM. A Eight altered mitochondria-related genes were revealed to have similar and significant trend by crossing data from the PCR Array and the CGGA database. B Their corresponding expression with SOD2 in GBM cohort of the CGGA database is illustrated in scatter plots in the left panel (SLC25A5 is not illustrated, p = 0.310). The levels of SOD2 and SH3GLB1 according to recurrent or primary GBM were shown in box plot in the right panel with statistical evaluation. C Expression of SH3GLB1 in the paired primary- and recurrent-tissues was revealed using IHC staining. Two in nine cases are represented here with the remaining shown in Supplementary Fig. S1C (D) The Kaplan–Meier graph of the SH3GLB1 gene expression that was higher or lower than the median level in the GBM dataset from the CGGA database. E Ingenuity pathway analysis results of the 14 paired recurrent versus treatment-naïve glioma RNA-seq dataset. The heatmap showed selected pathways that are related to mitochondria-based functions in the “Molecular and Cellular Functions” category. Note that two grade III diseases and two having IDH1 mutations were among the samples
The CGGA database suggested higher SH3GLB1 expression in recurrent data (Fig. 1B right panel). Supportively, we found the protein expression enhanced in eight of nine recurrent tumor tissues than in primary ones (Fig. 1C and Supplementary Fig. S1C). In both the CGGA and the TCGA databases, survival curves of cases with higher SH3GLB1 expression showed worse prognosis (Fig. 1D and Supplementary Fig. S1D). Given that SH3GLB1 can alter mitochondrial functions [18], IPA for the RNA transcriptome of paired recurrent and treatment-naïve high-grade glioma samples was used to pinpoint related pathways. Among major mitochondria-related functional pathways, the OXPHOS pathway was predicted to similarly activate in thirteen of fourteen recurrent tumors (Fig. 1E). We noticed increased levels of multiple individual genes particularly in complexes I, III, IV, Fe/S cluster, and was supported by the CGGA database (Supplementary Fig. S2).
SH3GLB1 is associated with tumor-initiating features in tumors
We performed scRNA transcriptomic analysis on five GBM tumors to study the intratumoral cell distribution. Among the sorted clusters based on their differences in gene expression, five were identified as tumors (number 1, 2, 4, 5, and 6; Fig. 2A and Supplementary Fig. S3). We observed that cluster 4 exhibited the most abundant markers for TICs, such as CD133, Olig2, SOX2, Bmi1, and Myc (Fig. 2B) [25]. Notably, clusters 1 and 4 exhibited significantly higher expressions of SH3GLB1 than clusters 2 and 6 (Fig. 2C), in which both possessed the lowest levels of TIC profiles. Next, hierarchical clustering by OXPHOS genes suggested that cluster 4 expressed distinct features from clusters 2 and 6, with clusters 1 and 5 being the intermediate types (Fig. 2D). Higher expression of genes was most commonly noted in cluster 4 in the entire complexes or in the individual complexes of I, IV, and the Fe/S clusters of OXPHOS (Fig. 2E). Additionally, the genes in complex III were generally expressed higher in cluster 1. These results demonstrated that SH3GLB1 and the altered OXPHOS genes were associated with TIC-feature cells, which could inherit resistance characteristics [5].

Higher SH3GLB1 levels are related to specific subsets of the tumor. A The tSNE plot of the scRNA transcriptome from five GBM tumor samples sorted into tumor clusters. B Common genes related to tumor-initiating cell features are shown in the heatmap plot. C Expression level of SH3GLB1 in tumor clusters is shown in the violin plot. D The statistical similarity matrix between the indicated clusters was based on the OXPHOS-related genes. E Levels of OXPHOS-related genes in the tumor clusters are shown in the heatmap by complexes
Sp1 in the resistant cells activates SH3GLB1 transcription
We previously demonstrated that upregulation of Sp1 plays crucial roles in the development of TMZ resistance in GBM [20]. By examining the data available from the CGGA database, a positive correlation between the expression of SH3GLB1 and Sp1 was found in GBM (Fig. 3A). A conserved binding site for Sp1 is present in the promoter region of endophilin family members [26]. We hereby studied TMZ-resistant GBM cells and found increased level of Sp1 binding on the promoter region of SH3GLB1, as compared to the drug-sensitive counterparts (Fig. 3B). The drug resistant cells also exhibited higher expression of Sp1 and SH3GLB1 comparing to the drug sensitive cells (Fig. 3C). Downregulation of Sp1 by siRNA decreased the expression of SH3GLB1 in U87MG-R and Pt#3-R cells (Fig. 3D). Inhibiting Sp1 by small molecule inhibitor, Mithramycin A, attenuated the upregulation effects of TMZ on SH3GLB1 expression (Fig. 3E). We observed higher SH3GLB1 and SP1 expression in CD133+ P1S cells than in their CD133− counterparts (Fig. 3F) [5].

