
Abstract
Prostate cancer is a leading cause of death worldwide and new estimates revealed prostate cancer as the leading cause of death in men in 2021. Therefore, new strategies are pertinent in the treatment of this malignant disease. Macroautophagy/autophagy is a “self-degradation” mechanism capable of facilitating the turnover of long-lived and toxic macromolecules and organelles. Recently, attention has been drawn towards the role of autophagy in cancer and how its modulation provides effective cancer therapy. In the present review, we provide a mechanistic discussion of autophagy in prostate cancer. Autophagy can promote/inhibit proliferation and survival of prostate cancer cells. Besides, metastasis of prostate cancer cells is affected (via induction and inhibition) by autophagy. Autophagy can affect the response of prostate cancer cells to therapy such as chemotherapy and radiotherapy, given the close association between autophagy and apoptosis. Increasing evidence has demonstrated that upstream mediators such as AMPK, non-coding RNAs, KLF5, MTOR and others regulate autophagy in prostate cancer. Anti-tumor compounds, for instance phytochemicals, dually inhibit or induce autophagy in prostate cancer therapy. For improving prostate cancer therapy, nanotherapeutics such as chitosan nanoparticles have been developed. With respect to the context-dependent role of autophagy in prostate cancer, genetic tools such as siRNA and CRISPR-Cas9 can be utilized for targeting autophagic genes. Finally, these findings can be translated into preclinical and clinical studies to improve survival and prognosis of prostate cancer patients.
Graphical abstract

Highlights
• Prostate cancer is among the leading causes of death in men where targeting autophagy is of importance in treatment;
• Autophagy governs proliferation and metastasis capacity of prostate cancer cells;
• Autophagy modulation is of interest in improving the therapeutic response of prostate cancer cells;
• Molecular pathways, especially involving non-coding RNAs, regulate autophagy in prostate cancer;
• Autophagy possesses both diagnostic and prognostic roles in prostate cancer, with promises for clinical application.
Similar content being viewed by others

Background
Prostate cancer is considered as one of the leading causes of death in men in the USA. According to the new estimates, prostate cancer is becoming the most common cause of cancer-related death, imposing a major public health concern, and novel strategies should be adopted for its prevention and cure. In 2021, it is estimated that prostate cancer will result in 31,000 deaths among 248,000 confirmed diagnoses [1, 2]. The risk of prostate cancer development is 11.6% in men [3, 4]. It should be noted that the 5-year survival rate is different among prostate cancer patients and is dependent on clinical stage, such that early stage detection results in a 5-year survival rate of more than 99%, although it is mostly incurable in advanced stages [5]. Therefore, it is of importance to develop medical tools for timely diagnosis of this malignant cancer. In most patients, prostate cancer is amenable to androgen-deprivation therapy [6,7,8,9]. However, castration-resistant prostate cancer (CRPC) develops as a result of androgen deprivation posing a major challenge in clinical treatment [10]. Different strategies have been developed for non-invasive diagnosis of prostate cancer including nanotechnology and liquid biopsies to identify circulating tumor cells, circulating nucleic acids, or exosomes, as well as other reliable biomarkers such as KLK3/prostate-specific antigen tests [11,12,13,14].
Among various factors involved in prostate cancer progression, genetic and epigenetic factors have received the major attention [15,16,17]. For instance, mutations in the AR (androgen receptor) gene leads to development of CRPC following therapy [18]. Epigenetic modifications can also lead to prostate cancer malignancy [19, 20]. Up to 20% of advanced prostate cancers have alterations and mutations in epigenetic regulators and chromatin remodelers [19, 21,22,23]. DNA methylation and demethylation, histone modification, REST (RE1 silencing transcription factor), and polycomb group proteins among others participate in prostate cancer development [18]. Furthermore, it has been reported that prostate cancer cells can develop therapy resistance via several molecular mechanisms including apoptosis inhibition [24,25,26,27]. Hence, it is important to reveal the molecular mechanisms in prostate cancer not only to understand progression of cancer cells, but also to determine the best response to therapy.
The aim of the current review is to provide a mechanistic viewpoint regarding the role of autophagy in prostate cancer. First, we provide a short introduction for the role of autophagy in cancer and how it affects different aspects of prostate cancer including growth, migration, and therapy response. Then, we specifically discuss the role of autophagy in prostate cancer with a focus on molecular pathways. Subsequently, we discuss anti-tumor compounds and nanotherapeutics for targeting autophagy in prostate cancer. This review should shed some light towards targeting autophagy in prostate cancer therapy.
Autophagy machinery and its role in oncology
Autophagy and related molecular pathways
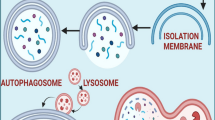
In cells, a wide variety of physiological processes perform unique roles for maintaining homeostasis and preventing the development of pathological events [28,29,30]. Autophagy is a physiological mechanism first identified in the 1950s with the emergence of electron microscopy [31, 32]. This mechanism is involved in degrading cellular components by forming autophagosomes and mediating their fusion with lysosomes, resulting in cargo decomposition followed by recycling [33]. There are three primary types of autophagy, namely chaperone-mediated autophagy (CMA), microautophagy and macroautophagy. Microautophagy is the least characterized, but describes a general term for a non-selective pathway that leads to the sequestration of cytoplasmic cargos directly at the limiting membrane of the lysosome through membrane invagination [34]. CMA is a selective form of autophagy which marks individual proteins for lysosomal degradation [35]. The major form of autophagy is macroautophagy in which transient double-membrane compartments known as phagophores are produced to engulf cargoes, resulting in their subsequent containment within autophagosomes, and degradation following fusion with lysosomes. This kind of autophagy plays a significant role in maintaining cellular homeostasis [36]. Recently, much attention has been directed to deciphering the role of autophagy in cancer, and experiments show that induction and inhibition of autophagy play a significant role in cancer progression [37,38,39,40,41,42,43].
Regardless of the nature of autophagy, it is essential to decipher its molecular mechanism not only for better designing future studies but also developing novel therapeutic agents [44, 45]. Overall, autophagy is divided into six steps including initiation, expansion, closure, fusion, degradation and recycling [46]. The ULK1 (unc-51 like autophagy activating kinase 1) complex containing ATG13 (autophagy related 13), ATG101 and RB1CC1 are involved in autophagy activation. The serine/threonine kinase ULK1, specifically, participates in phosphorylation of phosphatidylinositol 3-kinase (PtdIns3K) complex I components including BECN1 and PIK3C3/VPS34 and mediates phagophore production at the endoplasmic reticulum (ER) [47]. In the expansion step, the ATG12–ATG5 complex is formed through action of the ATG7 and ATG10 enzymes and is recruited to the phagophore membrane. After translation as precursor forms, MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) and GABARAP proteins (representing two subfamilies referred to as Atg8-family proteins due to homology with yeast Atg8) are cleaved by ATG4. These proteolytically processed proteins are then covalently attached to phosphatidylethanolamine at the phagophore membrane in an ATG3- and ATG7-dependent process like, and involving, the generation of the ATG12–ATG5 conjugate. The next step is expansion of the phagophore for engulfing the cargo, followed by maturation of the autophagosome. At this step, LC3-II is separated from the surface of the autophagosome to complete the maturation step and allow fusion directly with a lysosome or after first fusing with an endosome [37, 48,49,50,51,52]. Fusion occurs with the help of molecular components that include tethering factors such as RAB7 and soluble N-ethylmaleimide-sensitive factor-activating membrane fusion protein (SNARE) proteins (Fig. 1) [53]. Finally, upon fusion with a lysosome, the contents are degraded by lysosomal enzymes, and the breakdown products are released back into the cytosol for reuse. This is the general pathway for autophagy induction and completion. Other experiments reveal the role of additional molecular pathways and signaling networks in autophagy. For instance, MTOR (mechanistic target of rapamycin kinase) is an inhibitor of autophagy. Upon MTOR activation, AMP levels are increased which lead to AMP-activated protein kinase (AMPK) activation. AMPK suppresses MTOR, inducing autophagy and increasing energy levels in the cell. In fact, complex signaling networks work with each other to ensure an appropriate function of autophagy to maintain cellular survival and homeostasis.

Autophagy and its regulation. The autophagy mechanism has different phases from initiation to elongation and finally, fusion with the lysosome. In each step, various molecular pathways are involved; AMPK, MTOR, and ATGs are the most well-known regulators of autophagy
Autophagy and cancer treatment
The role of autophagy in cancer is controversial and no consensus has been reached regarding the precise involvement of autophagy in cancer progression and inhibition. Autophagy can be considered as an ideal target in cancer therapeutics to ameliorate cancer mortality and morbidity. However, autophagy serves as a double-edged sword to either enhance or suppress cancer progression. A recent paper reviews the history of autophagy and cancer, noting that the role of autophagy is context dependent [37]. As autophagy plays a dual regulatory role in cancer, different experiments have investigated its participation in cancer and have provided directions for its inhibition or induction. Autophagy and apoptosis are two major branches in programmed cell death (PCD) in cancer cells. When autophagy exerts a pro-survival role, its stimulation results in a decrease in apoptosis of cancer cells, paving the way for cancer progression [54]. However, when autophagy acts as a tumor suppressor, induction of both autophagy and apoptosis can lead to cancer suppression. For instance, it was reported that MTOR inhibition by AMPK in pancreatic cancer cells stimulates autophagy in favor of potentiating apoptosis [55]. Identification of molecular pathways regulating autophagy paves the way for cancer treatment. Hypoxic conditions enhance carcinogenesis via activating pro-survival autophagy by triggering PAK1 (p21 (RAC1) activated kinase 1), which leads to subsequent phosphorylation of ATG5 [56]. These studies clearly confirm the role of autophagy in cancer proliferation [57]. Now, the question that arises is whether there is any connection between autophagy and cancer metastasis. Experimentally, the answer is positive, and autophagy can dually enhance or decrease cancer migration and invasion in various contexts [58,59,60]. Autophagy can significantly promote cancer metastasis via epithelial-to-mesenchymal induction. Upon autophagy inhibition, the levels of mesenchymal markers including VIM (vimentin) and CDH2/N-cadherin undergo downregulation and further confirm the role of autophagy in cancer metastasis [61].
In addition to proliferation and migration, increasing evidence demonstrates the role of autophagy in regulating immune system function [62, 63]. In ensuring tumor progression, autophagy prevents major histocompatibility complex class I (MHCI) expression and restricts anti-tumor T cell immunity against pancreatic cancer cells, resulting in immune evasion [64]. Finally, autophagy can regulate the response of cancer cells to chemotherapy; a recently conducted experiment revealed that autophagy inhibition enhances the sensitivity of gastric cancer cells to chemotherapy [65]. Overall, most available evidence agrees with the following statements:
-
A)
Autophagy is a critical regulator of cell proliferation and metastasis;
-
B)
autophagy can modulate the response of cancer cells to therapy;
-
C)
anti-tumor immunity is tightly regulated by autophagy;
-
D)
in order to develop a novel therapeutic regimen for cancer, the exact role of autophagy should be defined and, based on its role, an inhibitor or activator be recommended [66,67,68,69,70].
Autophagy and prostate cancer
Cancer proliferation
Autophagy increases proliferation
One of the primary effects of autophagy is promoting the proliferation and survival rate of prostate cancer cells. This is multifaceted and a result of interaction with various molecular pathways. For example, AR signaling is responsible for prostate cancer progression by inducing autophagy [71, 72]. TFEB as a transcription factor is involved in regulating lysosomal biogenesis and function. AR stimulates TFEB expression in favor of autophagy induction. Furthermore, other upstream mediators of autophagy including ATG4B, ATG4D, ULK1 and ULK2 are regulated by AR leading to prostate cancer progression. Further investigation reveals that AR-mediated autophagy induction is vital for proliferation and viability of prostate cancer cells and is correlated with poor prognosis [73]. Another experiment reveals the role of AR signaling in autophagy regulation in prostate cancer cells. Transcriptional regulation of GABARAPL1 (GABA type A receptor associated protein like 1), as an autophagy modulator, can be mediated via androgen, and the proliferation of prostate cancer cells is delayed by inhibiting autophagy. Androgen deprivation leads to GABARAPL1 downregulation, subsequent induction of autophagy (GABARAPL1 is autophagy repressive in this context) and increased survival and proliferation of prostate cancer cells [74]. More investigations are warranted to delineate the role of androgen deprivation therapy in prostate cancer with autophagy.
Hypoxia is a hallmark of the tumor microenvironment and can enhance cancer progression [75]. It was reported that O2 deprivation affects DNA replication, metastasis, and angiogenesis in favor of cancer progression [76, 77]. Hypoxic conditions lead to activation of HIF1A/HIF-1α (hypoxia inducible factor 1 subunit alpha) that subsequently enhances the proliferation and survival of prostate cancer cells [78,79,80,81]. A dual autophagy regulatory role has been found for HIF1A in cancer [82]. HIF1A exerts a tumor-promoting role in prostate cancer via affecting autophagy. At the transcriptional level, HIF1A binds to the promoter of Atg5 to increase its expression, resulting in autophagy induction. The HIF1A-ATG5-autophagy axis is vital for tumor growth in nude mice [83]. ATG5 is a potential target for upstream mediators in autophagy induction and enhancing prostate cancer progression [84].
Different biological functions are considered for FGFs (fibroblast growth factors) including growth, apoptosis, differentiation, angiogenesis, development, and metabolic regulation [85]. FGF21 is secreted by liver and is associated with metabolic homeostasis [86]. Downregulation of FGF21 is correlated with decreased proliferation and survival suggesting an anti-tumor activity. Interestingly, through the inhibition of the PI3K-AKT-MTOR axis, FGF21 induces autophagy leading to a decrease in prostate cancer progression [87].
Autophagy decreases proliferation
Given the double-edged sword role of autophagy in biological events in cells, autophagy activation can reduce proliferation and survival rate of prostate cancer cells. Metabolic reprogramming in cancer cells can also influence proliferation and progression. Mitochondria play a significant role in this case. As dynamic structures, mitochondria shape, connectivity and subcellular distribution can be altered to enhance prostate cancer progression, a process modulated by DNM1L/DRP1 (dynamin 1 like) [88, 89]. A recent study has shown that DNM1L upregulation by AR signaling in prostate cancer cells affects metabolism and tumorigenesis and its downregulation results in autophagy induction and proliferation inhibition [90]. AURKA (aurora kinase A) is a serine/threonine kinase with potential roles in genetic stability by regulating spindle assembly, centrosome separation, and chromosome segregation [91, 92]. AURKA overexpression is vital for prostate cancer tumorigenesis and can function to allow tumor cells to evade therapy [93, 94]. AURKA inhibits the tumor-suppressor aspect of autophagy by suppressing AKT phosphorylation [95]. Therefore, decreasing AURKA expression can induce autophagy and suppress proliferation of prostate cancer cells. Although oxidative stress is a major pathway involved in apoptosis, it is suggested that triggering oxidative stress can also mediate autophagic cell death in prostate cancer cells [96]. The anti-tumor activity of autophagy is not only related to cell death but also other molecular pathways that are responsible for prostate cancer growth [97]. For instance, it was reported that HSD17B4 (hydroxysteroid 17-beta dehydrogenase 4) overexpression promotes proliferation and malignancy of prostate cancer cells. Acetylation of HSD17B4 promotes its degradation by CMA and leads to a subsequent decrease in prostate cancer progression [98]. In this case, autophagy does not participate in cell death, but leads to degradation of a factor that enhances prostate cancer progression.
Multiple molecular pathways have been reported to regulate cell fate in cancer cells including MAPK8/JNK1/c-Jun N-terminal kinase (mitogen-activated protein kinase 8) [99, 100]. MAPK8/JNK1 activation favors cancer proliferation and enhances stem cell-like features of cancer cells [101, 102]. MAPK8/JNK1 inhibitors have been developed for cancer therapy [103,104,105,106] and specifically for prostate cancer cells, as MAPK8/JNK1 inhibits autophagy and promotes survival and proliferation of cancer cells in an androgen-independent manner. Thus, MAPK8/JNK1 inhibition results in autophagy induction and subsequent decrease in prostate cancer proliferation. The mechanism of action is suggested by an upregulation in BECN1/beclin-1 and ATG5 [107]. Overall, the following conclusions can be drawn from the studies (Fig. 2, Table 1):
-
1)
Pre-clinical studies on prostate cancer reveal the dual role of autophagy in survival and proliferation;
-
2)
autophagy not only can affect cell death, but also can influence molecular pathways involved in prostate cancer proliferation. Specifically, HSD17B4 degradation is induced by CMA;
-
3)
autophagy can function as either a pro-survival or pro-death mechanism, and in this review, we explore targeting autophagy to suppress prostate cancer progression.

