
Abstract
The cohesin complex controls faithful chromosome segregation by pairing sister chromatids after DNA replication until mitosis. In addition, it is crucial for hierarchal three-dimensional organization of the genome, transcription regulation and maintaining DNA integrity. The core complex subunits SMC1A, SMC3, STAG1/2, and RAD21 as well as its modulators, have been found to be recurrently mutated in human cancers. The mechanisms by which cohesin mutations trigger cancer development and disease progression are still poorly understood. Since cohesin is involved in a range of chromosome-related processes, the outcome of cohesin mutations in cancer is complex. Herein, we discuss recent discoveries regarding cohesin that provide new insight into its role in tumorigenesis.
Similar content being viewed by others
Background
Cohesin belongs to the family of SMC (Structural Maintenance of Chromosomes) complexes that organize chromosomal DNA topology in all living organisms, from bacteria to eukaryotes.
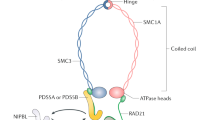
The core of the cohesin complex is composed of four subunits, SMC1A, SMC3, RAD21/Scc1, and STAG (Stromal antigen)/SA/Scc3, with a ring-shaped structure. SMC1A and SMC3 are characterized by a globular flexible hinge domain bordered by two coiled-coil domains, which fold back on themselves at the hinge, forming a long antiparallel alpha-helical coiled-coil arm that conveys the C- and N-termini together. This latter holds the Walker A box, which binds ATP, whereas the C-terminal contains the Walker B, binding to DNA. SMC1A and SMC3 dimerize at the hinge domains on one side, forming a V-shaped structure through hydrophobic interactions, and RAD21 closes the ring by connecting the SMC1A and SMC3 head domains on the other side. The fourth subunit, STAG1 or STAG2, binds to cohesin by contacting RAD21 and SMC subunits. STAG1/2 are essential for the association of cohesin with DNA (Fig. 1, Table 1) [1].

Structure of the cohesin complex. The cohesin ring is composed of SMC1A, SMC3, RAD21, and STAG1/2. SMC proteins are long polypeptides that fold back on themselves to form a coiled-coil domain with a hinge domain at one end and an ATPase domain at the other. SMC1A and SMC3 form a V-shaped structure by interaction of their hinge domains. The N- and C-terminus of RAD21 interact with SMC3 and SMC1A respectively. The STAG1/2 subunit interacts with RAD21.The NIPBL/MAU2 dimer loads cohesin onto DNA, whereas WAPL/PDS5 release cohesin from chromosomes
Cohesin is highly concentrated at the centromeric regions, while it has a frequency of only 10 kbp in yeast and up to 100 kbp in the higher organisms, along the chromosome arms [2, 3]. Cohesin activity during the cell cycle is regulated by interaction with several regulatory factors (Table 1). During the G1 phase in yeast or at the end of telophase of the previous cell cycle in mammalian cells, the cohesin complex is loaded onto chromatin in cooperation with the activity of the auxiliary factors NIPBL (Nipped-B like)-MAU2. The interaction of cohesin with sister chromatids is established by ESCO 1/2 (Establishment of cohesion 1 homolog 1/2) Eco1/Ctf7 that acetylates the SMC3 subunit during the S-phase. PDS5A/B (Precocious Dissociation of Sisters 5) are also involved in this process. In fact, they interact with cohesin for its establishment and maintenance [4,5,6,7].
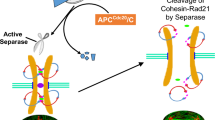
Cohesin dissociation from chromatin requires the activity of WAPL (Wings Apart-like Protein 1) which interacts directly with RAD21 and STAG1/2 [8, 9]. Recently, it has been shown that WAPL is deubiquitinated by USP37 (Ubiquitin-specific peptidase 37) during mitosis, thereby regulating chromosomal segregation, cohesion and mitotic progression [10, 11]. Finally, once chromosomes are correctly bioriented on the mitotic spindle at anaphase, cohesin is completely removed from chromosomes by the endopeptidase ESPL1 (Extra spindle pole bodies-like 1)/SEPARASE protein that cleaves RAD21 [12, 13]. This cleavage permits opening of the cohesin ring, causing it to dissociate from chromosomes and causing sister chromatid separation [12]. SEPARASE is activated by the proteolysis of its inhibitory partner PTTG1 (Pituitary tumor transforming gene 1) /SECURIN and the simultaneous degradation of CDK1’s subunit cyclin B [14, 15].
The cohesion of sister chromatids and resultant correct chromosome segregation are the best-known functions of cohesin. However, over the last few years increasing experimental evidence has brought to light its key roles in regulating gene expression by mediating functional connections between promoters and their distal enhancers [16, 17], in promoting DNA repair by homologous recombination and non-homologous end joining [18,19,20], in controlling fork replication stability [21, 22] and facilitating the recruitment of proteins involved in the activation of the intra-S and G2/M checkpoints [23, 24].
Germline pathogenic variants in cohesin core genes and associated factors are responsible for a class of human rare diseases collectively called cohesinopathies or DTRs (disorders of transcriptional regulation) [25]. Variants in NIPBL, SMC1A, SMC3, HDAC8, RAD21, BRD4, ANKRD11, ESCO2 and AFF4 genes are indeed associated with Cornelia de Lange syndrome, Roberts syndrome, and CHOPS syndrome (Cognitive, Heart defects, Obesity, Pulmonary and Short stature), the most frequently encountered and investigated diseases linked to cohesin dysfunction [26,27,28,29,30,31,32,33,34]. Of note, these diseases are characterized by gene expression dysregulation and impairment in DNA repair [35,36,37,38,39,40,41]. Somatic variants and gene dysregulation are instead associated with several types of cancer [42,43,44] including CRC (colorectal carcinoma) [45,46,47,48], breast cancer [49, 50], lung carcinoma [51], UBC (urothelial bladder carcinoma) [52,53,54,55,56], Ewing’s sarcoma [57,58,59], glioblastoma [60, 61], melanoma [62] and myeloid neoplasms [63,64,65,66].
The evolving realization that cohesin participates in a growing assortment of chromosome and chromatin-related processes suggests that its contribution to cancer development is complex. In this review, we summarize recent advances in the understanding of cohesin function in cancer pathogenesis.