Sp1 promotes SH3GLB1 expression. A Genetic expression of SH3GLB1 and Sp1 from the GBM dataset of the CGGA database is shown in scatter plots with correlation assessment. B Schematic graph suggesting the potential Sp1 binding site on the SH3GLB1 promoter. Enhanced binding of Sp1 to the SH3GLB1 promoter is shown by chromatin immunoprecipitation assay in the resistant cells. C Western blot analysis showing enhanced Sp1 and SH3GLB1 expression in the resistant cells. D Western blot analysis showing reduced SH3GLB1 expression in the resistant cells with siSp1. E Western blot analysis showing that mithramycin A (MA) alleviated the TMZ-induced enhancement of Sp1 and SH3GLB1. F Sorted CD133+ and CD133− subsets from patient-derived primary GBM cells. The former exhibit higher Sp1, LC3B-II and lower p62 in the Western blot analysis. N = 3 in each group, *p < 0.05
SH3GLB1 is associated with autophagy in response to TMZ in resistant cells
Autophagy has been learned to contribute to chemotherapy-resistance that is related to TIC-feature cancer cells [27]. Notably, increased LC3B-II and decreased p62 were revealed in the CD133+ P1S cells (Fig. 3F). To demonstrate SH3GLB1 to be crucial for autophagy in resistant cells, we first found SH3GLB1 co-localized with LC3, suggesting that its association with autophagosomes (Fig. 4A). Next, application of shSH3GLB1 in the resistant cells resulted in increased p62 expression and reduced LC3B-II expression (Fig. 4B). Conversely, SH3GLB1 overexpression in the parental cells decreased p62 expression and increased LC3B-II expression (Fig. 4C).

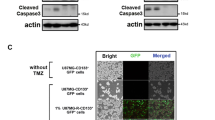
SH3GLB1 downregulation reduces TMZ-enhanced autophagy. A The SH3GLB1 protein (Red) is colocalized (white arrow) with autophagosomes labeled by LC3-EGFP (Green). The statistic graph showing the numbers of autophagic puncta (dot). Scale bar = 50 μm (B) Resistant cells treated with shSH3GLB1 exhibits attenuated autophagy levels. C The parental cells with SH3GLB1 overexpression exhibit enhanced autophagy levels. D Western blot analysis showing the expression of LC3, p62, Atg5–12 complex, and OXPHOS complexes from cells with or without SH3GLB1 knockdown that were cotreated with chloroquine (CQ). E Western blot analysis showing increased levels of cleaved caspase 3 in recurrent GBM spheroid cultures with siSH3GLB1 and TMZ cotreatment for 24 hours. F Autophagy enhanced by evidence of altered LC3B-II and p62 expression after TMZ treatment for 24 hours was attenuated in siSH3GLB1 cells. G Attenuated detection of induced acridine orange–stained acidic vesicular organelles is shown following TMZ treatment (24 hours) and siSH3GLB1 knockdown. The statistical graph obtained by flow cytometry is shown in the right panel (Scale bar = 200 μm; *p < 0.05; N = 3 in each group; R: resistance)
The functional relevance of autophagy was next investigated. Dual blockade of autophagy with shSH3GLB1 and chloroquine attenuated the Atg5–12 conjugate expression and OXPHOS in complexes I and III (Fig. 4D). ShSH3GLB1 significantly promoted caspase 3 cleavage (activation) in response to TMZ treatment in CD133+ P1S, suggesting that its role is related to TIC features (Fig. 4E). Likewise, SH3GLB1 knockdown in the CD133+ cells attenuated TMZ-induced autophagy (Fig. 4F). This also resulted in reduced formation of acidic vesicular organelles (Fig. 4G). Together, these findings revealed that SH3GLB1-related autophagy plays important roles in resistance cells.