The mechanism of autophagy in prostate cancer cell proliferation and survival. Due to the dual role of autophagy, it can both promote and inhibit proliferation and viability of cancer cells. This figure provides a summary of molecular pathways involved in cancer progression regulation by autophagy
Cancer metastasis
Metastasis is defined as a process in which certain subpopulations of cancer cells detach and disseminate from their primary tissues or site to secondary sites [116,117,118,119]. This process is mediated via blood or lymph vasculature [120] through complex interactions with extracellular matrix (ECM), remodeling, epithelial-to-mesenchymal transition (EMT), angiogenesis, or basement membrane invasion among others to reestablish tumor colony formation in metastasis sites [121,122,123,124,125,126,127]. Recent experiments have also demonstrated the role of molecular pathways in migration and invasion of prostate cancer cells. For instance, one study suggests that the metastasis of prostate cancer cells into the bone is mediated via complex formations between CDK19 and CCNL1, and subsequent phosphorylation of POLR2/polymerase II at serine 2 [128]. PLEC (plectin) is another factor that induces prostate cancer metastasis to major organs of the body such as liver, lung, kidney and bone [129]. In contrast, molecular pathways that inhibit prostate cancer metastasis include the role of WFDC2 (WAP four-disulfide core domain 2), which downregulates SNAI/snail expression and inhibits EGFR (epidermal growth factor receptor) [130]. These studies have highlighted the fact that metastasis is a critical challenge in the treatment of prostate cancer, and a variety of complex molecular pathways and mechanisms are involved in this process. In this section, the role of autophagy in prostate cancer metastasis is discussed.
Metastatic prostate cancer cells display induction in autophagy. Thus, there is a vital role for autophagy in metastasis and migration. Tumor microenvironment (TME) contains a variety of non-malignant cells such as inflammatory cells, cancer-associated fibroblasts and vascular cells [131]. It has been reported that among the TME constituents, endothelial cells play a significant role in prostate cancer metastasis. After androgen-deprivation therapy (ADT), apoptosis occurs in prostatic microvasculature, although the endothelial cells undergo immediate regeneration [132]. An increase in microvascular infiltration is associated with prostate cancer metastasis through AR signaling [133, 134]. In vivo and in vitro experiments showed that autophagy is associated with prostate cancer metastasis. For this to occur, endothelial cells inhibit AR signaling to provide autophagy induction. Subsequently, autophagy activation leads to a rise in migration and invasion of prostate cancer cells through focal adhesion protein disassembly [135].
With respect to the dual role of autophagy, metastasis of prostate cancer cells can also be suppressed. HADC6 (histone deacetylase 6) has a close relationship with autophagy in cells. The autophagy maturation by HDAC6 induces cancer suppression [136]. Furthermore, HDAC6 can inhibit autophagy via TUBA/α-tubulin deacetylation [136]. In prostate cancer cells, microtubule acetylation results in induction of autophagosome formation and autophagy flux to suppress migration and invasion. However, SQSTM1/p62 acts as a tumor-promoting factor, inducing HDAC6 expression to suppress microtubule acetylation-mediated autophagy, leading to prostate cancer metastasis [137]. This experiment reveals that the role of autophagy in suppressing prostate cancer metastasis and triggering autophagy flux is of importance in this case.
As more reports have surfaced, more molecular mechanisms involved in prostate cancer metastasis are revealed. One of the most important mechanisms in cancer metastasis is EMT occurring due to decrease in CDH1/E-cadherin levels and increase in CDH2/N-cadherin and VIM levels [138,139,140]. Upon EMT induction, cancer cells acquire a mesenchymal phenotype via losing their cellular polarity and adhesion to basement membrane [138, 139, 141]. TGFB1/TGF-β (transforming growth factor beta 1) is considered as main inducer of EMT via upregulation of NFKB/NF-κB (nuclear factor kappa B) and HIF1A [142,143,144]. Furthermore, TWIST, ZEB proteins and SNAI are among other factors involved in EMT induction [145]. Similarly, in prostate cancer cells, different molecular pathways such as STAT3 (signal transducer and activator of transcription 3) and microRNAs (miRNAs) participate in EMT regulation [146, 147]. Conversely, autophagy can regulate EMT in cancer cells [148]. In prostate cancer cells, SGK1 (serum/glucocorticoid regulated kinase 1) as a tumor-promoting factor, increases migratory ability in vivo and in vitro via EMT induction. Silencing SGK1 impairs prostate cancer metastasis via autophagy induction and subsequent suppression of EMT. It was indicated that autophagy inhibition of EMT in prostate cancer occurs via SNAI downregulation [149]. These studies highlight the fact that autophagy is in close association with prostate cancer metastasis and vital mechanisms such as EMT can be affected in this way. Conversely, upstream mediators target autophagy in regulating metastasis of prostate cancer cells (Fig. 3).

The mechanism of autophagy in prostate cancer metastasis. In addition to proliferation, migration of prostate cancer cells is regulated by autophagy. As shown, upstream mediators can induce EMT-mediated metastasis of prostate cancer cells, and autophagy is capable of suppressing EMT and invasion
Therapeutic resistance
In the field of cancer therapy, especially prostate cancer treatment, drug resistance remains an increasing challenge [150, 151]. A variety of molecular pathways and mechanisms can result in drug resistance development such as activation of tumor-promoting factors and inhibition of tumor-suppressor factors [152,153,154]. In respect to emergence of genetic tools such as small interfering RNA (siRNA), short-hairpin RNA (shRNA) and the CRISPR-Cas9 system for gene regulation, molecular mechanisms involved in drug resistance can be targeted in providing cancer sensitization to chemotherapy [155,156,157,158]. Autophagy, owing to its dual role, can either suppress or induce chemoresistance [159]. In some cases, inhibition of autophagy by MIR375 and WWOX (WW domain containing oxidoreductase) can promote chemosensitivity of cancer cells [160, 161]. Noteworthy, TME provides an environment favoring autophagy induction and chemoresistance [162]. Conversely, there is also evidence demonstrating that autophagy induction participates in elevated efficacy of chemotherapy in cancer elimination [163, 164]. In addition to chemotherapy, autophagy affects the response of cancer cells to radiotherapy. In this case, autophagy can also induce or inhibit radio-resistance [165,166,167]. Due to the aggressive behavior of prostate cancer cells, these malignant cells obtain resistance to radio- and chemotherapy [168, 169]. In this section, a mechanistic discussion of autophagy in the therapy response of prostate cancer cells is provided to direct the next experiments towards modulating this important molecular pathway in effective treatment of prostate cancer.
Autophagy as a pro-survival mechanism
Analysis of NPRL2/nitrogen permease regulator-like 2 (NPR2 like, GATOR1 complex subunit) demonstrates modulatory impacts on autophagy. NPRL2 can induce autophagy via MTOR signaling inhibition [170,171,172], and autophagy activation can promote resistance of cancer cells to everolimus [173, 174]. In CRPC cells, NPRL2 shows an increase in expression and is associated with proliferation and drug resistance features. Upon upregulation, NPRL2 suppresses MTOR signaling to stimulate autophagy, resulting in everolimus resistance via reducing apoptotic cell death [175]. This observation demonstrates that apoptosis and autophagy as two major arms of PCD are in close relationship and autophagy, as a tumor-promoting factor, can alleviate apoptosis induction in prostate cancer cells.
One of the important findings is the activation of autophagy by chemotherapeutic agents in mediating resistance. In fact, this unwanted response of chemotherapeutic agents negatively affects their anti-tumor activity. It has been reported that autophagy induction by NPRL2 leads to docetaxel resistance [176]. Exposing CRPC cells to docetaxel is associated with autophagy activation via BECN1 upregulation. Autophagy inhibition via MTOR signaling induction significantly enhances therapeutic efficacy of docetaxel in prostate cancer treatment [177]. TGFB is an upstream mediator of autophagy, and TGFB1 fucosylation induces autophagy [178]. It seems that TGFB and autophagy interaction is of importance for docetaxel resistance of prostate cancer cells. In this way, mesenchymal stem cells (MSCs) along with docetaxel induce autophagy to diminish proliferation inhibition and apoptosis induction in prostate cancer cells. It was reported that autophagy inhibition suppresses docetaxel resistance. In autophagy induction, MSCs secrete TGFB1 and preventing TGFB1 secretion inhibits autophagy, leading to increased docetaxel sensitivity of prostate cancer cells [179]. KLF5 is a component of another molecular pathway capable of regulating autophagy and docetaxel sensitivity of prostate cancer cells. As a transcription factor, KLF5 is widely distributed in various organs such as liver, kidney, prostate and colon with vital roles in regulating proliferation and tumorigenesis [180]. The KLF5 gene is located on chromosome 13q.21 and genomic hybridization analysis has demonstrated its deletion in 39% of prostate cancer cases [181]. There is a negative relationship between autophagy and KLF5 in cancer, so that KLF5 provides poor prognosis and autophagy inhibition [182]. However, a recent study depicted a tumor-suppressor role for KLF5 in prostate cancer. KLF5 downregulation is associated with unfavorable prognosis and decreased sensitivity of prostate cancer cells to docetaxel. Furthermore, In vivo and in vitro studies have demonstrated KLF5 downregulation upon docetaxel exposure. In fact, docetaxel reduces KLF5 expression to prevent cell death induction in prostate cancer cells. Examination of molecular pathways demonstrates that KLF5 inhibits BECN1 and HDAC3 (histone deacetylase 3) cooperation to suppress autophagy and promote docetaxel sensitivity in the context of prostate cancer. Besides, docetaxel exposure downregulates KLF5 expression via an AMPK-MTOR-RPS6KB/p70S6K axis to increase BECN1 expression, leading to autophagy induction and chemoresistance features of prostate cancer cells [183]. AMPK upregulation stimulates protective autophagy to provide survival and viability of prostate cancer cells against AD therapy [184].
Cisplatin (CP) is another well-known chemotherapeutic agent applied in prostate cancer therapy. Increasing cell stiffness, decreasing migration, and apoptosis induction result from treatment [185, 186]. However, activation in certain cell signaling cascades such as activation of the WNT-CTNNB1/beta-catenin and AKT pathways can result in CP resistance of prostate cancer cells [187, 188]. AMBRA1 (autophagy and beclin 1 regulator 1) can induce autophagy via regulating BECN1 and BCL2 [189, 190]. In prostate cancer cells. AMBRA1 undergoes overexpression and protects prostate cancer cells against CP-mediated apoptosis, and CASP3 (caspase 3)-mediated PARP (poly(ADP-ribose) polymerase) cleavage. By increasing autophagy, AMBRA1 prevents apoptosis in prostate cancer cells and enhances their colony formation, leading to CP resistance [191].
ASAH1/acid ceramidase (N-acylsphingosine amidohydrolase 1) is an enzyme localized in lysosomes capable of metabolizing ceramide, an intermediate in sphingolipid synthesis. Ceramide is involved in the regulation of molecular pathways such as apoptosis, cell cycle arrest and inflammation. It has been reported that changes in ceramide metabolism can provide drug resistance feature of cancer cells [192, 193]. Prostate cancer cells demonstrate overexpression of ASAH1 converting ceramide to sphingosine and sphingosine-1-phosphate, inhibiting apoptosis [194]. ASAH1 upregulation also enhances lysosomal density and results in autophagy induction. Autophagy inhibition can promote a therapeutic response of prostate cancer cells to C (6) ceramide. It is possible that autophagy can provide resistance of prostate cancer cells to SRC-family kinase inhibitors. SRC is a large family of nonreceptor tyrosine kinases, and its overexpression occurs in various cancers, particularly prostate cancer. Activation of SRC signaling is associated with recurrence of prostate cancer [195]. Besides, SRC mediates metastasis of prostate cancer cells in hypoxic condition [196]. Therefore, SRC inhibitors have been developed for prostate cancer therapy, but drug resistance has restricted their potential. It seems that autophagy is involved in resistance of prostate cancer cells to SRC inhibitors. SRC inhibition does not affect autophagy, showing that autophagy induction is independent of SRC upregulation. Using chloroquine as an autophagy inhibitor enhances sensitivity of prostate cancer cells to SRC inhibitors [197]. In fact, the anti-tumor activity of some compounds is boosted when protective autophagy is inhibited [198].
STAT3 mainly possesses a tumor-promoting role in cancer, and its downregulation is beneficial for cancer treatment [140, 199,200,201,202]. Conversely, STAT3 can function as an upstream mediator of autophagy in cancer [203,204,205]. Following docetaxel chemotherapy, autophagy activation occurs via upregulation of BECN1 and induction of the BECN1-PIK3C3/VPS34-ATG14 complex. It seems that STAT3 is a negative regulator of autophagy in prostate cancer cells upon docetaxel chemotherapy. Autophagy induction is vital for resistance to docetaxel chemotherapy. Enhancing the expression level of STAT3 suppresses autophagy that may promote docetaxel sensitivity of prostate cancer cells [206]. Similar to STAT3, HMGB1 (high mobility group box 1) can regulate a wide variety of biological mechanisms such as differentiation, autophagy and migration [207, 208]. HMGB1 overexpression promotes prostate cancer invasion via EMT induction [209]. Besides, HMGB1 can trigger chemoresistance feature of prostate cancer cells via activating downstream targets such as MYC/c-Myc signaling [209]. The association between HMGB1 and autophagy is in favor of triggering gemcitabine resistance of prostate cancer cells. In this way, exposing prostate cancer cells to gemcitabine significantly enhances the expression level of HMGB1. Downregulating HMGB1 via shRNA diminishes autophagy. Therefore, HMGB1 induces autophagy to protect prostate cancer cells against gemcitabine-mediated cell death [210]. In hormone-refractory prostate cancer cells, autophagy induction is of importance for cancer progression [211]. Although androgen deprivation is a promising therapy for prostate cancer, providing such conditions for prostate cancer cells induces protective autophagy to prevent apoptosis [212].
Autophagy as a pro-death mechanism
When autophagy possesses an anti-tumor role, its activation significantly enhances sensitivity of cancer cells to therapy. In the previous section, it was discussed that autophagy induction promotes docetaxel resistance in prostate cancer cells. Autophagy stimulation can also be helpful in docetaxel sensitivity via promoting apoptosis. TPD52/PrLZ/prostate leucine zipper (tumor protein D52) localized on chromosome 8q21.1 has specific expression in prostate tissues and it demonstrates upregulation in advanced prostate cancer [213]. In respect to the tumor-promoting role of TPD52, its activation remarkably promotes progression of prostate cancer cells [214]. A recent experiment has shown involvement of TPD52 in docetaxel resistance of prostate cancer cells via inhibiting autophagy and subsequent docetaxel-mediated apoptosis. Exposing prostate cancer cells to docetaxel inhibits TPD52 and STK11/LKB1 (serine/threonine kinase 11) interaction to promote STK11 expression. Then, STK11 as an upstream mediator of AMPK signaling, induces autophagy to potentiate apoptosis in cancer cells. However, TPD52 upregulation in drug resistant-prostate cancer cells leads to STK11 and AMPK downregulation and subsequent inhibition of autophagy [215].
PTAFR (platelet activating factor receptor) belongs to the G-protein-coupled receptor (GPCR) family with vital roles in cancer progression via regulating molecular pathways such as STAT3 and PI3K-AKT signaling networks [216]. PTAFR plays a significant role in mediating the radio-resistance feature of prostate cancer cells. In this way, PTAFR destabilizes BECN1 to suppress autophagy. Interfering with the PTAFR-BECN1 complex is of importance in enhancing the radio-sensitivity of prostate cancer cells (Fig. 4, Table 2) [225].

Autophagy regulates the response of prostate cancer cells to therapy. An increasing challenge in prostate cancer therapy is therapy resistance. On the one hand, autophagy activation as a tumor-promoting factor, can inhibit apoptosis and mediate chemoresistance. On the other hand, tumor-suppressor autophagy can sensitize prostate cancer cells to chemotherapy via triggering apoptosis
Molecular pathways regulating autophagy in prostate cancer
MicroRNAs
A variety of molecular pathways can function as upstream mediators of autophagy and miRNAs are among them. MiRNAs are a conserved class of non-coding RNAs (ncRNAs) 18–22 nucleotides in length. Although miRNAs do not encode proteins, they participate in the regulation of many protein-coding genes (up to 60%). Furthermore, miRNAs downregulate expression of target gene via targeting messenger RNA (mRNA) [226,227,228]. MiRNAs are supposed to be expressed in all tissues [145, 229,230,231]. For affecting expression of a target gene, first miRNAs cooperate with specific proteins, known as AGO (argonaute) proteins, and then they are embedded in an RNA-induced silencing complex (RISC) to bind to the 3/−untranslated region (3′-UTR) of mRNA, suppressing expression via inhibiting translation, inducing mRNA cleavage or enhancing mRNA stability [232]. Increasing evidence demonstrates a role for miRNAs in the regulation of various molecular pathways and mechanisms in physiological and pathological conditions [233, 234]. MiRNAs are potential regulators of autophagy in cancers, affecting proliferation, metastasis, and therapy response [235, 236]. In this section, a mechanistic discussion of autophagy regulation by miRNAs in prostate cancer is provided to shed more light on their interaction and contribution to cancer progression in malignant cells.