Cohesin, topologically associated domains and CCCTC-binding factor
Mammalian genomes are organized at multiple levels. In fact, DNA forms complexes with many proteins at different levels of what is known as the higher order chromatin organization in order to efficiently compact itself. The cohesin is an architectural protein complex involved in gene compartmentalization, enhancer/promoter communication and in organizing the genome into regions called TADs (Topologically Associated Domains). The precise nature and definition of TADs remains a matter of debate. TADs appear to play a double action: to increase the possibilities that regulatory elements meet each other within a single domain, and to segregate physical interactions across boundaries, thus decreasing the chance that detrimental interactions occur [67]. In mammalian cells, TADs range in size from a few 100kbs to 5Mbs in size (with an average of 1MB). The findings that they exhibit a high degree of conservation between cell types and species suggested that TADs represent the fundamental unit of physical organization of the genome [68]. TAD boundaries strongly correlate with replication-timing domains [69] and are enriched for insulator elements such as CTCF (CCCTC-binding factor) [68, 70]. CTCF is an 11-zinc finger DNA-binding protein conserved across most animals, but absent from plants, C. elegans and yeast [71]. All interactions mediated by CTCF require the cohesin complex [72,73,74]. In fact, CTCF directly interacts with the cohesin, and it has been proposed that cohesin extrudes DNA loops until it is arrested by CTCF bound to DNA in a certain orientation or other barrier proteins [75,76,77,78]. These loops facilitate the interactions between enhancers and promoters (Fig. 2) [79, 80]. In this process, loop domains prevent enhancers from forming incorrect interactions with targets that are placed in a different loop domain [81, 82]. In the absence of WAPL, PDS5A and PDS5B proteins, cohesin forms extended loops, presumably by passing CTCF sites [74, 83]. In detail, CTCF blocks the cohesin complex by acting as a "boundary" if the 3’ ends of the CTCF binding motifs are oriented towards the interior of the TAD [84]. However, in addition to its function as a translocation barrier, CTCF possesses a distinct loop stabilizing activity, which is realized through direct interaction with RAD21-STAG subunits. In fact, the N-terminal segment of CTCF directly engages the RAD21-STAG subcomplex through the CES (Conserved and Essential Surface) domain [78].

Schematic illustration of the normal and mutated loop-extrusion mechanism. A Hypothetical structure of CTCF defined chromatin loop. CTCF stabilizes cohesin in the depicted conformation. B Example of abnormal loop formation mechanism
Cohesin and DNA repair
Genomic integrity is continually threatened by endogenous and exogenous damaging factors such as oxidative damage during metabolism, bases hydrolysis, X-rays, ultraviolet light, and various chemicals. Every day, human cells experience approximately 70,000 DNA lesions, about 75% of them SSBs (Single-Strand Breaks) [85, 86]. SSBs can also be converted to DSBs (Double-Strand Breaks) which, although less much frequent, are highly deleterious. Unrepaired DSBs can generate chromosome translocations, deletions, and insertions, which in turn could lead to genome instability and cancer development. During their evolution cells have acquired highly conserved mechanisms to detect and repair these lesions, thereby restoring genome integrity.
The cohesin complex facilitates the recruitment of proteins involved in cell cycle checkpoints and is also required for DNA damage-induced intra-S phase and G2/M checkpoints in mammalian cells [23]. In fact, cohesin subunits are substrates of ATM (Ataxia Telangiectasia Mutated) and ATR (Ataxia Telangiectasia and Rad3 related) protein kinases activated by specific damaged DNA. ATM phosphorylates SMC1A at Ser957 and Ser966 residues at the intra S-phase checkpoint following irradiation [87, 88]. Instead, ATR phosphorylates SMC1A at Ser957 in response to replication stress [24]. Intriguingly, both human and murine cells carrying mutated or non-phosphorylable SMC1A sites showed decreased cell survival as well as defects in DNA repair [88, 89]. DSBs are repaired by two distinct pathways called HR (Homologous Recombination) and NHEJ (Nonhomologous End-Joining). During HR, the DSB is repaired by exchanges of equivalent regions of DNA between homologous chromosomes, whereas NHEJ reunites the ends without the use of a template. This means that HR-mediated repair is high-fidelity HR, and it is mainly active during the S and G2 phases, whereas NHEJ frequently leaves deletions or insertions at the breakpoint and therefore tends to be error prone.
Cohesin recruitment is fundamental for efficient DSBs repair by HR and this function depends on its ability to mediate cohesion between sister chromatids [19]. Experimental evidence suggests that DSBs allow the establishment of de novo sister chromatid cohesion in G2 cells, implicating damage-recruited cohesin in holding the broken chromatid near its undamaged sister template [90, 91]. Moreover, specific recruitment at damaged sites was observed in laser-induced DNA-damage [92]. In human cells, it was recently shown that a DSB unidirectionally blocks cohesin translocation, creating a pattern reminiscent of a TAD boundary. Inside this TAD, cohesin complexes anchored at DSBs extrude chromatin, while ATM phosphorylates chromatin as it passes through the cohesin ring [93]. These findings indicate that genome organization mediated by cohesin is critical for the response to DNA damage.
Instead, NHEJ is active during the cell cycle, and it is the principal pathway during the G1 phase, when there is no immediate close template for homologous repair. The recruitment of DNA–PKcs (DNA-dependent Protein Kinase catalytic subunit) and Ku70/80 to DNA ends triggers the NHEJ cascade, which is followed by enrolment of the XRCC4–ligase IV complex. This process also requires several DNA damage sensors or adaptors, such as ATM, γH2AX, 53BP1, MDC1, RNF168, and the MRE11–RAD50–NBS1 complex. In mammalian cells, the end-joining of the DSEs (Double-Strand DNA ends) is essential in CSR (Class Switch Recombination) and in V(D)J recombination, as well as for repair of DSBs generated by irradiation [94]. It has been hypothesized that cohesin represses the end-joining of distant DSEs specifically in the S/G2 phases while it allows the end-joining of close ends, even in the S/G2 phases [20]. CSR is initiated by recruitment of AID (Activation-Induced cytidine Deaminase) and the subsequent generation of DSBs. As a consequence, AID associates with subunits of cohesin and these breaks activate the DNA damage response and are resolved through the NHEJ pathway [95].
Cohesin alterations in human cancer
Cancer genome and exome sequencing has revealed that cohesin subunits undergo a wide spectrum of somatic mutations in cancer. According to the COSMIC (Catalogue of Somatic Mutations in Cancer) database (https://cancer.sanger.ac.uk/cosmic) both cohesin core and associated factor genes are involved in cancer (Table 2, as of February 2022). STAG1 (5%), NIPBL (4.9%), STAG2 (3.4%) and PDS5B (3.4%) are the most frequently mutated in cancer. In addition, STAG2, STAG1, SMC1A, and RAD21 are also reported in the Cancer Genes Census catalogue (https://cancer.sanger.ac.uk/census) [96], which contains mutations that have been causally implicated in cancer, suggesting that dysfunction of these genes may trigger the tumorigenesis.
In somatic vertebrate cells, two versions of cohesin cohabit, cohesin-STAG1 and cohesin-STAG2, [97]. STAG1 and STAG2 are composed of about 1250 amino acids and share about 75% in homology in their core region while the N- and C-terminal domains are more divergent [98]. Cohesin-STAG2 is more abundant than cohesin-STAG1 in HeLa and Xenopus somatic cells; on the contrary Xenopus eggs contain more cohesin-STAG2 [99, 100]. The two versions of cohesin complex play different biological functions. In fact, knockout mouse models indicate that STAG1 plays a pivotal part in telomeric cohesion whereas STAG2 plays a prominent role in cohesion at chromosome arms or in centromeric regions [101, 102].