SH3GLB1 affects mitochondrial function and TMZ susceptibility
As above mentioned, SH3GLB1-induced autophagy affects assembly of OXPHOS complexes (Fig. 4D). Downregulation of SH3GLB1 by shRNA attenuated TMZ-enhanced expression of Atg5–12 conjugate and the levels of assembly in OXPHOS complexes I and III (Fig. 5A). Using a live-cell metabolic assay, a switch of metabolic phenotype (OCR/ECAR) was observed in U87MG-R and Pt#3-R cells with SH3GLB1 downregulation (Supplementary Fig. S4A). Notably, SH3GLB1 suppression alleviated TMZ-enhanced mitochondrial respiration, such as basal respiration, ATP production, and proton leakage, suggesting that SH3GLB1 regulates OXPHOS and mitochondrial activity in TMZ-resistant cells (Fig. 5B). The pattern was similarly found in respect of the maximal and the nonmitochondrial respirations but not the spare respiration capacity (Supplementary Fig. S4B). In addition, SH3GLB1 downregulation alleviated TMZ-enhanced ΔΨm, suggesting the protein is a determinant factor for mitochondria polarization in the resistant cells (Fig. 5C and Supplementary Fig. S4C).

SH3GLB1 enhances mitochondrial functions and TMZ resistance. A Western blot analysis showing the expression of LC3, p62, Atg5–12 complex, and OXPHOS complexes from cells with or without shSH3GLB1 that were treated with TMZ. B OCR was measured using a Seahorse Analyzer. The individual OCR parameters were measured using the indicated reagents (left). The summary graphs of mitochondrial respiration are illustrated at right and in the Supplementary Fig. S4B. C Altered dynamics of ΔΨm were detected using JC-1 dye in U87MG-R cells treated with TMZ (24 hours) and shSH3GLB1. The positive control with carbonyl cyanide m-chlorophenyl hydrazone (CCCP, 10 μM for 24 hours) indicates ΔΨm impairment. The results are illustrated in the bar graph. (D and E) The results of cell density assays are illustrated with (D) control (black) or shSH3GLB1 (gray); (E) vector (black) or SH3GLB1-overexpressing plasmid (gray) that were treated with TMZ for 72 hours. N = 3 in each group, *p < 0.05
Inhibition of cell growth was also observed when TMZ was administered to shSH3GLB1 resistant cells (Fig. 5D). Conversely, SH3GLB1 overexpression attenuated the effect of TMZ on U87MG and Pt#3 cells (Fig. 5E). The inhibitory effect of a complex 1 inhibitor, IACS-010759, was reversed by SH3GLB1 overexpression (Supplementary Fig. S5A). Similarly, TMZ-resistant cells also exhibited decreased susceptibility to IACS-010759, which was reversed by SH3GLB1 knockdown (Supplementary Fig. S5B). These results suggested that SH3GLB1 and the related OXPHOS play an important role in the induction of TMZ resistance in GBM cells.
Downregulation of SH3GLB1 reinstates the TMZ treatment effect in xenograft GBM models
To verify the roles of SH3GLB1 in TMZ-resistant cells in vivo, a xenograft animal model was applied with luciferase-carrying U87MG-R cells implanted into the subcutaneous area of the flank. The results revealed slower growth rate of tumors in the shSH3GLB1 group that received TMZ treatment (Fig. 6A and Supplementary Fig. S6A ~ F). Analysis of the tumors showed reduced levels of the enhanced LC3B-II in the TMZ/shSH3GLB1 group and the decreased levels of assembly in OXPHOS complex I in shSH3GLB1 groups (Fig. 6B). These findings were compatible to the results of in vitro studies. Finally, the study with the orthotopic mouse model showed prolonged survival in the shSH3GLB1 group administrated with TMZ, demonstrating the significance of SH3GLB1 in respect of disease treatment (Fig. 6C).

SH3GLB1-knockdown resistant cells regain treatment benefit from TMZ in the xenograft mouse model. A The bioluminescent signal from the luciferase-carrying U87MG-R cells in tumor carrying mice was detected using an IVIS imaging system and plotted (N = 6 in each group, *p < 0.05). B The protein immunoblot of the tumor showing SH3GLB1 and the proteins related to autophagy and OXPHOS (N = 3 in each group). C Kaplan-Meier plot showing the survival curves of the orthotopic animals in each group (N = 6 in each group, *p < 0.05). D The working scheme of SH3GLB1-regulated OXPHOS for TMZ resistance
Discussion
SH3GLB1 has been linked to TMZ susceptibility owing to its role in cell proliferation and invasion [28]. In this study, we confirmed its role in mitochondrial metabolism, which could lead to the induction of TMZ resistance. SH3GLB1 enhancement was associated with cells in clusters 1, 4, and 5 (Fig. 2C). These clusters expressed genetic signatures of oligodendrocyte-progenitor-like and neural-progenitor-like subsets regarding of their cellular states, differentiating them from clusters 2 and 6 (Supplementary Fig. S3B) [29]. They also showed enriched CD133, SOX2, OCT4, and OLIG2 (Fig. 2B), which were compatible to the report that these genes often biased toward both cellular states [30]. Accordingly, a higher level of SH3GLB1 and enhanced autophagy were inherited in CD133+ tumor cells (Fig. 3F) [27, 31]. Pre-existing clones with TIC-features can endure drug toxicities and cause recurrence [32]. Given the tumor-propagating capability in these two cellular states [30], the tumor-initiating potentials here can confer substantial effects in causing failure of disease control. Supportively, the increased SH3GLB1 expression in recurrent clinical samples suggested its association with the resistance (Fig. 1C). This is because TMZ has generally been applied through the National Health Insurance in Taiwan. Studies on cell lines further confirmed SH3GLB1 to be the critical factor for cell resistance against TMZ. These also excluded interference from irradiation of the tumor samples, which is often used concomitantly with the drug to treat GBM. Notably, we identified Sp1-mediated promotor regulation (Fig. 3). Accumulation of the transcriptional factor contributes to the TIC-resistant features of SOD2, thereby promoting ROS regulation [33]. Simultaneously, SH3GLB1 is enhanced for metabolic regulation, which is supported by the data here. SH3GLB1 is crucial for normal brain function, as well as aiding in recovery from injury [34]. It is therefore not surprising that it can enhance the disease progression of the brain tumor, leading to more serious clinical outcomes.