Molecular pathways regulating autophagy are affected by miRNAs in prostate cancer. It is worth mentioning that miRNAs with a tumor-suppressing role undergo downregulation during cancer progression. It has been reported MIR26B is present at a low level in prostate cancer cells. This miRNA downregulates the expression level of ULK2 by binding to its 3′-UTR to suppress autophagy, leading to prostate cancer progression impairment [237]. In contrast, when autophagy possesses a tumor-suppressing role, its inhibition occurs by tumor-promoting miRNAs. MIR146B is considered as a tumor-suppressing factor, which inhibits proliferation and invasion of prostate cancer cells, and induces apoptosis [238]. However, a recent report has revealed the tumor-promoting role of MIR146B and its upregulation in prostate cancer, demonstrating a double-edged sword role of this miRNA [239]. PI3K affects autophagy via regulating MTOR. Of note, PTEN (phosphatase and tensin homolog) is an upstream and negative regulator of PI3K-AKT signaling [240, 241]. PTEN downregulates PI3K-AKT to prevent proliferation and invasion of cancer cells and promote their therapy response [242,243,244,245,246,247]. MIR146B downregulates expression of PTEN to induce AKT-MTOR signaling. Then, autophagy inhibition occurs to ensure progression of prostate cancer [239]. RELN (reelin) is another regulator of PI3K-AKT signaling, and in contrast to PTEN, RELN induces this pathway [248]. By downregulating AR expression, MIR381 suppresses proliferation and progression of prostate cancer cells [249]. It is worth noting that there is a close association among MIR381, RELN and the PI3K-AKT-MTOR axis in prostate cancer cells. In respect to the tumor-suppressing role of MIR381, this factor downregulates RELN to inhibit PI3K-AKT-MTOR signaling, leading to autophagy and apoptosis induction [250].
MIR493 is another miRNA with its role in prostate cancer not fully elucidated. Although one independent study has demonstrated the anti-tumor activity of MIR493 in prostate cancer via upregulation of N6-methyladenosine levels [251], another study revealed that MIR493 stimulates AKT signaling to enhance prostate cancer proliferation [252]. Therefore, more studies are required to reveal role of this miRNA in prostate cancer. However, MIR493 also exerts a tumor-suppressing role via autophagy regulation in prostate cancer. MIR493 upregulates PHLPP2 (PH domain and leucine rich repeat protein phosphatase 2) to promote expression of BECN1 and ATG7, resulting in autophagy and reduced ability of prostate cancer cells in colony formation [253]. Studies demonstrate that upstream mediators of autophagy such as MTOR and certain ATGs are tightly regulated by miRNAs in prostate cancer. For instance, MIR139 induces MTOR signaling, while downregulating BECN1 to inhibit autophagy flux, resulting in subsequent apoptosis induction in prostate cancer cells [254].
More importantly, miRNAs targeting autophagy can regulate the response of prostate cancer cells to therapy. In previous sections, it was shown how autophagy can affect the therapy response of cancer cells. Radio-resistance is an increasing in treatment for prostate cancer. Activation of DNA repair mechanisms, and downregulating tumor-suppressing miRNAs such as MIR501-3p participate in the radio-resistance feature of prostate cancer cells [255, 256]. Identification of miRNAs regulating autophagy in prostate cancer and its association with radiotherapy response are of importance in developing novel therapeutics to overcome this issue. Exposing prostate cancer cells (DU145 and LNCaP cells) to irradiation induces protective autophagy via TP53INP1 (tumor protein p53 inducible nuclear protein 1). MIR205 diminishes the radio-resistance of prostate cancer cells via downregulating TP53INP1, inhibiting autophagy and causing a subsequent impairment in viability and survival [257]. MIR32 is another factor that upregulates BECN1 and LC3-II, while downregulating MTOR to induce autophagy and provide radio-resistance in prostate cancer cells [258].
One of the hallmarks of cancer is hypoxia in the tumor microenvironment. The hypoxic condition is induced due to low vasculature and the presence of cancer cells with a rapid growth rate. Therefore, molecular pathways such as those involving HIF1α and angiogenesis are activated to ensure cancer progression and growth. In prostate cancer cells, SP1 (Sp1 transcription factor) induces hypoxia-mediated autophagy to promote cancer progression. It is noteworthy that SP1 regulates PKM2 (pyruvate kinase M1/2) in this manner. As a tumor-suppressing factor, MIR361-5p inhibits SP1 and its downstream target PKM2 to impair autophagy and cancer progression of prostate cancer cells [259]. It seems that the expression level of miRNAs with anti-tumor activity decreases in hypoxic conditions; in these conditions, MIR30A and MIR205 undergo downregulation to promote prostate cancer progression. Then, autophagy activation occurs to protect these malignant cells against irradiation. However, enhancing levels of MIR30A and MIR205 downregulate the level of TP53INP1 via binding to its 3′-UTR to suppress autophagy, resulting in an increased radio-sensitivity of prostate cancer cells [260].
MIR212 can dually induce or inhibit prostate cancer progression. This dual role of miRNAs has made it rather complicated to target miRNAs in prostate cancer therapy. For instance, MIR212 downregulates MAPK expression to suppress prostate cancer proliferation [261], and at the same time, this miRNA can induce NFKB signaling in prostate cancer development [262]. On a related note, there is a connection between SIRT1 (sirtuin 1) and autophagy [263], and SIRT1 can function as an autophagy inducer [264]. A significant decrease occurs in levels of circulatory MIR212 in the serum of prostate cancer patients. Investigation of molecular pathways demonstrates downregulation of SIRT1 by MIR212 and subsequent inhibition of autophagy that are of importance for suppressing prostate cancer progression [265].
ERN1/IRE1 (endoplasmic reticulum to nucleus signaling 1) is a marker of ER stress and its upregulation can also be associated with autophagy [266, 267]. During starvation, ER stress occurs and upon ERN1 upregulation autophagy is activated to restore homeostasis. In prostate cancer cells, MIR200C-3p induces autophagy via ERN1 upregulation [268]. With respect to the tumor-promoting role of autophagy in prostate cancer, the interaction among miRNAs and ER stress-related markers in regulating autophagy should be highlighted. Considering everything together, the following bullet points can be concluded:
-
1)
Both tumor-suppressing and tumor-promoting miRNAs can regulate autophagy in affecting prostate cancer progression;
-
2)
upstream mediators of autophagy such as SIRT1, BECN1, ATGs and LC3-II are regulated by miRNAs in prostate cancer;
-
3)
there is a close relationship between ER stress and autophagy markers that can be regulated by miRNAs;
-
4)
The tumor microenvironment of prostate cancer cells affects the expression level of miRNAs in favor of regulating autophagy for progression and survival;
-
5)
miRNA replacement therapy and, small interfering RNAs (siRNAs) can be utilized for affecting the expression level of miRNAs and their downstream targets, respectively to provide conditions for autophagy regulation and suppressing progression of prostate cancer cells.
LncRNAs
As transcripts with more than 200 nucleotides in length, lncRNAs are another key member of ncRNAs with vital physiological roles in development and differentiation [269,270,271,272]. Despite their expression in various major organs of the body such as heart, kidney, and liver, it seems that expression of lncRNAs is tissue specific and is maintained at a low level of expression [273,274,275,276]. LncRNAs do not encode proteins, but they have potential and important functions like protein-coding genes [277]. Genomic imprinting, apoptosis, differentiation, splicing, cell cycle regulation and epigenetic regulation are among the vital roles of lncRNAs in cells [278,279,280]. Dysregulation of lncRNAs is a common phenomenon in cancer due to their role in regulating important biological processes [281]. In prostate cancer, lncRNAs can regulate lymph node metastasis, proliferation, and therapy response via affecting different molecular pathways, especially miRNAs [282,283,284]. In this section, the role of lncRNAs in regulating autophagy and affecting prostate cancer progression is discussed.
SNHG1 (small nucleolar RNA host gene 1) is a new emerging lncRNA located on chromosome 11 that plays a key role in cancer. The role of SNHG1 in prostate cancer is suggested to be tumor-promoting, so that SNHG1 can increase metastasis via EMT induction [285]. In addition, SNHG1 can enhance proliferation and survival of prostate cancer cells via AKT2 upregulation [286]. About promoting prostate cancer progression, SNHG1 can affect autophagy. In this way, SNHG1 induces PI3K-AKT signaling to induce MTOR expression, resulting in autophagy inhibition, paving the way for survival and proliferation of prostate cancer cells. It is likely that SNHG1 indirectly affects PI3K-AKT signaling, so that first, SNHG1 promotes the expression level of EZH2 (enhancer of zeste 2 polycomb repressive complex 2 subunit) to affect PI3K-AKT signaling. Silencing SNHG1 or EZH2 is associated with decreased viability of prostate cancer cells. Further investigation reveals regulation of WNT-CTNNB1β-catenin signaling by the SNHG1-EZH2 axis, but it has not been determine whether WNT-CTNNB1/β-catenin will affect autophagy in these cancer cells [287]. Identification of these kinds of lncRNAs is of importance for their knockdown in further experiments for prostate cancer treatment. For instance, overexpression of the lncRNA PRRT3-AS1 occurs in prostate cancer cells and tissues. Silencing PRRT3-AS1 upregulates the expression level of PPARG/PPARγ (peroxisome proliferator activated receptor gamma) that, in turn, inhibits MTOR signaling, leading to autophagy induction and a decrease in viability and survival of prostate cancer cells. In vivo experiments on xenografts in nude mice also confirmed PRRT3-AS1 silencing and decreased tumor growth [288].
Similar to miRNAs, lncRNAs can target autophagy in affecting the response of prostate cancer cells. The role of the lncRNA HULC (hepatocellular carcinoma up-regulated long non-coding RNA) in prostate cancer has been examined in a few studies [289]. Interestingly, currently performed experiments revealed the role of HULC as a tumor-promoting factor in prostate cancer. Overexpression of HULC enhances prostate cancer metastasis via EMT induction and is correlated with poor prognosis [290]. It has been reported that silencing HULC promotes sensitivity of prostate cancer cells to radiotherapy and induces apoptosis and cell cycle arrest (G0/G1 phase). However, HULC overexpression significantly reduces sensitivity to radiotherapy. With regards to the radio-resistance feature of prostate cancer cells, HULC induces MTOR signaling, while it diminishes levels of BECN1, leading to autophagy inhibition. Silencing HULC triggers autophagy and sensitizes prostate cancer cells to irradiation-mediated apoptosis [291]. Although a few studies have demonstrated autophagy regulation by lncRNAs in prostate cancer, the following points can be concluded:
-
1)
The experiments about lncRNA-mediated autophagy regulation have shown a tumor-suppressor role of autophagy, so that lncRNAs that inhibit autophagy increase prostate cancer progression, and further studies can explore the tumor-promoting role of autophagy and its modulation by lncRNAs;
-
2)
studies have shown that lncRNAs indirectly affect autophagy in prostate cancer via regulating other molecular pathways such as PARP, PI3K-AKT-MTOR, and EZH2. In respect to regulation of miRNAs by lncRNAs via sponging, it can be explored whether lncRNAs can affect miRNAs in targeting autophagy in prostate cancer.
CircRNAs
Circular RNAs (circRNAs) have a different structure compared to miRNAs and lncRNAs. These ncRNAs form covalently closed loop structures lacking 5′-3′ polarities and polyadenylated tails [292,293,294]. This special structure of circRNAs makes them more stable than linear RNA molecules, and protects them against degradation by RNA exonucleases [295]. Like miRNAs and lncRNAs, deregulation of circRNAs is observed in prostate cancer. CircRNAs can be considered as potential biomarkers for prostate cancer diagnosis, and they participate in proliferation, migration, and therapeutic response [169, 296, 297]. To date, just one study has evaluated the role of circRNAs in autophagy regulation in prostate cancer, showing that it is still a long journey to reveal the precise role of circRNAs. The circRNA CCNB2 (cyclin B2) undergoes upregulation in prostate cancer and its silencing promotes sensitivity to irradiation. In inducing the radio-resistance feature of prostate cancer cells, circ-CCNB2 downregulates the expression level of MIR30B-5p via sponging. Then, an increase in the expression level of KIF18A (kinesin family member 18A) occurs to stimulate autophagy, leading to radio-resistance [298]. Overall, fewer studies have been performed regarding the role of circRNAs in prostate cancer compared to miRNAs and lncRNAs. However, based on the capacity of circRNAs to affect miRNA regulation via sponging, and the complicated molecular pathways involved in prostate cancer progression, specific attention should be directed to circRNAs (Fig. 5, Table 3).

MiRNAs, lncRNAs and circRNAs as regulators of autophagy in prostate cancer. As molecular pathways involved in autophagy regulation by non-coding RNAs have been identified, genetic tools can be utilized for affecting the non-coding RNA-autophagy axis in prostate cancer therapy
Other upstream mediators
Previous sections revealed a role for ncRNAs in autophagy regulation in prostate cancer. However, there are other upstream mediators of autophagy in prostate that should be highlighted in directing further research for evaluating other molecular pathways. In this section, our aim is to provide a discussion of some molecular pathways regulating autophagy. KDM4B is a lysine methyltransferase with potential role in AR signaling [305]. KDM4B can induce histone modification to enhance transcriptional activity of AR. Furthermore, KDM4B can increase AR stability via preventing its ubiquitination [306]. Due to the signaling role of AR in prostate cancer progression, KDM4B should have a critical role in this malignant tumor. KDM4B overexpression occurs in prostate cancer cells and is associated with their proliferation. In addition, autophagy activation occurs by KDM4B overexpression in prostate cancer cells. Importantly, KDM4B indirectly induces autophagy via triggering nuclear translation of CTNNB1/β-catenin, leading to prostate cancer progression [307]. CTNNB1/β-catenin is a vital member of WNT signaling and its nuclear translation leads to WNT signaling activation and further induction of other molecular pathways [308]. Although previous experiments revealed a role of CTNNB1/β-catenin in autophagy induction in prostate cancer cells, this pathway can also suppress autophagy in prostate cancer. Nuclear accumulation of CTNNB1/β-catenin caused by nitric oxide (NO) leads to autophagy inhibition in prostate cancer cells [309].
In the section related to prostate cancer metastasis, it was mentioned that SGK1 can mediate autophagy and EMT in prostate cancer cells [149]. SGK1 can also participate in regulating autophagy by affecting the MTOR-FOXO/FOXO3A axis. Briefly, FOXO3 is a Forkhead transcription factor and due to its pro-apoptotic role, is involved in cell cycle arrest and apoptosis [310, 311]. Mechanistically, SGK1 induces MTOR signaling to inhibit FOXO3 phosphorylation, leading to a decrease in apoptosis and autophagy in prostate cancer cells. Silencing SGK1 inhibits MTOR signaling, providing conditions for FOXO3 phosphorylation, and subsequent induction of apoptosis and autophagy in prostate cancer cells [312].
MAPK/JNK is capable of regulating apoptosis after exposure to different stresses [313], and it can also regulate autophagy [314, 315]. In prostate cancer cells, MAPK/JNK signaling induces autophagy as a protective mechanism against celecoxib-mediated apoptosis [316]. This study demonstrates how autophagy and apoptosis interact with each other in prostate cancer cells. As MAPK/JNK-mediated autophagy prevents apoptosis in prostate cancer cells, its inhibition is of importance. Previously, it was shown that KDM4B interaction with AR signaling occurs in prostate cancer. PARK7/DJ-1 is another factor that is vital for AR function and can induce AR signaling [317, 318]. PARK7 prevents the function of the AR inhibitor PIAS2/PIASxa/ARIP3 to induce AR signaling [318]. PARK7 undergoes upregulation in prostate cancer cells and reducing its expression significantly diminishes proliferation and survival. In this way, PARK7 overexpression inhibits MAPK/JNK signaling to prevent BECN1-BCL2 dissociation, resulting in autophagy inhibition and paving the way for prostate cancer progression [319].
Additional experiments have revealed that AR signaling plays a significant role in autophagy induction and prostate cancer progression. SIRT7 (sirtuin 7) is a member of the NAD+-dependent deacetylases and its role in cancer is controversial [320, 321]. SIRT7 is responsible for the aggressive behavior of prostate cancer cells, and its downregulation suppresses migration and invasion of cancer cells [322]. SMAD4 is an inhibitor of AR signaling, and its degradation is driven by SIRT7 [323,324,325]. In prostate cancer cells, SIRT7 undergoes upregulation to inhibit SMAD4, leading to activation of AR signaling, subsequent induction of autophagy and enhancing cancer progression [71]. Overall, different upstream mediators of autophagy such as LC3-II, ATG3, BECN1, EIF4EBP1, SQSTM1 and SIRT1 can be regulated by upstream mediators of autophagy such as TNFAIP8. Based on the role of autophagy in prostate cancer (pro-survival or pro-death), its inhibition or induction can be performed to pave the way into prostate cancer therapy [324, 326].