STAG2 is a frequent target of inactivating mutations in human cancers, which are only partially compensated for by its paralogue, STAG1 [103, 104]. The first evidence of its involvement in tumorigenesis was carried out from focal deletions on the X chromosome observed in glioblastoma [105]. Later, point mutations were identified in UBC [52,53,54,55, 106], melanoma [105], myelodysplastic syndrome, acute myeloid leukemia [63, 64] and Ewing's sarcoma [57, 58]. STAG2 mutations are usually frameshift, nonsense, or splice site mutations leading to absence of proteins [107] though gene deletion and changes in methylation status have also been reported [54, 108, 109]. Approximately 85% of STAG2 mutations are truncating and often result in loss of expression, indicating STAG2 as a tumor suppressor gene [104]. The downregulation of STAG2 in HeLa cells by siRNA has led to the suggestion that impairment of cohesin-STAG2 might be associated with chromosome imbalance [110]. However, cancer cell lines with inactivated STAG2 were genomically stable though they exhibited decreased cell viability and altered cell cycle. As a consequence, the role of STAG2 in triggering the aneuploidy associated cancer is still debated [43, 52, 55, 106, 111,112,113].
According to genomics datasets, 2087 mutations (as of February 2022) have been identified in STAG1 coding sequences in 41564 tested samples. STAG1 is frequently mutated in bladder cancer [52, 56], Ewing's sarcoma [59] and myeloid malignancies [64, 65]. About 80% of mutations are missense [114] and two hotspots, c.346G>A and c.419G>A, have been detected. Both are described as pathogenic by using the FATHMM prediction algorithm.
As STAG2, SMC1A maps in X chromosome in a region which escapes X inactivation. SMC1A variants have been detected in brain, blood and bladder cancer [52, 65, 115,116,117] but it is frequently mutated in CRC [45,46,47]. CRC is the third most common cancer diagnosed in the population and the second leading cause of death from cancer. CRC progresses through a series of histopathologic and clinical stages ranging from dysplastic crypts to malignant cancers. Most of the SMC1A mutations identified in CRC samples are missense [45,46,47]. The transfection of human primary fibroblasts with vectors carrying some of the SMC1A mutations identified in CRC has resulted in chromosome aneuploidy, abnormal anaphases, and micronuclei formation [45] suggesting that SMC1A might be responsible for the typical chromosomal instability observed in most cases of CRC. In addition, colorectal tissues acquire extra copies of SMC1A gene, and its expression was stronger in carcinoma than normal mucosa and adenoma [46, 48]. The increased expression of SMC1A was positively associated with worse clinico-pathologic variables, including increased tumor, node and metastase (TNM) stages [48].
In addition to CRC, SMC1A mutations are associated with other human cancers. Interestingly, SMC1A mutations have adverse prognostic relevance in acute myeloid leukemia (AML) resulting in significantly shorter overall survival [118]. Mutations are distributed along the length of the coding sequence but are enriched at several hotspots, preferentially at highly conserved residues within the hinge and ATPase domains [119].
Finally, querying the COSMIC database, 871 of 63,847 (1.4%) cancer samples tested harbored somatic mutations in the RAD21 coding region. These mutations have been mainly identified in haematological malignancies [120]. Instead, overexpression of RAD21 was observed in gastric tumors [121], prostate carcinomas [122], CRC [123], and breast cancer [124].
Effects of cohesin dysfunction in cancer
Genome instability is a marker of cancer cells. The notion that chromosomal instability may contribute to cancer development was postulated by Boveri more than 100 years ago and later the “aneuploidy first” hypothesis was proposed [125,126,127]. Mutations and dysregulation of cohesin and cohesin regulatory genes make them powerful driver events that provoke genome instability and cancer progression. The first obvious evidence results from its canonical role. Alterations in cohesin activity lead to impaired chromosome segregation which in turn causes chromosome imbalance, i.e., chromosome gain or loss. The recent query of TCGA (The Cancer Genome Atlas), the largest database of human cancer mutations, showed that half of all driver events are chromosome- and arm-level gains and losses [128]. Each time a chromosome is gained or lost, the dosages of hundreds or thousands of genes are affected. Chromosome segregation impairment can cause trisomy and consequently over-expression of proto-oncogene or transcription factors whose dysregulation play a role in cancer development (Fig. 3). For example, gains of whole chromosome 6 or 6p have been detected in bladder, colorectal, ovarian and hepatocellular carcinomas. It is worth noting that E2F3 and ID4 genes, which code for transcription factors, are located on chromosome 6p [129,130,131,132,133].

Chromosome imbalance and cancer. A Altered segregation of chromosomes harbor a proto-oncogene can lead to gene gain and proto-oncogene over-expression. B Knudson’s hypothesis foresees that two hits are required for the inactivation of a tumor suppressor gene. The first hit is an inactivating mutation on the suppressor gene. The second hit is the chromosome loss caused by cohesin dysfunction
According to Knudson’s hypothesis, tumor suppressor genes are inactivated by two sequential mutational events or two hits [134]. Cohesin could contribute to one of these hits by chromosome missegregation leading to LOH (loss of heterozygosity) and tumorigenesis (Fig. 3). For instance, in retinoblastoma, one recessive allele of the RB1 gene may be inherited or result from an early somatic mutation, and the loss of chromosome 13 carrying the RB1 gene is a frequent second genetic change that leads to LOH of RB1 [135, 136].
About 85% of CRC is chromosomally unstable, with a worse prognosis. Of note, CRC development is characterized by the gain of several chromosomes containing cohesin genes, such as HDAC8, RAD21, SMC1A and STAG2 [46]. This finding suggests that cohesin mutations could contribute to generating chromosomal imbalances necessary for a growth advantage and the fully malignant transformation. However, this notion is still under debate. In fact, although STAG2 is significantly mutated in UBC [137], its alterations occur in the absence of chromosomal instability [52]. Again, no clear association of cohesin mutations and aneuploidy has been reported in myeloid malignancies [107]. Therefore, the role of cohesin dysfunction in cancer development is possibly not only related to cohesion defects and genomic instability, but mutated cohesin may contribute to disease pathology by altering genome structure and gene expression. Aberrant DNA looping could cause misregulation of proto-oncogenes or tumor suppressor genes during tumorigenesis or alter expression of developmental regulators during development and differentiation [81, 138,139,140,141,142,143].
Cohesin mutations affect the dynamic binding of cohesin onto chromatin and impair the recruitment of Pol II (RNA polymerase II) to both promoter and elongation sites [35, 39]. This data is further supported by the recent findings that cancer-associated mutations identified in SMC1A, STAG1 and STAG2 genes result in changes to gene expression and genome organization. Mutations interfere with cohesin localization to promoters and enhancers resulting in transcription dysregulation. In addition, mutated cohesin impairs the ability to organize chromatin into loops and the communication between regulatory elements such as enhancers and promoters [144, 145].