Traditionally, SH3GLB1 is known as a tumor-suppressor [35]. However, it has also been reported to affect cancer in other ways [36]. SH3GLB1 can promote cell death by inducing apoptosis through Bax, or by redirecting to necrosis through autophagy and GSK3β inhibition [37, 38]. It has been reported that downregulation of SH3GLB1 contributed to tumorigenesis by upregulating mitochondrial function in melanoma cells. However, this reaction was not associated with autophagy, and it was shown that the cells had equal sensitivity to anti-tumor treatments, which were not affected by SH3GLB1 levels [39]. Nonetheless, under certain circumstances, SH3GLB1 can enhance tumor migration and metastasis [40]. Additionally, results from the current study showed that SH3GLB1 enhances tumor resistance in TMZ conditions, with reduced activation of caspase 3. Further studies revealed that SH3GLB1 repressed the N-terminal epitope 6A7 of Bax in GBM models, suggesting the anti-apoptosis effect (data not shown). The protein-induced autophagy and reprogrammed OXPHOS in the resistant cells contributed toward resisting the anti-tumor effects of TMZ, highlighting the contribution of our study to the existing literatures.
Despite the general consensus that inhibition of autophagy process enhances the anti-tumor effect of TMZ in glioma cells [41], CQ clinical trials have not reached a clear conclusion in terms of treatment outcome [42, 43]. Autophagy exerts various pro- and anti-tumor effects, therefore, it is important to dissect and study the impacts in detail to better understand the complexity of this phenomenon. The scRNA transcriptome of clinical samples revealed that SH3GLB1 was most prevalent in cells with the most altered OXPHOS genes (Fig. 2). On a cellular level, a decrease in autophagy by CQ and shSH3GLB1 resulted in reduced expression of OXPHOS genes (Fig. 4D). The mechanism of SH3GLB1-autophagy in regulating cell metabolism was not elucidated here. However, it has been reported that the fatty acid production catabolized by autophagy mediated the OXPHOS reaction [10]. We then determined that SH3GLB1-related autophagy is functionally associated with the development of TMZ resistance, by regulating OXPHOS. This warrants exploration of potential anti-autophagy strategies to restore the anti-tumor effects of TMZ as well as a detailed investigation of the underlying metabolism.
Expression of individual OXPHOS genes varied in these clinical data as well as in the CGGA database (Supplementary Fig. S2). Genes encoding the core subunits of the complexes can affect the malignant cells in opposing manners. For example, low NDUFS1 and high NDUFS8 expression levels in lung cancer were found to predict poor overall survival [44]. In our study, higher levels of NDUFAF7 (complex I) and ATP5F1A (complex V) were related to more severe clinical prognoses, and were clearly enhanced in TIC-feature clusters (Fig. 2E and Supplementary Fig. S7A–B). However, more positive clinical prognoses were observed when NDUFB10 (complex I), NDUFB2 (complex I), SDHA (complex II), UQCRFS1 (complex III), UQCRB (complex III), COX7C (complex IV), IBA57 (Fe/S clusters), and CoQ3 (CoQ synthesis) were higher (Supplementary Fig. S7C-J), but had no tendency related to TIC-features in our data (Fig. 2E). Alterations in single genes can be functionally redundant and cause discrepancies, leading to incompatible results. Therefore, a functional assessment related to mitochondrial complexes would be more representative [45]. We found that SH3GLB1 promoted metabolism during TMZ treatment by altering ΔΨm and mitochondrial respiration (Fig. 5). Notably, SH3GLB1 did not consistently affect ECAR (Supplementary Fig. S4A), suggesting that resistant cells are more prone to utilize OXPHOS than glycolysis [9]. In summary, the network is functionally important for TIC features in TMZ-resistant cells [46]. Therefore, the OXPHOS profiles for the resilient tumor subsets provide important information.