Anti-tumor compounds targeting autophagy in prostate cancer
As autophagy plays a significant role in proliferation, metastasis and therapy response of prostate cancer cells, experiments have focused on its targeting this process to affect the progression of cancer cells. In this section, a mechanistic discussion of the role of anti-tumor compounds in autophagy regulation in prostate cancer is provided. Diosgenin is a naturally occurring steroid compound isolated from Dioscorea nipponica and has demonstrated high anti-tumor activity in prostate cancer inhibition [327]. Neural precursor cell express NEDD4 (NEDD4 E3 ubiquitin protein ligase), an E3 ligase with a tumor-promoting role in prostate cancer. Diosgenin downregulates NEDD4 expression to stimulate apoptosis and cell cycle arrest [328]. Diosgenin can promote NEDD4 E3 ubiquitin protein ligase Ca2+ levels and flux. Furthermore, this compound causes Ca2+-independent cell death in prostate cancer cells [329]. Diosgenin administration significantly reduces proliferation and viability of prostate cancer cells via inducing autophagy. In triggering autophagy, diosgenin suppresses PI3K-AKT-MTOR signaling as an upstream mediator of autophagy [330]. In this case, autophagy possesses a tumor-suppressing role. However, when autophagy has a tumor-promoting role, its induction has a negative impact on efficacy of anti-tumor compounds. In prostate cancer suppression, abiraterone downregulates the expression levels of LC3, ATG5 and BECN1 to inhibit autophagy. It is worth mentioning that co-administration of 3-methyladenine as an autophagy inhibitor, enhances the potential of abiraterone in prostate cancer suppression via inducing a further decrease in LC3, ATG5 and BECN1 levels compared to abiraterone alone. This combination remarkably diminishes proliferation and cell viability of prostate cancer cells and stimulates cell cycle arrest at the G2/M phase [331].
Eriocalyxin B (EriB), derived from Isodon eriocalyx var. laxiflora, is a medicinal plant with diverse pharmacological activities in which anti-tumor activity is among them. EriB is a promising candidate in cancer therapy capable of reducing proliferation and cell viability as well as inducing cell cycle arrest and apoptosis via targeting molecular pathways such as STAT3 [332,333,334]. Through inhibition of the AKT-MTOR axis, EriB stimulates both apoptosis and autophagy in prostate cancer cells. However, with respect to the tumor-promoting role of autophagy in this case, autophagy inhibition by chloroquine potentiates cytotoxicity of EriB against prostate cancer cells [335]. In fact, protective autophagy prevents apoptosis in prostate cancer cells by anti-tumor agents. Along these lines, the autophagy inhibitor bafilomycin A1 (BafA1) inhibits autophagy to augment the anti-tumor activity of withaferin A on prostate cancer cells [336].
Due to vital biological roles, it is of great importance to preserve the iron balance in cells [337, 338]. For iron release in cells, iron should first bind to plasma TF (transferrin), and then, iron-bound TF enters cells through endocytosis by binding to TFRC (transferrin receptor) located on the cell surface. When iron levels are low in cells, iron regulatory proteins (IRPs) bind to the iron response elements (IREs) of ferritin and TFRC mRNAs. Therefore, TFRC, ferritin and ACO1/iron regulatory protein 1 (aconitase 1) demonstrate the status of the iron level in cells [339]. Increasing evidence demonstrates the role of iron deprivation in apoptosis and autophagy induction in cancer cells [340, 341]. For autophagy induction, curcumin follows a same route. For this purpose, curcumin as a naturally occurring compound derived from Curcuma longa, binds to ferric ammonium citrate (FAC). Furthermore, curcumin promotes the expression levels of TFRC and ACO1, demonstrating iron deprivation. However, autophagy induction in these prostate cancer cells enhances their survival and using an autophagy inhibitor potentiates the anti-tumor activity of curcumin [342].
Pathological events in the ER can be evoked courtesy of accumulation of misfolded proteins, oxidative stress induction and impaired calcium homeostasis [343]. Increasing evidence has validated autophagy activation during ER stress to improve ER homeostasis and prevent development of various pathologies. However, when stress is beyond the capacity of autophagy to ameliorate, this condition results in both apoptotic and autophagic cell death [344]. δ-tocoterinol (δ-TT) is a potent anti-tumor agent in prostate cancer suppression. In this way, δ-TT increases the expression level of ER stress markers such as HSPA5/BiP, ElF2A, ERN1 and DDIT3/CHOP. Then, activation of ER stress paves the way for autophagy induction through upregulation of SQSTM1 [345].
One of the important aspects is the close association between levels of autophagy and reactive oxygen species (ROS) [346,347,348,349]. Like autophagy, ROS play a dual role in cancer, as tumor-promoting or tumor-suppressing factors [67, 79, 349,350,351,352]. Normally, activation of PI3K-AKT signaling upregulates MTOR expression to induce autophagy. Excessive ROS generation inhibits PI3K-AKT signaling to provide MTOR downregulation, resulting in autophagy [353]. Exposing prostate cancer cells to salinomycin induces both apoptosis and autophagy. In triggering apoptosis and autophagy, salinomycin inhibits the PI3K-AKT-MTOR axis, while it induces MAPK14/p38 and MAPK/ERK. Salinomycin also enhances ROS levels. It seems that ROS inhibits autophagy in prostate cancer, because autophagy increases upon the use of an ROS scavenger. More importantly, autophagy inhibition increases ROS production in prostate cancer cells [354].
To promote the potential of anti-tumor agents in prostate cancer treatment, combination cancer therapy is applied. A combination of chloroquine as an autophagy inhibitor and palladium (II) barbiturate complex results in inhibition of PI3K-AKT signaling, MTOR and MAPK14/p38 upregulation to inhibit protective autophagy and potentiate apoptosis induction in prostate cancer cells [355]. Most of the studies demonstrate induction of protective autophagy using anti-tumor compounds [356]. Therefore, if pre-clinical findings are going to be applied in a clinical regimen, using autophagy inhibitors such as chloroquine is recommended.
In the previous section, it was shown that autophagy is tightly regulated by miRNAs in prostate cancer. Identification of miRNAs and targeting their expression by transfection can pave the way to autophagy regulation and promoting cytotoxicity of anti-tumor compounds against prostate cancer. Celastrol is a bioactive component of Tripterygium wilfordii Hook [357] with the capacity of suppressing prostate bone metastasis via decreasing VEGF (vascular endothelial growth factor) secretion [358]. To potentiate the anti-tumor activity of celastrol against prostate cancer cells, nanoparticles such as polymeric and liposomal nanocarriers have been designed [359,360,361]. New experiments have shown that autophagy and miRNA interaction can determine the response of prostate cancer cells to celastrol therapy. Exposing prostate cancer cells to celastrol stimulates protective autophagy via downregulating MIR17HG (miR-17-92a-1 cluster host gene) expression. Further investigation reveals the role of AR as an upstream mediator of MIR17HG, so that silencing AR decreases MIR17HG expression in prostate cancer cells. Therefore, it seems that celastrol inhibition of AR and its downstream target MIR17HG induces protective autophagy [362]. Another experiment reveals the role of AR in regulating MIR101. Celastrol administration inhibits the AR-MIRNA101 axis to induce protective autophagy. Using miRNA replacement therapy and promoting MIR101 expression inhibits autophagy, potentiating the cytotoxicity of celastrol against prostate cancer cells [363]. Overall, the following points can be concluded (Fig. 6, Table 4):
-
1)
Anti-tumor compounds are extensively applied in prostate cancer therapy, and autophagy is one of their targets;
-
2)
most of the administered anti-tumor compounds are phytochemicals;
-
3)
most of the experiments are in agreement with the fact that autophagy induction by anti-tumor compounds functions as a shield against apoptosis;
-
4)
a variety of molecular pathways such as those involving BECN1, ATGs, miRNAs and AR are regulated by anti-tumor compounds in autophagy regulation;
-
5)
using autophagy inhibitors such as chloroquine for autophagy inhibition and enhancing anti-tumor activity in prostate cancer therapy is recommended [391,392,393,394,395,396,397,398].

Anti-tumor compounds regulate autophagy in prostate cancer therapy. To provide effective prostate cancer therapy, anti-tumor agents (most being phytochemicals) have been developed for affecting autophagy. Various steps of autophagy and its related molecular pathways are modulated by anti-tumor agents in prostate cancer treatment
Gene therapy
In earlier sections, it was discussed that anti-tumor compounds are efficient in autophagy regulation and suppressing prostate cancer progression. Now, the question that comes into mind is whether there any genetic tools with efficiency in autophagy regulation. As experiments have identified upstream mediators of autophagy in prostate cancer, and due to the presence of genetic tools such as siRNA, shRNA and the CRISPR-Cas9 system, molecular pathways can be targeted in autophagy regulation that affect the progression of prostate cancer cells. To date, siRNA has been extensively applied in autophagy regulation and cancer therapy. For instance, co-application of ATG7 siRNA and docetaxel is beneficial in breast cancer therapy [399]. SiRNA is the most well-known genetic tool utilized for affecting autophagy in prostate cancer. The siRNA application helps in inhibiting or inducing autophagy and determining its exact role—as a tumor promoter or tumor suppressor [223]. Furthermore, siRNA has helped reveal the apoptosis and autophagy interaction in prostate cancer. Previously, it was shown that tumor-suppressor autophagy can enhance the sensitivity of prostate cancer cells to apoptosis. Thus, does autophagy precede apoptosis in prostate cancer or vice versa? Overexpression of BIRC5/survivin prevents apoptosis in prostate cancer cells and is a positive factor in their progression and survival. BIRC5/survivin downregulation using siRNA sensitizes prostate cancer cells to apoptosis and autophagy. Suppressing early or later events of autophagy alleviates apoptosis induction in prostate cancer cells, showing that for apoptosis, the first step is autophagy induction. In fact, autophagy precedes apoptosis [400]. Noteworthy, siRNA has also been advantageous in elucidating the ER stress and autophagy interaction in prostate cancer. Exposure to cadmium is associated with prostate cancer tumorigenesis. Cadmium increases ROS generation to induce ER stress, leading to autophagy impairment and paving the way for prostate cancer development. ATF4 downregulation by siRNA inhibits ER stress and autophagy impairment, showing that ER stress stimulation is vital for triggering defects in autophagy [401]. Overall, experiments agree with the fact that upstream mediators of autophagy can be suppressed using siRNA, and this tool is also beneficial in revealing the relationship of autophagy with other molecular pathways and mechanisms in prostate cancer [402,403,404,405,406]. ShRNA is another tool that can be utilized for autophagy induction and suppressing prostate cancer progression. The function of shRNA is similar to siRNA, so that after entering cells, shRNA generates hairpin RNA that translocates to the cytoplasm, undergoing cleavage by DICER enzyme, producing siRNA and subsequent incorporation in the RNA-induced silencing complex (RISC) for gene silencing [407]. Overexpression of IGF1R (insulin like growth factor 1 receptor) promotes prostate cancer growth and progression. For exerting its carcinogenesis impact, IGF1R inhibits autophagy. IGF1R inhibition using shRNA results in autophagy induction through promoting expression levels of LC3B, leading to decreased prostate cancer progression [408]. More experiments are required to demonstrate the potential of other genetic tools such as the CRIPSR-Cas9 system in autophagy regulation and suppressing prostate cancer progression.
Nanotherapeutics and biological vectors
Effective treatment of cancer depends on combining different disciplines to augment the capacity of current therapeutic strategies. In the previous section, it was shown that autophagy plays a significant role in different aspects of prostate cancer cells such as proliferation, metastasis, and therapy response. To modulate autophagy, anti-tumor compounds have been utilized with promising results in prostate cancer treatment. Furthermore, gene therapy has been applied in regulating autophagy and prostate cancer progression. However, each of the strategies suffer from some drawbacks. Most of the anti-tumor compounds applied in prostate cancer treatment via autophagy regulation, are phytochemicals and poor bioavailability is the major issue related to plant derived-natural compounds, limiting their therapeutic impacts [409]. SiRNA application is restricted in autophagy regulation and prostate cancer treatment due to its degradation and off-target effects [410, 411]. Therefore, there is a need for development of novel strategies to improve the efficacy of biology-based methods. In this case, bioengineering comes into view and a wide variety of experiments have applied nanotechnological strategies for suppressing proliferation and metastasis of prostate cancer cells, gene, and drug delivery, and finally imaging [412,413,414,415,416,417]. Nanoparticles can regulate autophagy in prostate cancer via affecting molecular pathways and lysosomes. It has been shown that exposing prostate cancer cells to silver nanocarriers impairs the integrity of the lysosomal membrane, diminishes the number of lysosomes, and prevents lysosomal protease activity, leading to autophagy flux inhibition. Furthermore, silver nanoparticles induce the AMPK-MTOR axis in suppressing autophagy in prostate cancer cells [418]. In fact, attention should be directed towards regulating autophagy by nanoparticles, while these carriers are applied for gene delivery in prostate cancer therapy [419]. Regardless of nanoparticle-mediated regulation of autophagy in prostate cancer, nanocarriers can provide a platform for gene delivery in prostate cancer treatment. In one experiment, chitosan nanoparticles have been prepared for MIR34A delivery to affect autophagy in prostate cancer treatment. MIR34A is a tumor-suppressor factor in prostate cancer and mediating its delivery by chitosan nanoparticles promotes its potential in cancer suppression. Upon MIR34A delivery, a significant increase occurs in apoptosis induction in prostate cancer cells and their viability and survival decrease. Further investigation revealed the relationship between autophagy and MIR34A cell death in prostate cancer cells. MIR34A-loaded chitosan nanoparticles stimulate autophagy in prostate cancer cells independent of BECN1, ATG4, ATG5 and ATG7, known as non-canonical autophagy. Autophagy induction by MIR34A negatively affects survival of prostate cancer cells, so that apoptosis inhibition does not block the anti-proliferative activity of MIR34A-mediatd autophagy [420]. Inorganic carbon nanomaterials have shown anti-tumor activity against prostate cancer cells via autophagy and apoptosis induction [421]. To date, a few studies have investigated nanoparticles and autophagy regulation in prostate cancer, but as autophagy is a vital process in prostate cancer, further experiments can focus how delivery of anti-tumor compounds by nanocarriers or their co-delivery with genetic tools can affect autophagy in favor of prostate cancer treatment.
Oncolytic adenoviral mutants are ideal candidates in the treatment of solid tumors, and their combination with irradiation or anti-tumor compounds leads to remarkable reduction in cancer progression [422, 423]. Due to high potency and selectivity, adenoviral mutants with deletions in the viral E1ACR2-region are extensively applied in cancer therapy in clinical treatment [424,425,426,427]. In some prostate cancer therapy, mitoxantrone is applied, but autophagy induction reduces its potential in cancer treatment. Application of oncolytic mutant Ad∆∆ (E1B19K- and E1ACR2-deleted) inhibits autophagy to promote the potential of mitoxantrone in prostate cancer apoptosis. Further inhibition of ATG7 also enhances the efficacy of mitoxantrone in prostate cancer apoptosis, demonstrating a protective role of autophagy in prostate cancer and its inhibition by this oncolytic virus (Fig. 7) [428].

The nanotherapeutics and biological vectors in regulating autophagy for prostate cancer therapy
Other kinds of autophagy
Mitophagy
Mitophagy is a special type of autophagy responsible for degradation of damaged mitochondria [429]. The mitochondrial quality is determined via its contents [430]. Recently, studies have focused on mitophagy in disorders such as neurodegenerative disorders in which some mitophagy proteins including PINK1 (PTEN induced kinase 1) or PRKN/Parkin undergo mutation [431,432,433]. Mitophagy plays a significant role in cancer [434]. Autophagy inhibition results in an increase in bone metastasis of breast cancer cells [435]. Noteworthy, the role of mitophagy in prostate cancer has been examined. At the first step, it seems that mitophagy induction is advantageous for sensitizing prostate cancer cells to apoptosis. Exposing prostate cancer cells to abiraterone and MDC3100 results in downregulation of mitochondrial proteins such as FXN (frataxin), ACO2 and TOMM20, mitochondrial swelling, mitochondrial depolarization and decreased mitochondrial DNA copy number. These effects result in mitophagy that is associated with apoptosis and reduced proliferation of prostate cancer cells [436]. Metabolic vulnerability of prostate cancer cells is a negative factor for their survival. CAV1 (caveolin 1) alters lipid metabolism in prostate cancer cells that, subsequently, stimulates mitophagy as a lethal process [437]. Although much emphasis was placed on the anti-tumor role of mitophagy, additional investigation reveals a tumor-promoting role of mitophagy in prostate cancer that should be highlighted in directing further experiments. It has been shown that autophagy and mitophagy induction by the integrin ITGA6/a6b1 and the receptor BNIP3 significantly enhances survival and viability of prostate cancer cells [438]. Therefore, more experiments are required to reveal the role of mitophagy in prostate cancer. Agents capable of impairing mitochondrial function can promote mitophagy induction. Retigeric acid B as a potent anti-tumor agent, enhances ROS generation in prostate cancer cells to impair the normal function of mitochondria, resulting in mitophagy and autophagy. Autophagy inhibition increases apoptosis in prostate cancer cells [439]. Although a few studies have examined the role of mitophagy in prostate cancer, more efforts are warranted to decipher how mitophagy participates in the therapy response of prostate cancer cells, and how this important mechanism can affect senescence induction.