STAG2 LOF (loss of function) occurs in about 20% of Ewing's sarcoma cases [58, 59]. It strongly alters the anchored dynamic loop extrusion process at boundary CTCF sites and dramatically decreases cis-promoter-enhancer interactions, which in turn leads to profound changes in the transcriptome. In addition, cells carrying inactivated STAG2 showed decreased DNA damage signaling and diminished telomere shortening that resulted in delayed senescence. It has been suggested that STAG2 LOF increases the chance that mutated cells acquire tumor-driving mutations by extending cell life span [146]. This notion is supported by the observation that transcription factors (MYC, NF-κB) or signaling pathways (epithelial-to-mesenchymal transition, TGF-β, and EGF) are impacted upon STAG2 LOF suggesting that these alterations may contribute to tumorigenesis [147].
DNA replication fork progression can be challenged by several factors, such as presence of DNA lesions, inappropriate origin firing, the presence of unresolved DNA secondary structures, deficiency of nucleotide pools available for DNA synthesis, and presence of DNA–RNA hybrid intermediates, leading to transient replication fork progression defects. This replication stress can lead to stalling of DNA polymerases, and prolonged stalling can result in fork breakage due to fork collapse or nucleolytic processing of replication intermediates [148, 149]. The presence of transcriptionally engaged Pol II without productive elongation (promoter-proximal paused Pol II) was first observed for the c-myc and c-fos genes in mammalian cells [150, 151]. Cohesin has been found to accumulate at stalled forks and its loading depends on chromatin remodeling by the histone acetyltransferase Gcn5 and the H3K4 methyltransferase Set1 [152, 153]. It has been hypothesized that cohesin could facilitate template switching to repair DNA lesions and promote efficient fork restart [153, 154]. The important role of cohesin in resolving replication stress is supported by the observation that its depletion increases Pol II pausing at cohesin binding genes indicating that it regulates its transition to elongation [155]. STAG1 is also involved in this process. In fact, cohesin-STAG1 is implicated in the interactions with the SEC (Super Elongation Complex) involved in mobilization of the paused polymerase [34]. It is interesting to note that alterations in transcriptional control at the level of elongation have been linked to leukemia and multiple myeloma pathogenesis [156, 157].
Conclusions
In conclusion, cohesin mutations are most commonly found in CRC, bladder cancer, myeloid leukemia, Ewing's sarcoma and glioblastoma. Originally, it was thought that altered cohesin activity was a major cause of aneuploidy in cancer. Instead, increasing evidence indicates that cohesin is a chromatin regulator mediating DNA repair, 3D genome organization, and transcriptional regulation, and changes in chromatin accessibility and transcription are the most striking consequences of cohesin dysfunction in cancer development. A better understanding of how cohesin controls these important biological processes could also lead to the development of novel therapeutic strategies.
Availability of data and materials
Not applicable.
Abbreviations
- AID:
-
Activation-induced cytidine deaminase
- AML:
-
Acute myeloid leukemia
- ATM:
-
Ataxia telangiectasia mutated
- ATR:
-
Ataxia telangiectasia and Rad3 related
- CES:
-
Conserved and essential surface
- CHOPS:
-
Cognitive, heart defects, obesity, pulmonary and short stature
- COSMIC:
-
Catalogue of somatic mutations in cancer
- CRC:
-
Colorectal carcinoma
- CSR:
-
Class switch recombination
- CTCF:
-
CCCTC-binding factor
- DNA–PKcs:
-
DNA-dependent protein kinase catalytic subunit
- DSBs:
-
Double-strand breaks
- DSEs:
-
Double-strand DNA ends
- DTRs:
-
Disorders of transcriptional regulation
- ESCO1/2:
-
Establishment of cohesion 1 homolog ½
- ESPL1:
-
Extra spindle pole bodies-like 1
- HR:
-
Homologous recombination
- LOF:
-
Loss of function
- LOH:
-
Loss of heterozygosity
- NHEJ:
-
Nonhomologous end-joining
- NIPBL:
-
Nipped-B like
- PDS5:
-
Precocious dissociation of sisters 5
- PTTG1:
-
Pituitary tumor transforming gene 1
- SEC:
-
Super elongation complex
- SMC:
-
Structural Maintenance of Chromosomes
- SSBs:
-
Single-strand breaks
- STAG:
-
Stromal antigen
- TADs:
-
Topologically associated domains
- TCGA:
-
The cancer genome atlas
- TCGA:
-
The cancer genome atlas
- TNM:
-
Tumor-nodes-metastases
- UBC:
-
Urothelial bladder cancer
- USP37:
-
Ubiquitin-specific peptidase 37
- WAPL:
-
Wings apart-like protein 1
References
Yatskevich S, Rhodes J, Nasmyth K. Organization of chromosomal DNA by SMC complexes. Annu Rev Genet. 2019;53:445–82.
Glynn EF, Megee PC, Yu HG, Mistrot C, Unal E, Koshland DE, DeRisi JL, Gerton JL. Genome-wide mapping of the cohesin complex in the yeast saccharomyces cerevisiae. PLoS Biol. 2004;2(9):E259.
Lengronne A, Katou Y, Mori S, Yokobayashi S, Kelly GP, Itoh T, Watanabe Y, Shirahige K, Uhlmann F. Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature. 2004;430(6999):573–8.
Rolef Ben-Shahar T, Heeger S, Lehane C, East P, Flynn H, Skehel M, Uhlmann F. Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science. 2008;321(5888):563–6.
Unal E, Heidinger-Pauli JM, Kim W, Guacci V, Onn I, Gygi SP, Koshland DE. A molecular determinant for the establishment of sister chromatid cohesion. Science. 2008;321(5888):566–9.
Zhang J, Shi X, Li Y, Kim BJ, Jia J, Huang Z, Yang T, Fu X, Jung SY, Wang Y, et al. Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell. 2008;31(1):143–51.
Panizza S, Tanaka T, Hochwagen A, Eisenhaber F, Nasmyth K. Pds5 cooperates with cohesin in maintaining sister chromatid cohesion. Curr Biol. 2000;10(24):1557–64.
Gandhi R, Gillespie PJ, Hirano T. Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Curr Biol. 2006;16(24):2406–17.
Kueng S, Hegemann B, Peters BH, Lipp JJ, Schleiffer A, Mechtler K, Peters JM. Wapl controls the dynamic association of cohesin with chromatin. Cell. 2006;127(5):955–67.
Yeh C, Coyaud E, Bashkurov M, van der Lelij P, Cheung SW, Peters JM, Raught B, Pelletier L. The deubiquitinase USP37 regulates chromosome cohesion and mitotic progression. Curr Biol. 2015;25(17):2290–9.