Alteration of metabolic profiles has been suggested as a potential strategy for cancer treatment. With the discovery of high-affinity complex I inhibitors, major advances in research is expected by uncovering the benefits of suppressing intracellular OXPHOS function. For example, potential synergistic therapy with other anticancer agents has been suggested in a breast cancer model [47]. Selection of suitable candidates is vital because modulating metabolism causes varying results [48]. For example, treatment resistance in tumors can depend on the deregulated metabolic reprogramming [49]. Additionally, SH3GLB1 levels can potentially serve as a GBM cell indicator as they antagonized the complex I inhibitor effect (Supplementary Fig. S5). In the resistant cells, shSH3GLB1 attenuated TMZ-enhanced OXPHOS significantly by blocking the assembly of complex 1 (Figs. 5A and 6B). Our findings warrant further exploration for strategies to maximize the effect of OXPHOS-related therapies.
Conclusions
We found that SH3GLB1 enrichment in TIC-feature subsets enhances autophagy, leading to increased ΔΨm and enhanced OXPHOS (Fig. 6D). The importance of SH3GLB1 in the promotion of TMZ resistance was also verified using animal experiments and clinical data. SH3GLB1 was regulated via Sp1, and together they contribute to cell evasion from TMZ cytotoxicity in GBM. It is noteworthy that the data could be replicated in other cell lines including A172 and pt.#5 (Supplementary Figs. S8 and S9), confirming the impact of the protein in cell metabolism. However, this study does not exclude the possibility that SH3GLB1 regulates the OXPHOS genes by other means. Nevertheless, valuable contributions in terms of SH3GLB1 have been highlighted. This deserves further attention regarding the effect of TMZ or novel OXPHOS modulation strategies in future studies.
Availability of data and materials
All data found in the present study are available within the article and supplementary data.
Abbreviations
- CCCP :
-
Carbonyl cyanide m-chlorophenyl hydrazone
- CGGA:
-
Chinese Glioma Genome Atlas
- ChIP:
-
Chromatin immunoprecipitation
- CQ:
-
Chloroquine
- ΔΨm:
-
Membrane potential
- ECAR:
-
Extracellular acidification rate
- GBM:
-
Glioblastoma
- IHC:
-
Immunohistochemistry
- IPA:
-
Ingenuity pathway analysis
- OCR:
-
Oxygen consumption rate
- OXPHOS:
-
Oxidative phosphorylation
- ROS:
-
Reactive oxygen species
- RSEM:
-
RNA-Seq by Expectation Maximization
- scRNA transcriptome:
-
Single-cell RNA transcriptome
- shRNA:
-
Short hairpin RNA
- siRNA:
-
Small interfering RNA
- SOD2:
-
Superoxide dismutase 2
- Sp1:
-
Specificity protein 1
- TICs:
-
Tumor-initiating cells
- TMZ:
-
Temozolomide
References
Dolecek TA, Propp JM, Stroup NE, Kruchko C. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2005-2009. Neuro-Oncol. 2012;14(Suppl 5):v1–49.
Zhang WB, Wang Z, Shu F, Jin YH, Liu HY, Wang QJ, et al. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem. 2010;285(52):40461–71.
Chamberlain MC. Temozolomide: therapeutic limitations in the treatment of adult high-grade gliomas. Expert Rev Neurother. 2010;10(10):1537–44.
Wick W, Weller M, van den Bent M, Sanson M, Weiler M, von Deimling A, et al. MGMT testing--the challenges for biomarker-based glioma treatment. Nat Rev Neurol. 2014;10(7):372–85.
Chien CH, Chuang JY, Yang ST, Yang WB, Chen PY, Hsu TI, et al. Enrichment of superoxide dismutase 2 in glioblastoma confers to acquisition of temozolomide resistance that is associated with tumor-initiating cell subsets. J Biomed Sci. 2019;26(1):77.
Quijano C, Trujillo M, Castro L, Trostchansky A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016;8:28–42.
Guerra F, Arbini AA, L. Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg. 2017;1858(8):686–99.