Lipophagy
Lipophagy is a form of autophagy first characterized in 2009. Compared with other types of autophagy, lipophagy has not been well elucidated, and the terminology primarily depicts the degradation of lipid droplets (LDs) by autophagy [440]. Lipophagy is like non-specific canonical autophagy in terms of macro- and micro-based mechanisms. Macrolipophagy involves delivery of LDs to lysosomes through autophagosomes and subsequent degradation. However, in microautophagy, lysosomes directly and transiently interact with LDs. Noteworthy, CMA is not directly involved in lipohagy [441]. To date, only two studies have examined role of lipophagy in prostate cancer. Typically, cancer cells undergo senescence upon therapy. Exposure of prostate cancer cells to Abrus agglutinin (AGG) leads to lipophagy-induced accumulation of fatty acids, and a reduction in the number of LDs. Using a lysosomal acid lipase inhibitor prevents lipophagy-mediated senescence, showing that lipophagy is vital for this process. Mechanistically, AGG stimulates cytoplasmic SIRT1 that in turn, deacetylases LAMP1 (lysosomal associated membrane protein 1) on lysine residues of the cytosolic domain, resulting in lipophagy-mediated senescence in prostate cancer cells [442]. Another study revealed that autophagy can contribute to LDs depletion in prostate cancer cells and promote prostate cancer growth. Autophagy inhibition using pharmacological or genetic interventions lead to a decrease in LDs depletion and cell growth. Although this study does not specifically examine lipophagy, it involved autophagy in the degradation of LDs and thus probably should be considered as lipophagy (Fig. 8) [443].

Other types of autophagy in prostate cancer
Role of autophagy in prognosis and diagnosis
Importantly, autophagy can be considered as a reliable biomarker for prostate cancer diagnosis and prognosis. The aggressive behavior of prostate cancer cells has forced studies to focus on detecting biomarkers [444, 445]. Such biomarkers can provide a diagnosis map for prostate cancer and can help practitioners in providing a prognosis. In this section, our aim is to show how autophagy can be utilized as a diagnostic and prognostic tool in prostate cancer patients. A recent experiment has evaluated 495 prostate cancer tissues in terms of expression of differentially expressed autophagy-related genes (DEARGs). These genes are responsible for autophagy regulation and subsequent impact on resistance of prostate cancer to platinum-containing compounds and EGFR tyrosine kinase inhibitors. Among them, five autophagy-related genes (ARGs) including ATG9B, DNAJB1, HSPB8, NKX2-3 and TP63 demonstrate remarkable association with prostate cancer development [446]. Further investigation demonstrates that autophagy can also be considered as a prognostic factor in prostate cancer. For this purpose, a recently conducted experiment has shown that overexpression of ARGs such as FAM215A, MYC and FADD can provide a poor prognosis and low overall survival rate of prostate cancer patients [447]. An interesting study performed in hospitals in Guangzhou, China revealed the reliability of MAP1S as biomarker in prostate cancer. MAP1S provides a connection between autophagic components with microtubules and mitochondria, en route to induction of autophagy. Further examination noted prostate cancer patients with high level of MAP1S demonstrate better prognosis compared to those with low level of MAP1S [448].
LRPPRC (leucine rich pentatricopeptide repeat containing) is considered as a negative regulator of autophagy and demonstrates upregulation in prostate cancer tissues. An analysis has shown that LRPPRC is overexpressed in 75% of prostate cancer patients, while it has low expression in 10% of prostate cancer patients. As an autophagy inhibitor, upregulation of LRPPRC reveals poor prognosis, low overall survival, and resistance to hormone therapy of prostate cancer [449]. Another experiment on 458 prostate cancer patients demonstrates association of ATG16L1 polymorphism (rs78835907) with recurrence and poor survival. In fact, ATG16L1 as an autophagy regulator, undergoes downregulation in patients with poor survival [450]. Previously, it was discussed that miRNAs can regulate autophagy in prostate cancer. A clinical study has confirmed miRNA and ARGs interaction in prostate cancer and their biomarker role. One of the strategies for prostate cancer immunotherapy is using PDCD1/PD-1 (programmed cell death 1) inhibitors. Briefly, PDCD1 participates in providing immune evasion and reducing anti-tumor immunity [451]. An ARG, known as NKX2-3 demonstrates overexpression in prostate cancer patients, and is associated with poor overall survival, lymph node metastasis and reduced capacity of PDCD1 inhibitors. Further studies reveal an interaction between MIR205 and NKX2-3 that determines the therapy response of prostate cancer patients [452]. Therefore, autophagy and its related genes are potential biomarkers for prostate cancer diagnosis and prognosis [446, 453, 454]. Figure 9 provides a schematic representation of autophagy mechanism involvement in various aspects of prostate cancer from proliferation and metastasis to therapy resistance and prognostic signature.

The autophagy mechanism signature in prostate cancer
Paving the way for clinical translation
Our aim in this review was to recap how autophagy participates in proliferation, metastasis, and therapy response of prostate cancer cells. Emphasis was put on molecular signaling pathways regulating autophagy and how they may be altered by anti-tumor compounds, gene therapy, and bioengineering strategies. To improve our understanding towards the role of autophagy in prostate cancer, a section examining autophagy as a diagnostic and prognostic tool was included. Noteworthy, previous steps for introducing autophagy into clinical treatment have been made, and, currently, autophagy is considered as a reliable biomarker for prostate cancer diagnosis. However, there is no clinical trial related to targeting autophagy for treatment of prostate cancer patients. For instance, chemotherapy failure is a common phenomenon in prostate cancer patients, owing to a significant role of autophagy in drug resistance and radio-resistance. Therefore, novel therapeutics can be developed in this case. In addition to anti-tumor compounds targeting autophagy, genetic tools such as siRNA, shRNA and CRISPR-Cas9 can be applied in this case. Further improvement can be made via introducing bioengineering for improving the capacity in autophagy regulation for treatment of prostate cancer patients. However, we are still at a beginning point and further studies will shed more light on targeting autophagy in prostate cancer therapy.
Conclusion and remarks
Effective management of prostate cancer requires a better understanding of molecular mechanisms involved in the onset and progression of prostate cancer. Among various mechanisms, autophagy possesses a dual role in cancer, and can either promote or suppress cancer progression. This review aimed to evaluate the role of autophagy in prostate cancer, as the most malignant tumor in men. Proliferation and metastasis of prostate cancer cells are tightly regulated by autophagy. Through manipulation of autophagy (downregulation or upregulation based on its role), proliferation and metastasis can be regulated for prostate cancer cells. Metabolic reprogramming is extensively adopted by prostate cancer cells in enhancing their progression. It seems that autophagy inhibition (for instance, by DNM1L upregulation), accelerates metabolic reprogramming in favor of prostate cancer progression. In this case, stimulating autophagy is of importance. Since autophagy determines growth and migration of prostate cancer cells, this molecular mechanism can be related to therapy response of prostate cancer cells. Many experiments have demonstrated the role of autophagy in chemotherapy and radiotherapy, as well as its modulation as an appropriate tool. During the initiation of prostate cancer, the autophagy machinery function as a pro-apoptotic; however, once it progresses, autophagy act as a proliferator, more specifically in CRPC and androgen ablation therapies.
Among the various molecular pathways regulating autophagy in prostate cancer, ncRNAs including miRNAs, lncRNAs and circRNAs have been investigated to a greater extent compared to other molecular pathways. It is worth mentioning that like autophagy, a ncRNA such as miRNA or lncRNA can act as a double-edged sword in cancer. This feature significantly complicates autophagy regulation in prostate cancer, and further efforts in developing novel therapeutics. According to current review, autophagy function in prostate cancer is context-dependent and it may action as pro-survival or pro-death mechanism in each stage of prostate cancer. Therefore, extensive research in future can reveal and highlight this action of autophagy.
The important hint is that experiments have focused on using anti-tumor compounds for autophagy regulation in prostate cancer treatment, and, also, using genetic tools such as siRNA and shRNA. However, these strategies are not completely effective in prostate cancer treatment, unless their efficiency is improved using nanoparticles. There are currently no experimental data about gene delivery by nanoparticles for autophagy regulation in prostate cancer treatment and future studies can focus on this aspect. Furthermore, autophagy has been applied as a biomarker for diagnosis and prognosis of prostate cancer. This is a milestone in progress in introducing autophagy into the clinic. Finally, for improving autophagy targeting in prostate cancer therapy in clinical treatment, the recommendations in section 3.10 should be considered.
There are some limitations related to current works that should be considered in future experiments. Most of the experiments have focused on phytochemicals, while their clinical application is restricted. A few small molecules have been examined for targeting autophagy in prostate cancer therapy. Hence, future studies should focus on developing new small molecular targeting autophagy regulators such as BECN1 and ATGs, among others. Although autophagy function in chemotherapy and radiotherapy responses of prostate cancer cells has been investigated, more anti-cancer agents should be tested. Furthermore, association between autophagy and apoptosis in chemoresistance/chemosensitivity should be highlighted, as apoptosis is the most well-known mechanism affected by anti-cancer agents. Another limitation is related to therapeutic targeting of genes for autophagy regulation in prostate cancer treatment. Based on the discussions, ncRNAs and various kinds of molecular pathways have capacity of autophagy regulation. However, genetic tools such as CRISPR-Cas9 system, siRNA and shRNA have not been fully employed in autophagy regulation in prostate cancer suppression. Therefore, at the first step, future experiments should focus on targeting specific genes regulating autophagy such as BECN1 and ATGs. Despite efforts for using nanoparticles and biological vectors in autophagy regulation and suppressing prostate cancer progression, there are still some limitations. The studies have only focused on carbon-based and metal-based nanomaterials for autophagy regulation in prostate cancer treatment. Other kinds of nanostructures such as lipid-based nanoparticles and polymeric nanostructures should also be employed. Furthermore, nanostructures can be utilized for improving potential of genetic tools in autophagy regulation for prostate cancer suppression.
Availability of data and materials
Not applicable.
Abbreviations
- CRPC:
-
Castration-resistance prostate cancer
- AR:
-
Androgen receptor
- REST:
-
RE1 silencing transcription factor
- CMA:
-
Chaperone-mediated autophagy
- ATG:
-
Autophagy related
- PI3K:
-
Phosphoinositide 3-kinase
- ER:
-
Endoplasmic reticulum
- MAP 1LC3/LC3:
-
Microtubule associated protein 1 light chain 3
- MTOR:
-
Mechanistic target of rapamycin kinase
- AMPK:
-
AMP-activated protein kinase
- LAMP:
-
Lysosomal associated membrane protein
- PCD:
-
Programmed cell death
- MHCI:
-
Major histocompatibility complex class I
- GABARAPL1:
-
GABA type A receptor associated protein like 1
- HIF1A/HIF-1α:
-
Hypoxia inducible factor 1 subunit alpha
- FGFs:
-
Fibroblast growth factors
- DNM1L/DRP1:
-
Dynamin 1 like
- HSD17B4:
-
Hydroxysteroid 17-beta dehydrogenase 4
- MAPK/JNK:
-
Mitogen-activated protein kinase
- ECM:
-
Extracellular matrix
- EMT:
-
Epithelial-to-mesenchymal transition
- WFDC2:
-
WAP four-disulfide core domain 2
- EGFR:
-
Epidermal growth factor receptor
- TME:
-
Tumor microenvironment
- ADT:
-
Androgen-deprivation therapy
- HDAC6:
-
Histone deacetylase 6
- TGFB/TGF-β:
-
Transforming growth factor beta
- NFKB/NF-κB:
-
Nuclear factor kappa B
- STAT3:
-
Signal transducer and activator of transcription 3
- miRNAs:
-
MicroRNAs
- SGK1:
-
Serum/glucocorticoid regulated kinase 1
- siRNA:
-
Small interfering RNA
- shRNA:
-
Short hairpin RNA
- WWOX:
-
WW domain containing oxidoreductase
- NPRL2:
-
NPR2 like, GATOR1 complex subunit
- MSCs:
-
Mesenchymal stem cells
- HDAC3:
-
Histone deacetylase 3
- CP:
-
Cisplatin
- AMBRA1:
-
Autophagy and beclin 1 regulator 1
- PARP:
-
Poly(ADP-ribose) polymerase
- ASAH1/acid ceramidase:
-
N-acylsphingosine amidohydrolase 1
- HMGB1:
-
High mobility group box 1
- TPD52/PrLZ/prostate leucine zipper:
-
tumor protein D52
- STK11/LKB1:
-
Serine/threonine kinase 11
- PTAFR:
-
Platelet activating factor receptor
- GPCR:
-
G-protein-coupled receptor
- ncRNAs:
-
Non-coding RNAs
- mRNA:
-
Messenger RNA
- 3’-UTR:
-
3’-untranslated region
- PTEN:
-
Phosphatase and tensin homolog
- RELN:
-
Reelin
- PHLPP2:
-
PH domain and leucine rich repeat protein phosphatase 2
- TP53INP1:
-
Tumor protein p53 inducible nuclear protein 1
- SP1:
-
Sp1 transcription factor
- PKM2:
-
Pyruvate kinase M1/2; SIRT1
sirtuin 1
- ERN1/IRE1:
-
Endoplasmic reticulum to nucleus signaling 1
- SNHG1 :
-
Small nucleolar RNA host gene 1
- EZH2:
-
Enhancer of zeste polycomb repressive complex 2 subunit
- PPARG/PPARγ:
-
Peroxisome proliferator activated receptor gamma
- HULC :
-
Hepatocellular carcinoma up-regulated long non-coding RNA
- circRNAs:
-
Circular RNAs
- circ-CCNB2 :
-
CircRNA cyclin B2
- KIF18A:
-
Kinesin family member 18A
- NO:
-
Nitric oxide
- MAPK:
-
Mitogen-activated protein kinase
- SIRT7:
-
Sirtuin 7
- NEDD4:
-
NEDD4 E3 ubiquitin protein ligase
- EriB:
-
Eriocalyxin B
- BafA1:
-
Bafilomycin A1
- TF:
-
Transferrin
- TFRC/TfR1:
-
Transferrin receptor
- IRPs:
-
Iron regulatory proteins
- IREs:
-
Iron response elements
- ACO1/IRP1:
-
Aconitase 1
- FACs:
-
Ferric ammonium citrate
- αTT:
-
α-tocotericol
- ROS:
-
Reactive oxygen species
- VEGF:
-
Vascular endothelial growth factor
- RISC:
-
RNA-induced silencing complex
- IGF1R:
-
Insulin like growth factor 1 receptor
- PINK1:
-
PTEN induced kinase 1
- CAV1:
-
Caveolin 1
- LDs:
-
Lipid droplets
- AGG:
-
Abrus agglutinin
- DEARGs:
-
Differentially expressed autophagy-related genes
- ARGs:
-
Autophagy-related genes
- LRPPRC:
-
Leucine rich pentatricopeptide repeat containing
- PDCD1/PD-1:
-
Programmed cell death 1
References
Swami U, et al. Advanced prostate cancer: treatment advances and future directions. Trends Cancer. 2020;6(8):702–15.
Siegel RL, et al. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7–33.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
Sung H, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Moul JW. The evolving definition of advanced prostate cancer. Rev Urol. 2004;6(Suppl 8):S10.
Lorenc T, et al. Exosomes in prostate cancer diagnosis, prognosis and therapy. Int J Mol Sci. 2020;21(6):2118.
Ingrosso G, et al. Current therapeutic options in metastatic castration-resistant prostate cancer. Semin Oncol. 2018;45(5–6):303–15.
Shanmugam MK, et al. Ursolic acid inhibits the initiation, progression of prostate cancer and prolongs the survival of TRAMP mice by modulating pro-inflammatory pathways. Plos One. 2012;7(3):e32476.
Jung YY, et al. LDL cholesterol promotes the proliferation of prostate and pancreatic cancer cells by activating the STAT3 pathway. J Cell Physiol. 2021;236(7):5253–64.
Kretschmer A, Tilki D. Biomarkers in prostate cancer - current clinical utility and future perspectives. Crit Rev Oncol Hematol. 2017;120:180–93.
Lu YT, et al. Current status of liquid biopsies for the detection and management of prostate cancer. Cancer Manag Res. 2019;11:5271–91.