Chauhan R, Bhat AA, Masoodi T, Bagga P, Reddy R, Gupta A, Sheikh ZA, Macha MA, Haris M, Singh M. Ubiquitin-specific peptidase 37: an important cog in the oncogenic machinery of cancerous cells. J Exp Clin Cancer Res. 2021;40(1):356.
Hauf S, Waizenegger IC, Peters JM. Cohesin cleavage by separase required for anaphase and cytokinesis in human cells. Science. 2001;293(5533):1320–3.
Uhlmann F, Lottspeich F, Nasmyth K. Sister-chromatid separation at anaphase onset is promoted by cleavage of the cohesin subunit Scc1. Nature. 1999;400(6739):37–42.
Funabiki H, Yamano H, Kumada K, Nagao K, Hunt T, Yanagida M. Cut2 proteolysis required for sister-chromatid seperation in fission yeast. Nature. 1996;381(6581):438–41.
Cohen-Fix O, Peters JM, Kirschner MW, Koshland D. Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev. 1996;10(24):3081–93.
Huang J, Li K, Cai W, Liu X, Zhang Y, Orkin SH, Xu J, Yuan GC. Dissecting super-enhancer hierarchy based on chromatin interactions. Nat Commun. 2018;9(1):943.
Dowen JM, Bilodeau S, Orlando DA, Hubner MR, Abraham BJ, Spector DL, Young RA. Multiple structural maintenance of chromosome complexes at transcriptional regulatory elements. Stem Cell Reports. 2013;1(5):371–8.
Brough R, Bajrami I, Vatcheva R, Natrajan R, Reis-Filho JS, Lord CJ, Ashworth A. APRIN is a cell cycle specific BRCA2-interacting protein required for genome integrity and a predictor of outcome after chemotherapy in breast cancer. EMBO J. 2012;31(5):1160–76.
Sjogren C, Nasmyth K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr Biol. 2001;11(12):991–5.
Gelot C, Guirouilh-Barbat J, Le Guen T, Dardillac E, Chailleux C, Canitrot Y, Lopez BS. The cohesin complex prevents the end joining of distant DNA double-strand ends. Mol Cell. 2016;61(1):15–26.
Cucco F, Palumbo E, Camerini S, D’Alessio B, Quarantotti V, Casella ML, Rizzo IM, Cukrov D, Delia D, Russo A, et al. Separase prevents genomic instability by controlling replication fork speed. Nucleic Acids Res. 2018;46(1):267–78.
Terret ME, Sherwood R, Rahman S, Qin J, Jallepalli PV. Cohesin acetylation speeds the replication fork. Nature. 2009;462(7270):231–4.
Watrin E, Peters JM. The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. Embo J. 2009;28(17):2625–35.
Musio A, Montagna C, Mariani T, Tilenni M, Focarelli ML, Brait L, Indino E, Benedetti PA, Chessa L, Albertini A, et al. SMC1 involvement in fragile site expression. Hum Mol Genet. 2005;14(4):525–33.
Izumi K. Disorders of transcriptional regulation: an emerging category of multiple malformation syndromes. Mol Syndromol. 2016;7(5):262–73.
Ansari M, Poke G, Ferry Q, Williamson K, Aldridge R, Meynert AM, Bengani H, Chan CY, Kayserili H, Avci S, et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J Med Genet. 2014;51(10):659–68.
Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, Pie J, Gil-Rodriguez C, Arnedo M, Loeys B, Kline AD, et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de lange syndrome with predominant mental retardation. Am J Hum Genet. 2007;80(3):485–94.
Deardorff MA, Wilde JJ, Albrecht M, Dickinson E, Tennstedt S, Braunholz D, Monnich M, Yan Y, Xu W, Gil-Rodriguez MC, et al. RAD21 mutations cause a human cohesinopathy. Am J Hum Genet. 2012;90(6):1014–27.
Kaiser FJ, Ansari M, Braunholz D, Concepcion Gil-Rodriguez M, Decroos C, Wilde JJ, Fincher CT, Kaur M, Bando M, Amor DJ, et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum Mol Genet. 2014;23(11):2888–900.
Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004;36(6):631–5.
Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, Russo S, Vezzoni P, Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet. 2006;38(5):528–30.
Olley G, Ansari M, Bengani H, Grimes GR, Rhodes J, von Kriegsheim A, Blatnik A, Stewart FJ, Wakeling E, Carroll N, et al. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange-like syndrome. Nat Genet. 2018;50(3):329–32.
Vega H, Waisfisz Q, Gordillo M, Sakai N, Yanagihara I, Yamada M, van Gosliga D, Kayserili H, Xu C, Ozono K, et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet. 2005;37(5):468–70.
Izumi K, Nakato R, Zhang Z, Edmondson AC, Noon S, Dulik MC, Rajagopalan R, Venditti CP, Gripp K, Samanich J, et al. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat Genet. 2015;47(4):338–44.
Revenkova E, Focarelli ML, Susani L, Paulis M, Bassi MT, Mannini L, Frattini A, Delia D, Krantz I, Vezzoni P, et al. Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum Mol Genet. 2009;18(3):418–27.
Liu J, Zhang Z, Bando M, Itoh T, Deardorff MA, Clark D, Kaur M, Tandy S, Kondoh T, Rappaport E, et al. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol. 2009;7(5):e1000119.
Mannini L, Menga S, Tonelli A, Zanotti S, Bassi MT, Magnani C, Musio A. SMC1A codon 496 mutations affect the cellular response to genotoxic treatments. Am J Med Genet A. 2012;158A(1):224–8.
Gimigliano A, Mannini L, Bianchi L, Puglia M, Deardorff MA, Menga S, Krantz ID, Musio A, Bini L. Proteomic profile identifies dysregulated pathways in Cornelia de Lange syndrome cells with distinct mutations in SMC1A and SMC3 genes. J Proteome Res. 2012;11(12):6111–23.
Mannini L, Lamaze FC, Cucco F, Amato C, Quarantotti V, Rizzo IM, Krantz ID, Bilodeau S, Musio A. Mutant cohesin affects RNA polymerase II regulation in Cornelia de Lange syndrome. Sci Rep. 2015;5:16803.
Vrouwe MG, Elghalbzouri-Maghrani E, Meijers M, Schouten P, Godthelp BC, Bhuiyan ZA, Redeker EJ, Mannens MM, Mullenders LH, Pastink A, et al. Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: evidence for impaired recombinational repair. Hum Mol Genet. 2007;16(12):1478–87.
Pallotta MM, Di Nardo M, Sarogni P, Krantz ID, Musio A. Disease-associated c-MYC downregulation in human disorders of transcriptional regulation. Hum Mol Genet. 2021. https://doi.org/10.1093/hmg/ddab348.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–9.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501.
Leiserson MD, Vandin F, Wu HT, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M, et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet. 2015;47(2):106–14.