Oliva CR, Moellering DR, Gillespie GY, Griguer CE. Acquisition of chemoresistance in gliomas is associated with increased mitochondrial coupling and decreased ROS production. Plos One. 2011;6(9):e24665.
Rabé M, Dumont S, Álvarez-Arenas A, Janati H, Belmonte-Beitia J, Calvo GF, et al. Identification of a transient state during the acquisition of temozolomide resistance in glioblastoma. Cell Death Dis. 2020;11(1):19.
Bosc C, Broin N, Fanjul M, Saland E, Farge T, Courdy C, et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat Commun. 2020;11(1):4056.
Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells. 2013;36(1):7–16.
Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961–7.
Buccarelli M, Marconi M, Pacioni S, De Pascalis I, D'Alessandris QG, Martini M, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018;9(8):841.
Takahashi Y, Meyerkord CL, Wang HG. Bif-1/endophilin B1: a candidate for crescent driving force in autophagy. Cell Death Differ. 2009;16(7):947–55.
Wan J, Cheung AY, Fu WY, Wu C, Zhang M, Mobley WC, et al. Endophilin B1 as a novel regulator of nerve growth factor/ TrkA trafficking and neurite outgrowth. J Neurosci. 2008;28(36):9002–12.
Wong AS, Lee RH, Cheung AY, Yeung PK, Chung SK, Cheung ZH, et al. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson's disease. Nat Cell Biol. 2011;13(5):568–79.
Wang DB, Kinoshita Y, Kinoshita C, Uo T, Sopher BL, Cudaback E, et al. Loss of endophilin-B1 exacerbates Alzheimer’s disease pathology. Brain. 2015;138(7):2005–19.
Takahashi Y, Young MM, Serfass JM, Hori T, Wang HG. Sh3glb1/Bif-1 and mitophagy: acquisition of apoptosis resistance during Myc-driven lymphomagenesis. Autophagy. 2013;9(7):1107–9.
Tsai YT, Lo WL, Chen PY, Ko CY, Chuang JY, Kao TJ, et al. Reprogramming of arachidonate metabolism confers temozolomide resistance to glioblastoma through enhancing mitochondrial activity in fatty acid oxidation. J Biomed Sci. 2022;29(1):21.
Chang KY, Hsu TI, Hsu CC, Tsai SY, Liu JJ, Chou SW, et al. Specificity protein 1-modulated superoxide dismutase 2 enhances temozolomide resistance in glioblastoma, which is independent of O (6)-methylguanine-DNA methyltransferase. Redox Biol. 2017;13:655–64.
Bowman RL, Wang Q, Carro A, Verhaak RGW, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology. 2017;19(1):139–41.
Zhao Z, Zhang KN, Wang Q, Li G, Zeng F, Zhang Y, et al. Chinese glioma genome atlas (CGGA): a comprehensive resource with functional genomic data from Chinese glioma patients. Genomics Proteomics Bioinformatics. 2021;19(1):1–12.
Couturier CP, Ayyadhury S, Le PU, Nadaf J, Monlong J, Riva G, et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat Commun. 2020;11(1):3406.
Arroyo JD, Jourdain AA, Calvo SE, Ballarano CA, Doench JG, Root DE, et al. A genome-wide CRISPR death screen identifies genes essential for oxidative phosphorylation. Cell Metab. 2016;24(6):875–85.
Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, Itinteang T. Cancer stem cell hierarchy in glioblastoma Multiforme. Front Surg. 2016;3:21.
Reutens AT, Begley CG. Endophilin-1: a multifunctional protein. Int J Biochem Cell Biol. 2002;34(10):1173–7.
Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Cell Death Differ. 2019;26(4):690–702.
Zhang H, Lu X, Wang N, Wang J, Cao Y, Wang T, et al. Autophagy-related gene expression is an independent prognostic indicator of glioma. Oncotarget. 2017;8(37):60987–1000.
Naftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178(4):835–49.
Suvà ML, Tirosh I. The glioma stem cell model in the era of single-cell genomics. Cancer Cell. 2020;37(5):630–6.
Vora P, Venugopal C, Salim SK, Tatari N, Bakhshinyan D, Singh M, et al. The rational development of CD133-targeting immunotherapies for glioblastoma. Cell Stem Cell. 2020;26(6):832–44.e6.
Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. 2018;18(11):669–80.
Chang KY, Huang CT, Hsu TI, Hsu CC, Liu JJ, Chuang CK, et al. Stress stimuli induce cancer-stemness gene expression via Sp1 activation leading to therapeutic resistance in glioblastoma. Biochem Biophys Res Commun. 2017;493(1):14–9.