Htoo KPP, et al. Colorimetric detection of PCA3 in urine for prostate cancer diagnosis using thiol-labeled PCR primer and unmodified gold nanoparticles. Clin Chim Acta. 2019;488:40–9.
Wu J, et al. Functionalized NIR-II semiconducting polymer nanoparticles for single-cell to whole-organ imaging of PSMA-positive prostate cancer. Small. 2020;16(19):e2001215.
Yamkamon V, et al. Urinary PCA3 detection in prostate cancer by magnetic nanoparticles coupled with colorimetric enzyme-linked oligonucleotide assay. EXCLI J. 2020;19:501–13.
Zhang J, et al. Nimbolide-induced oxidative stress abrogates STAT3 signaling Cascade and inhibits tumor growth in transgenic adenocarcinoma of mouse prostate model. Antioxid Redox Signal. 2016;24(11):575–89.
Liu Y, et al. Inhibition of the deubiquitinase USP9x induces pre-B cell homeobox 1 (PBX1) degradation and thereby stimulates prostate cancer cell apoptosis. J Biol Chem. 2019;294(12):4572–82.
Zhang J, et al. Cardamonin represses proliferation, invasion, and causes apoptosis through the modulation of signal transducer and activator of transcription 3 pathway in prostate cancer. Apoptosis. 2017;22(1):158–68.
Ge R, et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Ann Oncol. 2020;31(4):470–9.
Cheng L, Zhang DY. Molecular genetic pathology. New York: Springer Science & Business Media; 2010.
Wang HT, et al. Neuroendocrine prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: factors associated with time to development of NEPC and survival from NEPC diagnosis—a systematic review and pooled analysis. J Clin Oncol. 2014;32(30):3383–90.
Robinson D, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28.
Beltran H, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer discovery. 2011;1(6):487–95.
Williamson SR, Eble JN, Cheng L. Molecular pathology of kidney tumors, in Molecular Surgical Pathology. New York: Springer; 2013. p. 171–212.
Jiang G, et al. p62 promotes proliferation, apoptosis-resistance and invasion of prostate cancer cells through the Keap1/Nrf2/ARE axis. Oncol Rep. 2020;43(5):1547–57.
Ding Y, et al. Neuropeptide Y nerve paracrine regulation of prostate cancer oncogenesis and therapy resistance. Prostate. 2021;81(1):58–71.
Sikka S, et al. Targeting PPARγ signaling Cascade for the prevention and treatment of prostate Cancer. PPAR Res. 2012;2012:968040.
Lee JH, et al. Capsazepine inhibits JAK/STAT3 signaling, tumor growth, and cell survival in prostate cancer. Oncotarget. 2017;8(11):17700–11.
Ichimiya T, et al. Autophagy and autophagy-related diseases: a review. Int J Mol Sci. 2020;21(23):8974.
Aventaggiato M, et al. Sirtuins’ control of autophagy and mitophagy in cancer. Pharmacol Ther. 2020;221:107748.
Bhardwaj M, et al. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin Cancer Biol. 2019;66:116-28. Elsevier.
Singh SS, et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2018;37(9):1142–58.
Deng S, et al. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer. 2019;125(8):1228–46.
De Duve C, et al. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 1955;60(4):604–17.
Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69(7):1125–36.
Tasset I, Cuervo AM. Role of chaperone-mediated autophagy in metabolism. FEBS J. 2016;283(13):2403–13.
Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27(6):421–9.
Wen X, Klionsky DJ. At a glance: a history of autophagy and cancer. Semin Cancer Biol. 2020;66:3–11.
Gremke N, et al. mTOR-mediated cancer drug resistance suppresses autophagy and generates a druggable metabolic vulnerability. Nat Commun. 2020;11(1):4684.
Ishaq M, et al. Autophagy in cancer: recent advances and future directions. Semin Cancer Biol. 2020;66:171–81.
Raj S, et al. Molecular mechanisms of interplay between autophagy and metabolism in cancer. Life Sci. 2020;259:118184.
Tanaka H, et al. Comparison of autophagy inducibility in various tyrosine kinase inhibitors and their enhanced cytotoxicity via inhibition of autophagy in cancer cells in combined treatment with azithromycin. Biochem Biophys Rep. 2020;22:100750.
Patel SM, et al. Potential of neem (Azadirachta indica L.) for prevention and treatment of oncologic diseases. Semin Cancer Biol. 2016;40-41:100–15.
Hwang ST, et al. Cycloastragenol can negate constitutive STAT3 activation and promote paclitaxel-induced apoptosis in human gastric cancer cells. Phytomedicine. 2019;59:152907.
Tavakol S, et al. Autophagy modulators: mechanistic aspects and drug delivery systems. Biomolecules. 2019;9(10):530.
Bordoloi D, et al. TIPE family of proteins and its implications in different chronic diseases. Int J Mol Sci. 2018;19(10):2974.
Acevo-Rodríguez PS, et al. Autophagy regulation by the translation machinery and its implications in cancer. Front Oncol. 2020;10:322.
Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–64.
Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207–15.
Takahashi Y, et al. An autophagy assay reveals the ESCRT-III component CHMP2A as a regulator of phagophore closure. Nat Commun. 2018;9(1):2855.
Zhang R, et al. Ion channels and transporters in autophagy. Autophagy. 2021:1–20. https://doi.org/10.1080/15548627.2021.1885147.
Liu J, et al. Autophagy-dependent Ferroptosis: machinery and regulation. Cell Chemical Biology. 2020;27(4):420–35.
Yin Z, et al. The roles of ubiquitin in mediating autophagy. Cells. 2020;9(9):2025.
Benvenuto M, et al. Polyphenol-mediated autophagy in cancer: evidence of in vitro and in vivo studies. Int J Mol Sci. 2020;21(18):6635.
Zhang HM, et al. MKL1/miR-5100/CAAP1 loop regulates autophagy and apoptosis in gastric cancer cells. Neoplasia. 2020;22(5):220–30.
Kong F, et al. Downregulation of METTL14 increases apoptosis and autophagy induced by cisplatin in pancreatic cancer cells. Int J Biochem Cell Biol. 2020;122:105731.
Feng X, et al. Hypoxia-induced acetylation of PAK1 enhances autophagy and promotes brain tumorigenesis via phosphorylating ATG5. Autophagy. 2020:1–20. https://doi.org/10.1080/15548627.2020.1731266.
Li JP, et al. miR-133a-3p/FOXP3 axis regulates cell proliferation and autophagy in gastric cancer. J Cell Biochem. 2020;121(5–6):3392–405.
Babaei G, Aziz SG, Jaghi NZZ. EMT, cancer stem cells and autophagy; the three main axes of metastasis. Biomed Pharmacother. 2021;133:110909.
Salimi L, et al. Synergies in exosomes and autophagy pathways for cellular homeostasis and metastasis of tumor cells. Cell Biosci. 2020;10:64.
Marsh T, Debnath J. Autophagy suppresses breast cancer metastasis by degrading NBR1. Autophagy. 2020;16(6):1164–5.
Alizadeh J, et al. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim Biophys Acta Mol Cell Res. 2018;1865(5):749–68.
de Souza ASC, et al. The role of autophagy in tumor immunology-complex mechanisms that may be explored therapeutically. Front Oncol. 2020;10:603661.
Chavez-Dominguez R, et al. The double-edge sword of autophagy in Cancer: from tumor suppression to pro-tumor activity. Front Oncol. 2020;10:578418.
Yamamoto K, et al. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy. 2020;16(8):1524–5.
Hou G, et al. Inhibition of autophagy improves resistance and enhances sensitivity of gastric cancer cells to cisplatin. Can J Physiol Pharmacol. 2020;98(7):449–58.
Zheng X, et al. PRDX2 removal inhibits the cell cycle and autophagy in colorectal cancer cells. Aging (Albany NY). 2020;12(16):16390–409.
Ueda S, et al. miR-27a ameliorates chemoresistance of breast cancer cells by disruption of reactive oxygen species homeostasis and impairment of autophagy. Lab Investig. 2020;100(6):863–73.
Gao S, Wang K, Wang X. miR-375 targeting autophagy-related 2B (ATG2B) suppresses autophagy and tumorigenesis in cisplatin-resistant osteosarcoma cells. Neoplasma. 2020;67(4):724–34.
Zamame Ramirez JA, Romagnoli GG, Kaneno R. Inhibiting autophagy to prevent drug resistance and improve anti-tumor therapy. Life Sci. 2021;265:118745.
Muscolino E, et al. Herpesviruses induce aggregation and selective autophagy of host signalling proteins NEMO and RIPK1 as an immune-evasion mechanism. Nat Microbiol. 2020;5(2):331–42.
Ding M, et al. SIRT7 depletion inhibits cell proliferation and androgen-induced autophagy by suppressing the AR signaling in prostate cancer. J Exp Clin Cancer Res. 2020;39(1):28.
Wadosky KM, et al. Riluzole induces AR degradation via endoplasmic reticulum stress pathway in androgen-dependent and castration-resistant prostate cancer cells. Prostate. 2019;79(2):140–50.
Blessing AM, et al. Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy. 2017;13(3):506–21.
Xie CW, et al. Gabarapl1 mediates androgen-regulated autophagy in prostate cancer. Tumour Biol. 2015;36(11):8727–33.
Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–75.
Young SD, Marshall RS, Hill RP. Hypoxia induces DNA overreplication and enhances metastatic potential of murine tumor cells. Proc Natl Acad Sci U S A. 1988;85(24):9533–7.
Young SD, Hill RP. Effects of reoxygenation on cells from hypoxic regions of solid tumors: analysis of transplanted murine tumors for evidence of DNA overreplication. Cancer Res. 1990;50(16):5031–8.
Xia L, et al. PRKAR2B-HIF-1α loop promotes aerobic glycolysis and tumour growth in prostate cancer. Cell Prolif. 2020;53(11):e12918.
Fan Y, et al. PLCε regulates metabolism and metastasis signaling via HIF-1α/MEK/ERK pathway in prostate cancer. J Cell Physiol. 2020;235(11):8546–57.
Ma Z, et al. Targeting hypoxia-inducible factor-1-mediated metastasis for cancer therapy. Antioxid Redox Signal. 2021;34(18):1484-97.
Shanmugam MK, et al. Potential role of natural compounds as anti-angiogenic agents in cancer. Curr Vasc Pharmacol. 2017;15(6):503–19.
Deng M, et al. HIF-1a regulates hypoxia-induced autophagy via translocation of ANKRD37 in colon cancer. Exp Cell Res. 2020;395(1):112175.
Yu K, et al. HIF1α promotes prostate cancer progression by increasing ATG5 expression. Anim Cells Syst (Seoul). 2019;23(5):326–34.
Zhang N, et al. PAX5-induced upregulation of IDH1-AS1 promotes tumor growth in prostate cancer by regulating ATG5-mediated autophagy. Cell Death Dis. 2019;10(10):734.
Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discov. 2009;8(3):235–53.
Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26(4):312–24.
Dai H, et al. FGF21 facilitates autophagy in prostate cancer cells by inhibiting the PI3K-Akt-mTOR signaling pathway. Cell Death Dis. 2021;12(4):303.
Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. 2016;212(4):379–87.
Senft D, Ze’ev AR. Regulators of mitochondrial dynamics in cancer. Curr Opin Cell Biol. 2016;39:43–52.
Lee YG, et al. Androgen-induced expression of DRP1 regulates mitochondrial metabolic reprogramming in prostate cancer. Cancer Lett. 2020;471:72–87.
Glover DM, et al. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81(1):95–105.
Zhou H, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20(2):189–93.
Jones D, et al. Aurora a regulates expression of AR-V7 in models of castrate resistant prostate cancer. Sci Rep. 2017;7:40957.
Buschhorn HM, et al. Aurora-a over-expression in high-grade PIN lesions and prostate cancer. Prostate. 2005;64(4):341–6.
Zhang S, et al. Aurora-a regulates autophagy through the Akt pathway in human prostate cancer. Cancer Biomark. 2017;19(1):27–34.
You D, et al. KML001 induces apoptosis and Autophagic cell death in prostate Cancer cells via oxidative stress pathway. Plos One. 2015;10(9):e0137589.
Chopra P, et al. Polo-like kinase inhibitors: an emerging opportunity for cancer therapeutics. Expert Opin Investig Drugs. 2010;19(1):27–43.
Huang H, et al. Acetylation-mediated degradation of HSD17B4 regulates the progression of prostate cancer. Aging (Albany NY). 2020;12(14):14699–717.
Dai X, et al. A novel benzimidazole derivative, MBIC inhibits tumor growth and promotes apoptosis via activation of ROS-dependent JNK signaling pathway in hepatocellular carcinoma. Oncotarget. 2017;8(8):12831–42.
Ko JH, et al. 2,5-Dihydroxyacetophenone induces apoptosis of multiple myeloma cells by regulating the MAPK activation pathway. Molecules. 2017;22(7):1157.
Kim C, et al. Formononetin regulates multiple oncogenic signaling cascades and enhances sensitivity to bortezomib in a multiple myeloma mouse model. Biomolecules. 2019;9(7):262.
Tewari D, et al. Targeting activator protein 1 signaling pathway by bioactive natural agents: possible therapeutic strategy for cancer prevention and intervention. Pharmacol Res. 2018;128:366–75.
Zhang P, et al. SH3RF3 promotes breast cancer stem-like properties via JNK activation and PTX3 upregulation. Nat Commun. 2020;11(1):2487.
Wu Q, et al. Selective inhibitors for JNK signalling: a potential targeted therapy in cancer. J Enzyme Inhib Med Chem. 2020;35(1):574–83.
Morgan EL, et al. E6-mediated activation of JNK drives EGFR signalling to promote proliferation and viral oncoprotein expression in cervical cancer. Cell Death Differ. 2020;28(5):1669-87.
Shishodia S, et al. Guggulsterone inhibits tumor cell proliferation, induces S-phase arrest, and promotes apoptosis through activation of c-Jun N-terminal kinase, suppression of Akt pathway, and downregulation of antiapoptotic gene products. Biochem Pharmacol. 2007;74(1):118–30.
Arisan ED, et al. Inhibition on JNK mimics silencing of Wnt-11 mediated cellular response in androgen-independent Prostate cancer cells. Biology (Basel). 2020;9(7):142.
Wang Z, et al. Clinicopathological signature of p21-activated kinase 1 in prostate cancer and its regulation of proliferation and autophagy via the mTOR signaling pathway. Oncotarget. 2017;8(14):22563–80.
Wang L, et al. Co-targeting hexokinase 2-mediated Warburg effect and ULK1-dependent autophagy suppresses tumor growth of PTEN- and TP53-deficiency-driven castration-resistant prostate cancer. EBioMedicine. 2016;7:50–61.
Lin C, et al. Inhibition of CAMKK2 impairs autophagy and castration-resistant prostate cancer via suppression of AMPK-ULK1 signaling. Oncogene. 2021;40(9):1690–705.
Huang YL, et al. Extrinsic sphingosine 1-phosphate activates S1P5 and induces autophagy through generating endoplasmic reticulum stress in human prostate cancer PC-3 cells. Cell Signal. 2014;26(3):611–8.
Zhou R, et al. p300/CBP-associated factor promotes autophagic degradation of δ-catenin through acetylation and decreases prostate cancer tumorigenicity. Sci Rep. 2019;9(1):3351.
Quan Z, et al. PLCɛ maintains the functionality of AR signaling in prostate cancer via an autophagy-dependent mechanism. Cell Death Dis. 2020;11(8):716.
Jiang Q, et al. Targeting androgen receptor leads to suppression of prostate cancer via induction of autophagy. J Urol. 2012;188(4):1361–8.
Fang F, et al. CD147 modulates autophagy through the PI3K/Akt/mTOR pathway in human prostate cancer PC-3 cells. Oncol Lett. 2015;9(3):1439–43.
Shanmugam MK, et al. Inhibition of CXCR4/CXCL12 signaling axis by ursolic acid leads to suppression of metastasis in transgenic adenocarcinoma of mouse prostate model. Int J Cancer. 2011;129(7):1552–63.
Shanmugam MK, et al. Thymoquinone inhibits bone metastasis of breast cancer cells through abrogation of the CXCR4 signaling axis. Front Pharmacol. 2018;9:1294.
Loh CY, et al. Signal Transducer and Activator of Transcription (STATs) proteins in cancer and inflammation: functions and therapeutic implication. Front Oncol. 2019;9:48.
Manu KA, et al. Emodin suppresses migration and invasion through the modulation of CXCR4 expression in an orthotopic model of human hepatocellular carcinoma. Plos One. 2013;8(3):e57015.
Adekoya TO, Richardson RM. Cytokines and chemokines as mediators of prostate cancer metastasis. Int J Mol Sci. 2020;21(12):4449.
Jin JK, Dayyani F, Gallick GE. Steps in prostate cancer progression that lead to bone metastasis. Int J Cancer. 2011;128(11):2545–61.
Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Rev Oncog. 2013;18(1–2):43–73.
Guan X. Cancer metastases: challenges and opportunities. Acta Pharm Sin B. 2015;5(5):402–18.
Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127(4):679–95.
Ashrafizadeh M, et al. Association of the Epithelial-Mesenchymal Transition (EMT) with cisplatin resistance. Int J Mol Sci. 2020;21(11):4002.
Cheng JT, et al. Insights into biological role of LncRNAs in Epithelial-Mesenchymal Transition. Cells. 2019;8(10):1178.
Ashrafizadeh M, et al. Role of microRNA/Epithelial-to-Mesenchymal Transition Axis in the metastasis of bladder cancer. Biomolecules. 2020;10(8):1159.
Wen S, et al. Long non-coding RNA NEAT1 promotes bone metastasis of prostate cancer through N6-methyladenosine. Mol Cancer. 2020;19(1):171.
Buckup M, et al. Plectin is a regulator of prostate cancer growth and metastasis. Oncogene. 2021;40(3):663–76.
Xiong Y, et al. WFDC2 suppresses prostate cancer metastasis by modulating EGFR signaling inactivation. Cell Death Dis. 2020;11(7):537.
Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591–6.
Godoy A, et al. Androgen deprivation induces rapid involution and recovery of human prostate vasculature. Am J Physiol Endocrinol Metab. 2011;300(2):E263–75.
Wang X, et al. Endothelial cells enhance prostate cancer metastasis via IL-6→androgen receptor→TGF-β→MMP-9 signals. Mol Cancer Ther. 2013;12(6):1026–37.
Tomić TT, et al. Castration resistant prostate cancer is associated with increased blood vessel stabilization and elevated levels of VEGF and Ang-2. Prostate. 2012;72(7):705–12.
Zhao R, et al. Endothelial cells promote metastasis of prostate cancer by enhancing autophagy. J Exp Clin Cancer Res. 2018;37(1):221.
Liang T, et al. HDAC6-mediated α-tubulin deacetylation suppresses autophagy and enhances motility of podocytes in diabetic nephropathy. J Cell Mol Med. 2020;24(19):11558–72.
Jiang X, et al. Metastatic prostate cancer-associated P62 inhibits autophagy flux and promotes epithelial to mesenchymal transition by sustaining the level of HDAC6. Prostate. 2018;78(6):426–34.
Lee JH, et al. Farnesol abrogates epithelial to mesenchymal transition process through regulating Akt/mTOR pathway. Pharmacol Res. 2019;150:104504.
Hwang ST, et al. Corilagin represses epithelial to mesenchymal transition process through modulating Wnt/β-Catenin signaling cascade. Biomolecules. 2020;10(10):1406.
Lee JH, et al. Brusatol suppresses STAT3-driven metastasis by downregulating epithelial-mesenchymal transition in hepatocellular carcinoma. J Adv Res. 2020;26:83–94.
Archer M, Dogra N, Kyprianou N. Inflammation as a driver of prostate cancer metastasis and therapeutic resistance. Cancers. 2020;12(10):2984.
López-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009;1(6–7):303–14.
Puar YR, et al. Evidence for the involvement of the master transcription Factor NF-κB in cancer initiation and progression. Biomedicines. 2018;6(3):82.
Ahn KS, et al. Genetic deletion of NAD(P)H:quinone oxidoreductase 1 abrogates activation of nuclear factor-kappaB, IkappaBalpha kinase, c-Jun N-terminal kinase, Akt, p38, and p44/42 mitogen-activated protein kinases and potentiates apoptosis. J Biol Chem. 2006;281(29):19798–808.
Ashrafizadeh M, et al. MicroRNAs and their influence on the ZEB family: mechanistic aspects and therapeutic applications in cancer therapy. Biomolecules. 2020;10(7):1040.
Wang Y, et al. miR-19a promotes the metastasis and EMT through CUL5 in prostate cancer cell line PC3. J buon. 2020;25(4):2028–35.
Gorrab A, et al. Leptin promotes prostate cancer proliferation and migration by stimulating STAT3 pathway. Nutr Cancer. 2020:1–11. https://doi.org/10.1080/01635581.2020.1792946.
Zhang Q, et al. LOXL2 upregulation in gliomas drives Tumorigenicity by activating autophagy to promote TMZ resistance and trigger EMT. Front Oncol. 2020;10:569584.
Liu W, et al. SGK1 inhibition-induced autophagy impairs prostate cancer metastasis by reversing EMT. J Exp Clin Cancer Res. 2018;37(1):73.
Manu KA, et al. Isorhamnetin augments the anti-tumor effect of capecitabine through the negative regulation of NF-κB signaling cascade in gastric cancer. Cancer Lett. 2015;363(1):28–36.
Manu KA, et al. Simvastatin sensitizes human gastric cancer xenograft in nude mice to capecitabine by suppressing nuclear factor-kappa B-regulated gene products. J Mol Med (Berl). 2014;92(3):267–76.
Raghunath A, et al. Dysregulation of Nrf2 in hepatocellular carcinoma: role in cancer progression and chemoresistance. Cancers (Basel). 2018;10(12):481.
Monisha J, et al. NGAL is downregulated in oral squamous cell carcinoma and leads to increased survival, proliferation, migration and chemoresistance. Cancers (Basel). 2018;10(7):228.
Jung YY, et al. Anti-myeloma effects of icariin are mediated through the attenuation of JAK/STAT3-dependent signaling Cascade. Front Pharmacol. 2018;9:531.
Tieu T, et al. Patient-derived prostate Cancer explants: a clinically relevant model to assess siRNA-based nanomedicines. Adv Healthc Mater. 2021;10(6):e2001594.
Ashrafizadeh M, et al. Progress in delivery of siRNA-Based therapeutics employing nano-vehicles for treatment of prostate cancer. Bioengineering (Basel). 2020;7(3):91.
Wei L, et al. Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat Commun. 2019;10(1):4681.
Kim E, et al. Prostate cancer cell death produced by the co-delivery of Bcl-xL shRNA and doxorubicin using an aptamer-conjugated polyplex. Biomaterials. 2010;31(16):4592–9.
Cristofani R, et al. Dual role of autophagy on docetaxel-sensitivity in prostate cancer cells. Cell Death Dis. 2018;9(9):889.
Zhao Y, et al. WWOX promotes apoptosis and inhibits autophagy in paclitaxel-treated ovarian carcinoma cells. Mol Med Rep. 2021;23(2):1.
Yang S, et al. MicroRNA-375 targets ATG14 to inhibit autophagy and sensitize hepatocellular carcinoma cells to Sorafenib. Onco Targets Ther. 2020;13:3557–70.
Anlaş AA, Nelson CM. Soft microenvironments induce Chemoresistance by increasing autophagy downstream of integrin-linked kinase. Cancer Res. 2020;80(19):4103–13.
Lin JH, et al. Ursolic acid promotes apoptosis, autophagy, and chemosensitivity in gemcitabine-resistant human pancreatic cancer cells. Phytother Res. 2020;34(8):2053–66.
Dubois C, et al. Co-targeting Mitochondrial Ca (2+) Homeostasis and autophagy enhances cancer cells’ chemosensitivity. iScience. 2020;23(7):101263.
Zheng W, et al. Inhibition of Cathepsin D (CTSD) enhances radiosensitivity of glioblastoma cells by attenuating autophagy. Mol Carcinog. 2020;59(6):651–60.
Chen Q, et al. ANXA6 contributes to Radioresistance by promoting autophagy via inhibiting the PI3K/AKT/mTOR signaling pathway in nasopharyngeal carcinoma. Front Cell Dev Biol. 2020;8:232.
Chen H, et al. MiR-450a-5p inhibits autophagy and enhances radiosensitivity by targeting dual-specificity phosphatase 10 in esophageal squamous cell carcinoma. Cancer Lett. 2020;483:114–26.
Ray J, et al. miR-191 promotes radiation resistance of prostate cancer through interaction with RXRA. Cancer Lett. 2020;473:107–17.
Chen L, et al. Identification of cofilin-1 as a novel mediator for the metastatic potentials and chemoresistance of the prostate cancer cells. Eur J Pharmacol. 2020;880:173100.
Wei Y, et al. The GATOR1 complex regulates metabolic homeostasis and the response to nutrient stress in Drosophila melanogaster. G3. 2016;6(12):3859–67.
Kwak SS, et al. Amino acid-dependent NPRL2 interaction with raptor determines mTOR complex 1 activation. Cell Signal. 2016;28(2):32–41.
Dutchak PA, et al. Regulation of hematopoiesis and methionine homeostasis by mTORC1 inhibitor NPRL2. Cell Rep. 2015;12(3):371–9.
Haas NB, et al. Autophagy inhibition to augment mTOR inhibition: a phase I/II trial of Everolimus and hydroxychloroquine in patients with previously treated renal cell carcinoma. Clin Cancer Res. 2019;25(7):2080–7.
Rosich L, et al. Counteracting autophagy overcomes resistance to everolimus in mantle cell lymphoma. Clin Cancer Res. 2012;18(19):5278–89.
Chen Z, et al. NPRL2 enhances autophagy and the resistance to Everolimus in castration-resistant prostate cancer. Prostate. 2019;79(1):44–53.
Luo S, et al. NPRL2 promotes docetaxel chemoresistance in castration resistant prostate cancer cells by regulating autophagy through the mTOR pathway. Exp Cell Res. 2020;390(2):111981.
Wang Q, et al. Inhibiting autophagy overcomes docetaxel resistance in castration-resistant prostate cancer cells. Int Urol Nephrol. 2018;50(4):675–86.
Jin S, et al. TGF-β1 fucosylation enhances the autophagy and mitophagy via PI3K/Akt and Ras-Raf-MEK-ERK in ovarian carcinoma. Biochem Biophys Res Commun. 2020;524(4):970–6.
Yu Y, et al. Mesenchymal stem cells desensitize castration-resistant prostate cancer to docetaxel chemotherapy via inducing TGF-β1-mediated cell autophagy. Cell Biosci. 2021;11(1):7.
Dong JT, Chen C. Essential role of KLF5 transcription factor in cell proliferation and differentiation and its implications for human diseases. Cell Mol Life Sci. 2009;66(16):2691–706.
Chen C, et al. KLF5 is frequently deleted and down-regulated but rarely mutated in prostate cancer. Prostate. 2003;55(2):81–8.
Jia X, et al. BAP1 antagonizes WWP1-mediated transcription factor KLF5 ubiquitination and inhibits autophagy to promote melanoma progression. Exp Cell Res. 2021;402(1):112506.
Jia J, et al. KLF5 downregulation desensitizes castration-resistant prostate cancer cells to docetaxel by increasing BECN1 expression and inducing cell autophagy. Theranostics. 2019;9(19):5464–77.
Chhipa RR, Wu Y, Ip C. AMPK-mediated autophagy is a survival mechanism in androgen-dependent prostate cancer cells subjected to androgen deprivation and hypoxia. Cell Signal. 2011;23(9):1466–72.
Raudenska M, et al. Cisplatin enhances cell stiffness and decreases invasiveness rate in prostate cancer cells by actin accumulation. Sci Rep. 2019;9(1):1660.
Gupta A, et al. Mdm2-P53 interaction inhibitor with cisplatin enhances apoptosis in Colon and Prostate Cancer cells in-vitro. Asian Pac J Cancer Prev. 2019;20(11):3341–51.
Zhou P, et al. miR-17-92 plays an oncogenic role and conveys chemo-resistance to cisplatin in human prostate cancer cells. Int J Oncol. 2016;48(4):1737–48.
Jiang H, et al. Knockdown of the long noncoding RNA HOTTIP inhibits cell proliferation and enhances cell sensitivity to cisplatin by suppressing the Wnt/β-catenin pathway in prostate cancer. J Cell Biochem. 2019;120(6):8965–74.
Strappazzon F, et al. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J. 2011;30(7):1195–208.
Fimia GM, et al. Unleashing the Ambra1-Beclin 1 complex from dynein chains: Ulk1 sets Ambra1 free to induce autophagy. Autophagy. 2011;7(1):115–7.
Liu J, et al. Ambra1 induces autophagy and desensitizes human prostate cancer cells to cisplatin. Biosci Rep. 2019;39(8).
Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004;4(8):604–16.
Norris JS, et al. Combined therapeutic use of AdGFPFasL and small molecule inhibitors of ceramide metabolism in prostate and head and neck cancers: a status report. Cancer Gene Ther. 2006;13(12):1045–51.
Turner LS, et al. Autophagy is increased in prostate cancer cells overexpressing acid ceramidase and enhances resistance to C6 ceramide. Prostate Cancer Prostatic Dis. 2011;14(1):30–7.
Lu Y, et al. CXCL1-LCN2 paracrine axis promotes progression of prostate cancer via the Src activation and epithelial-mesenchymal transition. Cell Commun Signal. 2019;17(1):118.
Dai Y, Sieman D. n, c-Src is required for hypoxia-induced metastasis-associated functions in prostate cancer cells. Onco Targets Ther. 2019;12:3519–29.
Wu Z, et al. Autophagy blockade sensitizes prostate Cancer cells towards Src family kinase inhibitors. Genes Cancer. 2010;1(1):40–9.
Vyas AR, et al. Chemoprevention of prostate cancer by d,l-sulforaphane is augmented by pharmacological inhibition of autophagy. Cancer Res. 2013;73(19):5985–95.
Yun Jung Y, et al. Fangchinoline diminishes STAT3 activation by stimulating oxidative stress and targeting SHP-1 protein in multiple myeloma model. J Adv Res. 2021;35:245-57.
Kim C, et al. β-Caryophyllene oxide inhibits constitutive and inducible STAT3 signaling pathway through induction of the SHP-1 protein tyrosine phosphatase. Mol Carcinog. 2014;53(10):793–806.
Lee JH, et al. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget. 2015;6(8):6386–405.
Lee JH, et al. Capillarisin inhibits constitutive and inducible STAT3 activation through induction of SHP-1 and SHP-2 tyrosine phosphatases. Cancer Lett. 2014;345(1):140–8.
Chen X, et al. Circular RNA circHIPK3 modulates autophagy via MIR124-3p-STAT3-PRKAA/AMPKα signaling in STK11 mutant lung cancer. Autophagy. 2020;16(4):659–71.
Garg M, et al. The pleiotropic role of transcription factor STAT3 in oncogenesis and its targeting through natural products for cancer prevention and therapy. Med Res Rev. 2020;41(3):1291-336.
Mohan CD, et al. Targeting STAT3 signaling pathway in cancer by agents derived from mother nature. Semin Cancer Biol. 2020. https://doi.org/10.1016/j.semcancer.2020.03.016.
Hu F, et al. Docetaxel-mediated autophagy promotes chemoresistance in castration-resistant prostate cancer cells by inhibiting STAT3. Cancer Lett. 2018;416:24–30.
Srinivasan M, et al. HMGB1 in hormone-related cancer: a potential therapeutic target. Horm Cancer. 2014;5(3):127–39.
Tang D, et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene. 2010;29(38):5299–310.
Lei X, et al. HMGB1 release promotes paclitaxel resistance in castration-resistant prostate cancer cells via activating c-Myc expression. Cell Signal. 2020;72:109631.
Zhang YX, et al. HMGB1-mediated autophagy confers resistance to gemcitabine in hormone-independent prostate cancer cells. Oncol Lett. 2017;14(5):6285–90.
Xu S, Weihua Z. Loss of EGFR induced autophagy sensitizes hormone refractory prostate cancer cells to adriamycin. Prostate. 2011;71(11):1216–24.
Boutin B, et al. Androgen deprivation and androgen receptor competition by bicalutamide induce autophagy of hormone-resistant prostate cancer cells and confer resistance to apoptosis. Prostate. 2013;73(10):1090–102.
Wang R, et al. PrLZ, a novel prostate-specific and androgen-responsive gene of the TPD52 family, amplified in chromosome 8q21.1 and overexpressed in human prostate cancer. Cancer Res. 2004;64(5):1589–94.
Wu R, et al. EphA3, induced by PC-1/PrLZ, contributes to the malignant progression of prostate cancer. Oncol Rep. 2014;32(6):2657–65.
Zeng J, et al. PrLZ increases prostate cancer docetaxel resistance by inhibiting LKB1/AMPK-mediated autophagy. Theranostics. 2018;8(1):109–23.
Chen J, et al. Platelet-activating factor receptor-mediated PI3K/AKT activation contributes to the malignant development of esophageal squamous cell carcinoma. Oncogene. 2015;34(40):5114–27.
Lin JZ, et al. FOXM1 contributes to docetaxel resistance in castration-resistant prostate cancer by inducing AMPK/mTOR-mediated autophagy. Cancer Lett. 2020;469:481–9.
Lamoureux F, et al. Blocked autophagy using lysosomotropic agents sensitizes resistant prostate tumor cells to the novel Akt inhibitor AZD5363. Clin Cancer Res. 2013;19(4):833–44.