Cucco F, Servadio A, Gatti V, Bianchi P, Mannini L, Prodosmo A, De Vitis E, Basso G, Friuli A, Laghi L, et al. Mutant cohesin drives chromosomal instability in early colorectal adenomas. Hum Mol Genet. 2014;23(25):6.
Sarogni P, Palumbo O, Servadio A, Astigiano S, D’Alessio B, Gatti V, Cukrov D, Baldari S, Pallotta MM, Aretini P, et al. Overexpression of the cohesin-core subunit SMC1A contributes to colorectal cancer development. J Exp Clin Cancer Res. 2019;38(1):108.
Barber TD, McManus K, Yuen KW, Reis M, Parmigiani G, Shen D, Barrett I, Nouhi Y, Spencer F, Markowitz S, et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc Natl Acad Sci U S A. 2008;105(9):3443–8.
Wang J, Yu S, Cui L, Wang W, Li J, Wang K, Lao X. Role of SMC1A overexpression as a predictor of poor prognosis in late stage colorectal cancer. BMC Cancer. 2015;15:90.
Koedoot E, van Steijn E, Vermeer M, Gonzalez-Prieto R, Vertegaal ACO, Martens JWM, Le Devedec SE, van de Water B. Splicing factors control triple-negative breast cancer cell mitosis through SUN2 interaction and sororin intron retention. J Exp Clin Cancer Res. 2021;40(1):82.
Oishi Y, Nagasaki K, Miyata S, Matsuura M, Nishimura SI, Akiyama F, Iwai T, Miki Y. Functional pathway characterized by gene expression analysis of supraclavicular lymph node metastasis-positive breast cancer. J Hum Genet. 2007;52(3):271–9.
Zhu HE, Li T, Shi S, Chen DX, Chen W, Chen H. ESCO2 promotes lung adenocarcinoma progression by regulating hnRNPA1 acetylation. J Exp Clin Cancer Res. 2021;40(1):64.
Balbas-Martinez C, Sagrera A, Carrillo-de-Santa-Pau E, Earl J, Marquez M, Vazquez M, Lapi E, Castro-Giner F, Beltran S, Bayes M, et al. Recurrent inactivation of STAG2 in bladder cancer is not associated with aneuploidy. Nat Genet. 2013;45(12):1464–9.
Solomon DA, Kim JS, Bondaruk J, Shariat SF, Wang ZF, Elkahloun AG, Ozawa T, Gerard J, Zhuang D, Zhang S, et al. Frequent truncating mutations of STAG2 in bladder cancer. Nat Genet. 2013;45(12):1428–30.
Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, Dean M, Huang Y, Jia W, Zhou Q, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. 2013;45(12):1459–63.
Taylor CF, Platt FM, Hurst CD, Thygesen HH, Knowles MA. Frequent inactivating mutations of STAG2 in bladder cancer are associated with low tumour grade and stage and inversely related to chromosomal copy number changes. Hum Mol Genet. 2014;23(8):1964–74.
Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507(7492):315–22.
Crompton BD, Stewart C, Taylor-Weiner A, Alexe G, Kurek KC, Calicchio ML, Kiezun A, Carter SL, Shukla SA, Mehta SS, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov. 2014;4(11):1326–41.
Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S, Patidar R, Hurd L, Chen L, Shern JF, et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014;10(7):e1004475.
Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A, Zhang Z, Lapouble E, Grossetete-Lalami S, Rusch M, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014;4(11):1342–53.
Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–77.
Bailey ML, O’Neil NJ, van Pel DM, Solomon DA, Waldman T, Hieter P. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol Cancer Ther. 2014;13(3):724–32.
Ryu B, Kim DS, Deluca AM, Alani RM. Comprehensive expression profiling of tumor cell lines identifies molecular signatures of melanoma progression. PLoS One. 2007;2(7):e594.
Kon A, Shih LY, Minamino M, Sanada M, Shiraishi Y, Nagata Y, Yoshida K, Okuno Y, Bando M, Nakato R, et al. Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet. 2013;45(10):1232–7.
Thota S, Viny AD, Makishima H, Spitzer B, Radivoyevitch T, Przychodzen B, Sekeres MA, Levine RL, Maciejewski JP. Genetic alterations of the cohesin complex genes in myeloid malignancies. Blood. 2014;124(11):1790–8.
Thol F, Bollin R, Gehlhaar M, Walter C, Dugas M, Suchanek KJ, Kirchner A, Huang L, Chaturvedi A, Wichmann M, et al. Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood. 2014;123(6):914–20.
Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, Potter NE, Heuser M, Thol F, Bolli N, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21.
Zhan Y, Mariani L, Barozzi I, Schulz EG, Bluthgen N, Stadler M, Tiana G, Giorgetti L. Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res. 2017;27(3):479–90.
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376–80.
Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515(7527):402–5.
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–80.
Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15(4):234–46.
Rao SSP, Huang SC, Glenn St Hilaire B, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID, et al. Cohesin loss eliminates all loop domains. Cell. 2017;171(2):305-320 e324.
Hadjur S, Williams LM, Ryan NK, Cobb BS, Sexton T, Fraser P, Fisher AG, Merkenschlager M. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460(7253):410–3.
Wutz G, Varnai C, Nagasaka K, Cisneros DA, Stocsits RR, Tang W, Schoenfelder S, Jessberger G, Muhar M, Hossain MJ, et al. Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 2017;36(24):3573–99.
Fudenberg G, Imakaev M, Lu C, Goloborodko A, Abdennur N, Mirny LA. Formation of chromosomal domains by loop extrusion. Cell Rep. 2016;15(9):2038–49.
Nichols MH, Corces VG. A CTCF code for 3D genome architecture. Cell. 2015;162(4):703–5.
Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J, et al. Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A. 2015;112(47):E6456-6465.
Li Y, Haarhuis JHI, Sedeno Cacciatore A, Oldenkamp R, van Ruiten MS, Willems L, Teunissen H, Muir KW, de Wit E, Rowland BD, et al. The structural basis for cohesin-CTCF-anchored loops. Nature. 2020;578(7795):472–6.
Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467(7314):430–5.
Merkenschlager M, Nora EP. CTCF and cohesin in genome folding and transcriptional gene regulation. Annu Rev Genomics Hum Genet. 2016;17:17–43.
Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015;161(5):1012–25.
Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, Bernstein BE. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529(7584):110–4.
Haarhuis JHI, van der Weide RH, Blomen VA, Yanez-Cuna JO, Amendola M, van Ruiten MS, Krijger PHL, Teunissen H, Medema RH, van Steensel B, et al. The cohesin release factor WAPL restricts chromatin loop extension. Cell. 2017;169(169):693-707 e614.
Nora EP, Goloborodko A, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017;169(5):930-944 e922.
Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168(4):644–56.
Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–33.
Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002;16(5):571–82.
Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16(5):560–70.
Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18(12):1423–38.
Strom L, Lindroos HB, Shirahige K, Sjogren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol Cell. 2004;16(6):1003–15.