Wang DB, Kinoshita C, Kinoshita Y, Sopher BL, Uo T, Lee RJ, et al. Neuronal susceptibility to beta-amyloid toxicity and ischemic injury involves histone deacetylase-2 regulation of endophilin-B1. Brain Pathol. 2019;29(2):164–75.
Ko YH, Cho YS, Won HS, An HJ, Sun DS, Hong SU, et al. Stage-stratified analysis of prognostic significance of Bax-interacting factor-1 expression in resected colorectal cancer. Biomed Res Int. 2013;2013:329839.
Kim SY, Oh YL, Kim KM, Jeong EG, Kim MS, Yoo NJ, et al. Decreased expression of Bax-interacting factor-1 (Bif-1) in invasive urinary bladder and gallbladder cancers. Pathol. 2008;40(6):553–7.
Etxebarria A, Terrones O, Yamaguchi H, Landajuela A, Landeta O, Antonsson B, et al. Endophilin B1/Bif-1 stimulates BAX activation independently from its capacity to produce large scale membrane morphological rearrangements. J Biol Chem. 2009;284(7):4200–12.
Yang J, Takahashi Y, Cheng E, Liu J, Terranova PF, Zhao B, et al. GSK-3beta promotes cell survival by modulating Bif-1-dependent autophagy and cell death. J Cell Sci. 2010;123(6):861–70.
Frangež Ž, Fernández-Marrero Y, Stojkov D, Seyed Jafari SM, Hunger RE, Djonov V, et al. BIF-1 inhibits both mitochondrial and glycolytic ATP production: its downregulation promotes melanoma growth. Oncogene. 2020;39(26):4944–55.
Zhang C, Liu F, Chen H, Li N, Luo Z, Guo W, et al. Bif-1 promotes tumor cell migration and metastasis via Cdc42 expression and activity. Clin Exp Metastasis. 2017;34(1):11–23.
Kanzawa T, Germano IM, Komata T, Ito H, Kondo Y, Kondo S. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11(4):448–57.
Rosenfeld MR, Ye X, Supko JG, Desideri S, Grossman SA, Brem S, et al. A phase I/II trial of hydroxychloroquine in conjunction with radiation therapy and concurrent and adjuvant temozolomide in patients with newly diagnosed glioblastoma multiforme. Autophagy. 2014;10(8):1359–68.
Compter I, Eekers DBP, Hoeben A, Rouschop KMA, Reymen B, Ackermans L, et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: a phase IB trial. Autophagy. 2021;17(9):2604–12.
Su CY, Chang YC, Yang CJ, Huang MS, Hsiao M. The opposite prognostic effect of NDUFS1 and NDUFS8 in lung cancer reflects the oncojanus role of mitochondrial complex I. Sci Rep. 2016;6:31357.
Shi Y, Lim SK, Liang Q, Iyer SV, Wang HY, Wang Z, et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature. 2019;567(7748):341–6.
El Hout M, Cosialls E, Mehrpour M, Hamaï A. Crosstalk between autophagy and metabolic regulation of cancer stem cells. Mol Cancer. 2020;19(1):27.
Evans KW, Yuca E, Scott SS, Zhao M, Paez Arango N, Cruz Pico CX, et al. Oxidative phosphorylation is a metabolic vulnerability in chemotherapy-resistant triple negative breast cancer. Cancer Res. 2021;81(21):5572–81.
Sica V, Bravo-San Pedro JM, Stoll G, Kroemer G. Oxidative phosphorylation as a potential therapeutic target for cancer therapy. Int J Cancer. 2020;146(1):10–7.
Morandi A, Indraccolo S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim Biophys Acta Rev Cancer. 2017;1868(1):1–6.
Acknowledgments
Not applicable.
Funding
This work was supported by grants from Ministry of Science and Technology (Taiwan, MOST 110–2314-B-400-030), National Health Research Institutes (Taiwan, CA-110-PP-19), and from National Cheng Kung University Hospital (Taiwan, NCKUH-11009024).
Author information
Authors and Affiliations
Contributions
C.H.C. designed the model, interpret the results, and wrote the manuscript. C.H.C., A.C.W., C.C.Lai, P.I.C., J.M.C. carried out the experiment. J.Y.C. conceived the original idea. W.B.Y., C.C.Lai, S.M.C., D.Y.H., S.H.C. performed the database analysis. J.S.L., C.Y.H., P.Y.C., collected patient samples. S.T.Y provides materials. W.A.L interpreted the results. C.C.Liu participated in experiment discussion. K.Y.C. supervised the project and was in charge of the overall direction. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The application of clinical tumor samples for IHC staining and scRNA transcriptome was approved by the IRB of National Cheng Kung University Hospital (A-ER-109-039) and National Health Research Institutes (EC1080202). The application of the 14 RNA-seq data were approved by the IRB of Chang Gung Medical Hospital (201800077B0).