Potts PR, Porteus MH, Yu H. Human SMC5/6 complex promotes sister chromatid homologous recombination by recruiting the SMC1/3 cohesin complex to double-strand breaks. EMBO J. 2006;25(14):3377–88.
Kim JS, Krasieva TB, LaMorte V, Taylor AM, Yokomori K. Specific recruitment of human cohesin to laser-induced DNA damage. J Biol Chem. 2002;277(47):45149–53.
Arnould C, Rocher V, Finoux AL, Clouaire T, Li K, Zhou F, Caron P, Mangeot PE, Ricci EP, Mourad R, et al. Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature. 2021;590(7847):660–5.
Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet. 2013;47:433–55.
Thomas-Claudepierre AS, Schiavo E, Heyer V, Fournier M, Page A, Robert I, Reina-San-Martin B. The cohesin complex regulates immunoglobulin class switch recombination. J Exp Med. 2013;210(12):2495–502.
Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA. The COSMIC cancer gene census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018;18(11):696–705.
Remeseiro S, Losada A. Cohesin, a chromatin engagement ring. Curr Opin Cell Biol. 2013;25(1):63–71.
Palidwor GA, Shcherbinin S, Huska MR, Rasko T, Stelzl U, Arumughan A, Foulle R, Porras P, Sanchez-Pulido L, Wanker EE, et al. Detection of alpha-rod protein repeats using a neural network and application to huntingtin. PLoS Comput Biol. 2009;5(3):e1000304.
Losada A, Yokochi T, Kobayashi R, Hirano T. Identification and characterization of SA/Scc3p subunits in the Xenopus and human cohesin complexes. J Cell Biol. 2000;150(3):405–16.
Holzmann J, Fuchs J, Pichler P, Peters JM, Mechtler K. Lesson from the stoichiometry determination of the cohesin complex: a short protease mediated elution increases the recovery from cross-linked antibody-conjugated beads. J Proteome Res. 2011;10(2):780–9.
Remeseiro S, Cuadrado A, Gomez-Lopez G, Pisano DG, Losada A. A unique role of cohesin-SA1 in gene regulation and development. EMBO J. 2012;31(9):2090–102.
Remeseiro S, Cuadrado A, Carretero M, Martinez P, Drosopoulos WC, Canamero M, Schildkraut CL, Blasco MA, Losada A. Cohesin-SA1 deficiency drives aneuploidy and tumourigenesis in mice due to impaired replication of telomeres. EMBO J. 2012;31(9):2076–89.
Arruda NL, Carico ZM, Justice M, Liu YF, Zhou J, Stefan HC, Dowen JM. Distinct and overlapping roles of STAG1 and STAG2 in cohesin localization and gene expression in embryonic stem cells. Epigenetics Chromatin. 2020;13(1):32.
Hill VK, Kim JS, Waldman T. Cohesin mutations in human cancer. Biochim Biophys Acta. 2016;1866(1):1–11.
Solomon DA, Kim T, Diaz-Martinez LA, Fair J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N, Ladanyi M, et al. Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science. 2011;333(6045):1039–43.
Kim JS, He X, Orr B, Wutz G, Hill V, Peters JM, Compton DA, Waldman T. Intact cohesion, anaphase, and chromosome segregation in human cells harboring tumor-derived mutations in STAG2. PLoS Genet. 2016;12(2):e1005865.
De Koninck M, Losada A. Cohesin mutations in cancer. Cold Spring Harb Perspect Med. 2016;6(12):a026476.
Shen CH, Kim SH, Trousil S, Frederick DT, Piris A, Yuan P, Cai L, Gu L, Li M, Lee JH, et al. Loss of cohesin complex components STAG2 or STAG3 confers resistance to BRAF inhibition in melanoma. Nat Med. 2016;22(9):1056–61.
Kim Y, Shi Z, Zhang H, Finkelstein IJ, Yu H. Human cohesin compacts DNA by loop extrusion. Science. 2019;366(6471):1345–9.
Canudas S, Smith S. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J Cell Biol. 2009;187(2):165–73.
Mullenders J, Aranda-Orgilles B, Lhoumaud P, Keller M, Pae J, Wang K, Kayembe C, Rocha PP, Raviram R, Gong Y, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med. 2015;212(11):1833–50.
Benedict B, van JJM Schie, Oostra AB, Balk JA, Wolthuis RMF, Riele HT, de Lange J. WAPL-dependent repair of damaged DNA replication forks underlies oncogene-induced loss of sister chromatid cohesion. Dev Cell. 2020;52(6):683-698 e687.
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–78.
Romero-Perez L, Surdez D, Brunet E, Delattre O, Grunewald TGP. STAG Mutations in cancer. Trends Cancer. 2019;5(8):506–20.
Cancer Genome Atlas Research N, Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–74.
Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630.
Cessna MH, Paulraj P, Hilton B, Sadre-Bazzaz K, Szankasi P, Cluff A, Patel JL, Hoda D, Toydemir RM. Chronic myelomonocytic leukemia with ETV6-ABL1 rearrangement and SMC1A mutation. Cancer Genet. 2019;238:31–6.
Opatz S, Bamopoulos SA, Metzeler KH, Herold T, Ksienzyk B, Braundl K, Tschuri S, Vosberg S, Konstandin NP, Wang C, et al. The clinical mutatome of core binding factor leukemia. Leukemia. 2020;34(6):1553–62.
Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017;45(D1):D777–83.
Cheng H, Zhang N, Pati D. Cohesin subunit RAD21: from biology to disease. Gene. 2020;758:144966.
Yun J, Song SH, Kang JY, Park J, Kim HP, Han SW, Kim TY. Reduced cohesin destabilizes high-level gene amplification by disrupting pre-replication complex bindings in human cancers with chromosomal instability. Nucleic Acids Res. 2016;44(2):558–72.
Porkka KP, Tammela TL, Vessella RL, Visakorpi T. RAD21 and KIAA0196 at 8q24 are amplified and overexpressed in prostate cancer. Genes Chromosomes Cancer. 2004;39(1):1–10.
Deb S, Xu H, Tuynman J, George J, Yan Y, Li J, Ward RL, Mortensen N, Hawkins NJ, McKay MJ, et al. RAD21 cohesin overexpression is a prognostic and predictive marker exacerbating poor prognosis in KRAS mutant colorectal carcinomas. Br J Cancer. 2014;110(6):1606–13.
Xu H, Yan M, Patra J, Natrajan R, Yan Y, Swagemakers S, Tomaszewski JM, Verschoor S, Millar EK, van der Spek P, et al. Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemotherapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res. 2011;13(1):R9.
Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10(7):478–87.
Duesberg P, Li R. Multistep carcinogenesis: a chain reaction of aneuploidizations. Cell Cycle. 2003;2:202–10.
Li R, Zhu J. Effects of aneuploidy on cell behaviour and function. Nat Rev Mol Cell Biol. 2022. https://doi.org/10.1038/s41580-021-00436-9.
Vyatkin AD, Otnyukov DV, Leonov SV, Belikov AV. Comprehensive patient-level classification and quantification of driver events in TCGA PanCanAtlas cohorts. PLoS Genet. 2022;18(1):e1009996.
Feber A, Clark J, Goodwin G, Dodson AR, Smith PH, Fletcher A, Edwards S, Flohr P, Falconer A, Roe T, et al. Amplification and overexpression of E2F3 in human bladder cancer. Oncogene. 2004;23(8):1627–30.
Oeggerli M, Tomovska S, Schraml P, Calvano-Forte D, Schafroth S, Simon R, Gasser T, Mihatsch MJ, Sauter G. E2F3 amplification and overexpression is associated with invasive tumor growth and rapid tumor cell proliferation in urinary bladder cancer. Oncogene. 2004;23(33):5616–23.
Wu Q, Hoffmann MJ, Hartmann FH, Schulz WA. Amplification and overexpression of the ID4 gene at 6p22.3 in bladder cancer. Mol Cancer. 2005;4(1):16.
Al-Mulla F, Keith WN, Pickford IR, Going JJ, Birnie GD. Comparative genomic hybridization analysis of primary colorectal carcinomas and their synchronous metastases. Genes Chromosomes Cancer. 1999;24(4):306–14.
Diep CB, Parada LA, Teixeira MR, Eknaes M, Nesland JM, Johansson B, Lothe RA. Genetic profiling of colorectal cancer liver metastases by combined comparative genomic hybridization and G-banding analysis. Genes Chromosomes Cancer. 2003;36(2):189–97.
Knudson AG. A personal sixty-year tour of genetics and medicine. Annu Rev Genomics Hum Genet. 2005;6:1–14.
Dimaras H, Corson TW, Cobrinik D, White A, Zhao J, Munier FL, Abramson DH, Shields CL, Chantada GL, Njuguna F, et al. Retinoblastoma. Nat Rev Dis Primers. 2015;1:15021.
Cavenee WK, Dryja TP, Phillips RA, Benedict WF, Godbout R, Gallie BL, Murphree AL, Strong LC, White RL. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma. Nature. 1983;305(5937):779–84.
Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43(9):875–8.
Hnisz D, Weintraub AS, Day DS, Valton AL, Bak RO, Li CH, Goldmann J, Lajoie BR, Fan ZP, Sigova AA, et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science. 2016;351(6280):1454–8.
Ji X, Dadon DB, Powell BE, Fan ZP, Borges-Rivera D, Shachar S, Weintraub AS, Hnisz D, Pegoraro G, Lee TI, et al. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016;18(2):262–75.
Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schopflin R, Kraft K, Kempfer R, Jerkovic I, Chan WL, et al. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 2016;538(7624):265–9.
Weischenfeldt J, Dubash T, Drainas AP, Mardin BR, Chen Y, Stutz AM, Waszak SM, Bosco G, Halvorsen AR, Raeder B, et al. Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat Genet. 2017;49(1):65–74.
Mouri K, Sagai T, Maeno A, Amano T, Toyoda A, Shiroishi T. Enhancer adoption caused by genomic insertion elicits interdigital Shh expression and syndactyly in mouse. Proc Natl Acad Sci U S A. 2018;115(5):1021–6.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–63.
Carico ZM, Stefan HC, Justice M, Yimit A, Dowen JM. A cohesin cancer mutation reveals a role for the hinge domain in genome organization and gene expression. PLoS Genet. 2021;17(3):e1009435.
Rittenhouse NL, Carico ZM, Liu YF, Stefan HC, Arruda NL, Zhou J, Dowen JM. Functional impact of cancer-associated cohesin variants on gene expression and cellular identity. Genetics. 2021;217(4):iyab025.
Daniloski Z, Smith S. Loss of tumor suppressor STAG2 promotes telomere recombination and extends the replicative lifespan of normal human cells. Cancer Res. 2017;77(20):5530–42.
Surdez D, Zaidi S, Grossetete S, Laud-Duval K, Ferre AS, Mous L, Vourc’h T, Tirode F, Pierron G, Raynal V, et al. STAG2 mutations alter CTCF-anchored loop extrusion, reduce cis-regulatory interactions and EWSR1-FLI1 activity in Ewing sarcoma. Cancer Cell. 2021;39(6):810-826 e819.
Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Annu Rev Pathol. 2015;10:425–48.
van Schie JJM, de Lange J. The interplay of cohesin and the replisome at processive and stressed DNA replication forks. Cells. 2021;10(12):3455.
Krumm A, Meulia T, Brunvand M, Groudine M. The block to transcriptional elongation within the human c-myc gene is determined in the promoter-proximal region. Genes Dev. 1992;6(11):2201–13.
Fort P, Rech J, Vie A, Piechaczyk M, Bonnieu A, Jeanteur P, Blanchard JM. Regulation of c-fos gene expression in hamster fibroblasts: initiation and elongation of transcription and mRNA degradation. Nucleic Acids Res. 1987;15(14):5657–67.
Delamarre A, Barthe A, de la Roche Saint-Andre C, Luciano P, Forey R, Padioleau I, Skrzypczak M, Ginalski K, Geli V, Pasero P, et al. MRX increases chromatin accessibility at stalled replication forks to promote nascent DNA resection and cohesin loading. Mol Cell. 2020;77(2):395-410 e393.
Tittel-Elmer M, Lengronne A, Davidson MB, Bacal J, Francois P, Hohl M, Petrini JHJ, Pasero P, Cobb JA. Cohesin association to replication sites depends on rad50 and promotes fork restart. Mol Cell. 2012;48(1):98–108.
Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D. Error-free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polalpha/Primase/Ctf4 Complex. Mol Cell. 2015;57(5):812–23.
Schaaf CA, Kwak H, Koenig A, Misulovin Z, Gohara DW, Watson A, Zhou Y, Lis JT, Dorsett D. Genome-wide control of RNA polymerase II activity by cohesin. PLoS Genet. 2013;9(3):e1003382.
Lin C, Smith ER, Takahashi H, Lai KC, Martin-Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC, Shilatifard A. AFF4, a component of the ELL/P-TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell. 2010;37(3):429–37.
Botrugno OA, Tonon G. Genomic instability and replicative stress in multiple myeloma: the final curtain? Cancers. 2021;14(1):25.
Funding
This work was supported by a grant from Italian Association for Cancer Research (AIRC, IG23284) to A.M.
Author information
Authors and Affiliations
Contributions
MDN, MMP and AM wrote the manuscript. MDN and MMP generated figures and tables. AM contributed to the concept & design and critically edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Di Nardo, M., Pallotta, M.M. & Musio, A. The multifaceted roles of cohesin in cancer. J Exp Clin Cancer Res 41, 96 (2022). https://doi.org/10.1186/s13046-022-02321-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02321-5