All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of National Health Research Institutes (NHRI-IACUC-108048-A).
Consent for publication
Not applicable.
Competing interests
The authors declared no potential competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
(A) Differentiation of mitochondria-related genes in A172 TMZ resistance cells (A172-R) with shSOD2 or shControl is shown in the heatmap graphs. A total of 84 genes was assessible from the assay. Among them, however, only 71 of the items were found in the database of mitochondria-related genes according to MitoCarta 2.0. (B) The correlation between SH3GLB1 and SOD2 was also shown in TCGA-GBM dataset and the 14 paired patients. (C) Nine cases showing expression of SH3GLB1 in the paired primary- and recurrent-tissues using IHC staining (100x and 400x magnification). (D) Kaplan-Meier curves of TCGA-GBMLGG (GBM and low-grade glioma) database showed higher SH3GLB1 levels caused poor survival. Figure S2. Ingenuity Pathway Analysis was applied for analysis of the major mitochondria-related molecular and cellular functional alterations in the transcriptome data from 14 RNA-seq data from paired recurrent and treatment-naïve high-grade glioma samples. As shown in the figure, sample numbers of down- (left column) or up-regulated (right column) genes. In each grid, the color is determined by the rank percentile, and the number represents sample that fulfills the criteria. The levels and significance of the genes ratio from CGGA glioma database (recurrent versus primary) are also shown aside. Note that none of the genes in complex II had CGGA database significance better than **, and the genes in complex V and CoQ synthesis was not higher in the resistant groups in the 14 paired samples. ***p < 0.001, **p < 0.01, *p < 0.05, NS: not significant. NA: data not available. Figure S3. (A) Five naïve glioblastoma tumor samples were used for single-cell transcriptome and were sorted into nine clusters according to their gene expression (Fig. 2A). The heatmap graph shows the common related genes of GBM and tumor-initiating cells among the clusters. Note that clusters 3, 7, 8, and 9 are identified as immunocytes because of the presence of the markers such as CD3E/G, GZMA, ITGAM, ITGAX. (B) The clusters reclassified by the studies of Neftel et al. (Cell, 2019) were divided into four cellular states. Figure S4. (A) Cell metabolic phenotype profile were measured using Seahorse Analyzer (OCR and ECAR). (B) The individual OCR parameters were measured using the indicated reagents and plotted in Fig. 5B, with the statistic graphs showing the parameters of maximal respiration, spare respiratory capacity, and nonmitochondrial respiration. (C) The parental cells transfected with SH3GLB1-overexpressing vector were stained with a JC-1 dye, showing the enhanced fluorescent ratio. Figure S5. (A) IACS-010759 effect to cell density for 72 hours of the parental cells with or without SH3GLB1 overexpression are shown in the bar graph. (B) IACS-010759 effect to cell density for 72 hours of the resistant cells with or without SH3GLB1 knockdown are shown in the bar graph. (*P < 0.05) Figure S6. (A) The U87MG-R luciferase-carrying cells (U87MG-R-luc-EGFP) were transfected with shSH3GLB1 used in animal experiment. SOD2 levels did not be affected by SH3GLB1 deficiency. (B) The tumor volume was measured at Day 20 and Day 23 according to the National Cancer Institute formula. (C) The body weight was recorded at the indicated day as the statistic graph. (*P < 0.05) (D) The image shows the tumor extracted from euthanized mice (scale bar = 1 cm) (E) The SH3GLB1 expression in the tissue using an IHC staining is shown (scale bar = 200 μm). (F) The body weight was recorded and shown in the statistical graph. Figure S7. Kaplan-Meier curves of GBM database from CGGA showed the survival of the indicated subunits genes of OXPHOS complex. Figure S8. A172 or A172-R cells are used in the results including roles of SH3GLB1 on Sp1 promoter, Sp1 expression, 36 autophagy levels, OXPHOS levels, cell density and OCR analysis. Figure S9. Pt#5 or Pt#5-R cells are used in the results including roles of SH3GLB1 on Sp1 promoter, Sp1 expression, autophagy levels, OXPHOS levels, cell density and OCR analysis.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chien, CH., Yang, WB., Chuang, JY. et al. SH3GLB1-related autophagy mediates mitochondrial metabolism to acquire resistance against temozolomide in glioblastoma. J Exp Clin Cancer Res 41, 220 (2022). https://doi.org/10.1186/s13046-022-02429-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02429-8