Zhu Q, et al. Knockdown of CFTR enhances sensitivity of prostate cancer cells to cisplatin via inhibition of autophagy. Neoplasma. 2017;64(5):709–17.
Frame FM, et al. Mechanisms of growth inhibition of primary prostate epithelial cells following gamma irradiation or photodynamic therapy include senescence, necrosis, and autophagy, but not apoptosis. Cancer Med. 2016;5(1):61–73.
Cao C, et al. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006;66(20):10040–7.
Li XR, et al. Knockdown of FBP1 enhances radiosensitivity in prostate cancer cells by activating autophagy. Neoplasma. 2020;67(5):982–91.
Wu Y, et al. Low-frequency ultrasound enhances chemotherapy sensitivity and induces autophagy in PTX-resistant PC-3 cells via the endoplasmic reticulum stress-mediated PI3K/Akt/mTOR signaling pathway. Onco Targets Ther. 2018;11:5621–30.
Qiao Y, et al. Autophagy inhibition by targeting PIKfyve potentiates response to immune checkpoint blockade in prostate cancer. Nat Cancer. 2021;2(9):978–93.
Yao B, et al. PAFR selectively mediates radioresistance and irradiation-induced autophagy suppression in prostate cancer cells. Oncotarget. 2017;8(8):13846–54.
Iacona JR, Lutz CS. miR-146a-5p: expression, regulation, and functions in cancer. Wiley Interdiscip Rev RNA. 2019;10(4):e1533.
Mirzaei S, et al. Regulation of nuclear factor-KappaB (NF-κB) signaling pathway by non-coding RNAs in cancer: inhibiting or promoting carcinogenesis? Cancer Lett. 2021;509:63–80.
Sailo BL, et al. FBXW7 in cancer: what has been unraveled thus far? Cancers (Basel). 2019;11(2):246.
Friedman RC, et al. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105.
Rishabh K, et al. MicroRNAs as modulators of oral tumorigenesis-a focused review. Int J Mol Sci. 2021;22(5):2561.
Abadi AJ, et al. Small in size, but large in action: microRNAs as potential modulators of PTEN in breast and lung cancers. Biomolecules. 2021;11(2):304.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33.
Bayraktar R, Van Roosbroeck K. miR-155 in cancer drug resistance and as target for miRNA-based therapeutics. Cancer Metastasis Rev. 2018;37(1):33–44.
Haneklaus M, et al. miR-223: infection, inflammation and cancer. J Intern Med. 2013;274(3):215–26.
Lv P, et al. miR-373 inhibits autophagy and further promotes apoptosis of cholangiocarcinoma cells by targeting ULK1. Kaohsiung J Med Sci. 2020;36(6):429–40.
Ashrafizadeh M, et al. MicroRNA-mediated autophagy regulation in cancer therapy: the role in chemoresistance/chemosensitivity. Eur J Pharmacol. 2020;892:173660.
John Clotaire DZ, et al. MiR-26b inhibits autophagy by targeting ULK2 in prostate cancer cells. Biochem Biophys Res Commun. 2016;472(1):194–200.
Ding HY, Qian WQ, Xu J. MicroRNA-146b acts as a potential tumor suppressor in human prostate cancer. J buon. 2016;21(2):434–43.
Gao S, et al. MiR-146b inhibits autophagy in prostate cancer by targeting the PTEN/Akt/mTOR signaling pathway. Aging (Albany NY). 2018;10(8):2113–21.
Lee JH, et al. Casticin-induced inhibition of cell growth and survival are mediated through the dual modulation of Akt/mTOR signaling cascade. Cancers (Basel). 2019;11(2):254.
Mohan CD, et al. Trisubstituted-Imidazoles induce apoptosis in human breast Cancer cells by targeting the oncogenic PI3K/Akt/mTOR signaling pathway. Plos One. 2016;11(4):e0153155.
Braglia L, et al. Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: still a potential druggable target? Biochim Biophys Acta Mol Cell Res. 1867;2020(9):118731.
Deng R, et al. BAP1 suppresses prostate cancer progression by deubiquitinating and stabilizing PTEN. Mol Oncol. 2021;15(1):279–98.
Zhang X, et al. IMP3 accelerates the progression of prostate cancer through inhibiting PTEN expression in a SMURF1-dependent way. J Exp Clin Cancer Res. 2020;39(1):190.
Singh SS, et al. Targeting the PI3K/Akt signaling pathway in gastric carcinoma: a reality for personalized medicine? World J Gastroenterol. 2015;21(43):12261–73.
Siveen KS, et al. Y-tocotrienol inhibits angiogenesis-dependent growth of human hepatocellular carcinoma through abrogation of AKT/mTOR pathway in an orthotopic mouse model. Oncotarget. 2014;5(7):1897–911.
Ong PS, et al. Judicious toggling of mTOR activity to combat insulin resistance and Cancer: current evidence and perspectives. Front Pharmacol. 2016;7:395.
Lee GH, D’Arcangelo G. New insights into reelin-mediated signaling pathways. Front Cell Neurosci. 2016;10:122.
Rui X, et al. MicroRNA-381 suppresses proliferation and invasion of prostate cancer cells through downregulation of the androgen receptor. Oncol Lett. 2019;18(2):2066–72.
Liao W, Zhang Y. MicroRNA-381 facilitates autophagy and apoptosis in prostate cancer cells via inhibiting the RELN-mediated PI3K/AKT/mTOR signaling pathway. Life Sci. 2020;254:117672.
Li J, et al. Downregulation of N (6)-methyladenosine binding YTHDF2 protein mediated by miR-493-3p suppresses prostate cancer by elevating N (6)-methyladenosine levels. Oncotarget. 2018;9(3):3752–64.
Deng J, et al. miR-493 promotes prostate cancer cells proliferation by targeting PHLPP2 and activating akt signaling pathway. Clin Lab. 2019;65(3).
Deng J, et al. MiR-493 induces cytotoxic autophagy in prostate cancer cells through regulation on PHLPP2. Curr Pharm Biotechnol. 2020;21(14):1451–6.
Nam RK, et al. Mir-139 regulates autophagy in prostate cancer cells through Beclin-1 and mTOR signaling proteins. Anticancer Res. 2020;40(12):6649–63.
Du S, et al. Circ-ZNF609 accelerates the Radioresistance of prostate Cancer cells by promoting the glycolytic metabolism through miR-501-3p/HK2 Axis. Cancer Manag Res. 2020;12:7487–99.
Fan L, et al. Histone demethylase JMJD1A promotes expression of DNA repair factors and radio-resistance of prostate cancer cells. Cell Death Dis. 2020;11(4):214.
Wang W, Liu J, Wu Q. MiR-205 suppresses autophagy and enhances radiosensitivity of prostate cancer cells by targeting TP53INP1. Eur Rev Med Pharmacol Sci. 2016;20(1):92–100.
Liao H, et al. microRNA-32 induces radioresistance by targeting DAB2IP and regulating autophagy in prostate cancer cells. Oncol Lett. 2015;10(4):2055–62.
Ling Z, et al. miR-361-5p modulates metabolism and autophagy via the Sp1-mediated regulation of PKM2 in prostate cancer. Oncol Rep. 2017;38(3):1621–8.
Xu CG, et al. MiR-30a and miR-205 are downregulated in hypoxia and modulate radiosensitivity of prostate cancer cells by inhibiting autophagy via TP53INP1. Eur Rev Med Pharmacol Sci. 2016;20(8):1501–8.
Hu B, Jin X, Wang J. MicroRNA-212 targets mitogen-activated protein kinase 1 to inhibit Proliferation and invasion of prostate Cancer cells. Oncol Res. 2018;26(7):1093–102.
Qu HW, et al. MicroRNA-212 participates in the development of prostate cancer by upregulating BMI1 via NF-κB pathway. Eur Rev Med Pharmacol Sci. 2018;22(11):3348–56.
Xu C, et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol. 2020;22(10):1170–9.
Jang KH, Hwang Y, Kim E. PARP1 impedes SIRT1-mediated autophagy during degeneration of the retinal pigment epithelium under oxidative stress. Mol Cells. 2020;43(7):632–44.
Ramalinga M, et al. MicroRNA-212 negatively regulates starvation induced autophagy in prostate cancer cells by inhibiting SIRT1 and is a modulator of angiogenesis and cellular senescence. Oncotarget. 2015;6(33):34446–57.
Zou J, et al. VEGF-A promotes angiogenesis after acute myocardial infarction through increasing ROS production and enhancing ER stress-mediated autophagy. J Cell Physiol. 2019;234(10):17690–703.
Deegan S, et al. A close connection between the PERK and IRE arms of the UPR and the transcriptional regulation of autophagy. Biochem Biophys Res Commun. 2015;456(1):305–11.
Sohn EJ. MicroRNA 200c-3p regulates autophagy via upregulation of endoplasmic reticulum stress in PC-3 cells. Cancer Cell Int. 2018;18:2.
Wu M, et al. The potential of long noncoding RNAs for precision medicine in human cancer. Cancer Lett. 2020;501:12-9.
Lim YWS, et al. The double-edged sword of H19 lncRNA: insights into cancer therapy. Cancer Lett. 2020;500:253-62.
Mishra S, et al. Long non-coding RNAs are emerging targets of phytochemicals for cancer and other chronic diseases. Cell Mol Life Sci. 2019;76(10):1947–66.
Chen X, Tang FR, Arfuso F, Cai WQ, Ma Z, Yang J, Sethi G. The emerging role of long non-coding RNAs in the metastasis of hepatocellular carcinoma. Biomolecules. 2020;10(1):66.
Dinger ME, et al. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. Plos Comput Biol. 2008;4(11):e1000176.
Ma Z, et al. The expanding roles of long non-coding RNAs in the regulation of cancer stem cells. Int J Biochem Cell Biol. 2019;108:17–20.
Ong MS, et al. ‘Lnc’-ing Wnt in female reproductive cancers: therapeutic potential of long non-coding RNAs in Wnt signalling. Br J Pharmacol. 2017;174(24):4684–700.
Pandya G, et al. The implication of long non-coding RNAs in the diagnosis, pathogenesis and drug resistance of pancreatic ductal adenocarcinoma and their possible therapeutic potential. Biochim Biophys Acta Rev Cancer. 1874;2020(2):188423.
Kansara S, et al. Mechanistic involvement of long non-coding RNAs in oncotherapeutics resistance in triple-negative breast cancer. Cells. 2020;9(6):1511.
Butler AA, Webb WM, Lubin FD. Regulatory RNAs and control of epigenetic mechanisms: expectations for cognition and cognitive dysfunction. Epigenomics. 2016;8(1):135–51.
Wapinski O, Chang HY. Long noncoding RNAs and human disease. Trends Cell Biol. 2011;21(6):354–61.
Clark MB, Mattick JS. Long noncoding RNAs in cell biology. Semin Cell Dev Biol. 2011;22:366-76. Elsevier.
Shen C, et al. Long non-coding RNAs: emerging regulators for chemo/immunotherapy resistance in cancer stem cells. Cancer Lett. 2021;500:244–52.
Pucci P, et al. LncRNA HORAS5 promotes taxane resistance in castration-resistant prostate cancer via a BCL2A1-dependent mechanism. Epigenomics. 2020;12(13):1123–38.
Cui R, et al. LncRNA AC245100.4 binds HSP90 to promote the proliferation of prostate cancer. Epigenomics. 2020;12(15):1257–71.
Zhao Z, Liang S, Sun F. LncRNA DLX6-AS1 promotes malignant phenotype and lymph node metastasis in prostate cancer by inducing LARGE methylation. Front Oncol. 2020;10:1172.
Meng XF, Liu AD, Li SL. SNHG1 promotes proliferation, invasion and EMT of prostate cancer cells through miR-195-5p. Eur Rev Med Pharmacol Sci. 2020;24(19):9880–8.
Xie M, Zhang Z, Cui Y. Long noncoding RNA SNHG1 contributes to the promotion of prostate cancer cells through regulating miR-377-3p/AKT2 Axis. Cancer Biother Radiopharm. 2020;35(2):109–19.
Chen J, et al. Long non-coding RNA SNHG1 regulates the Wnt/β-catenin and PI3K/AKT/mTOR signaling pathways via EZH2 to affect the proliferation, apoptosis, and autophagy of prostate cancer cell. Front Oncol. 2020;10:552907.
Fan L, Li H, Wang W. Long non-coding RNA PRRT3-AS1 silencing inhibits prostate cancer cell proliferation and promotes apoptosis and autophagy. Exp Physiol. 2020;105(5):793–808.
Wang C, et al. Interactome analysis reveals that lncRNA HULC promotes aerobic glycolysis through LDHA and PKM2. Nat Commun. 2020;11(1):3162.
Zheng P, et al. High lncRNA HULC expression is associated with poor prognosis and promotes tumor progression by regulating epithelial-mesenchymal transition in prostate cancer. Arch Med Sci. 2018;14(3):679–86.
Chen C, et al. LncRNA HULC mediates radioresistance via autophagy in prostate cancer cells. Braz J Med Biol Res. 2018;51(6):e7080.
Meng S, et al. CircRNA: functions and properties of a novel potential biomarker for cancer. Mol Cancer. 2017;16(1):1–8.
Verduci L, et al. The circRNA–microRNA code: emerging implications for cancer diagnosis and treatment. Mol Oncol. 2019;13(4):669–80.
Yin Y, et al. Emerging roles of circRNA in formation and progression of cancer. J Cancer. 2019;10(21):5015.
Suzuki H, Tsukahara T. A view of pre-mRNA splicing from RNase R resistant RNAs. Int J Mol Sci. 2014;15(6):9331–42.
Zheng Y, et al. Circ_KATNAL1 regulates prostate cancer cell growth and invasiveness through the miR-145-3p/WISP1 pathway. Biochem Cell Biol. 2020;98(3):396–404.
Li H, et al. Circ_0062020 knockdown strengthens the radiosensitivity of prostate cancer cells. Cancer Manag Res. 2020;12:11701–12.
Cai F, et al. Knockdown of Circ_CCNB2 sensitizes prostate cancer to radiation through repressing autophagy by the miR-30b-5p/KIF18A Axis. Cancer Biother Radiopharm. 2020. https://doi.org/10.1089/cbr.2019.3538.
Gu H, et al. Hypoxia-responsive miR-124 and miR-144 reduce hypoxia-induced autophagy and enhance radiosensitivity of prostate cancer cells via suppressing PIM1. Cancer Med. 2016;5(6):1174–82.
Guo YJ, Liu JX, Guan YW. Hypoxia induced upregulation of miR-301a/b contributes to increased cell autophagy and viability of prostate cancer cells by targeting NDRG2. Eur Rev Med Pharmacol Sci. 2016;20(1):101–8.
Ma Y, et al. Biphasic regulation of autophagy by miR-96 in prostate cancer cells under hypoxia. Oncotarget. 2014;5(19):9169–82.
Pennati M, et al. miR-205 impairs the autophagic flux and enhances cisplatin cytotoxicity in castration-resistant prostate cancer cells. Biochem Pharmacol. 2014;87(4):579–97.
Liao H, et al. Methylation-induced silencing of miR-34a enhances chemoresistance by directly upregulating ATG4B-induced autophagy through AMPK/mTOR pathway in prostate cancer. Oncol Rep. 2016;35(1):64–72.
Wang X, et al. Dysregulation of long non-coding RNA SNHG12 alters the viability, apoptosis, and autophagy of prostate cancer cells by regulating miR-195/CCNE1 axis. Int J Clin Exp Pathol. 2019;12(4):1272–83.
Coffey K, et al. The lysine demethylase, KDM4B, is a key molecule in androgen receptor signalling and turnover. Nucleic Acids Res. 2013;41(8):4433–46.
Yang J, Harris AL, Davidoff AM. Hypoxia and hormone-mediated pathways converge at the histone demethylase KDM4B in cancer. Int J Mol Sci. 2018;19(1):240.
Sha J, et al. Upregulated KDM4B promotes prostate cancer cell proliferation by activating autophagy. J Cell Physiol. 2020;235(3):2129–38.
Ashrafizadeh M, et al. Resveratrol targeting the Wnt signaling pathway: a focus on therapeutic activities. J Cell Physiol. 2020;235(5):4135–45.
Lin R, et al. Regulation of autophagy of prostate cancer cells by β-catenin signaling. Cell Physiol Biochem. 2015;35(3):926–32.
Brunet A, et al. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol. 2001;21(3):952–65.
Dehner M, et al. Wnt signaling inhibits Forkhead box O3a-induced transcription and apoptosis through up-regulation of serum- and glucocorticoid-inducible kinase 1. J Biol Chem. 2008;283(28):19201–10.
Liu W, et al. SGK1 inhibition induces autophagy-dependent apoptosis via the mTOR-Foxo3a pathway. Br J Cancer. 2017;117(8):1139–53.
Verheij M, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380(6569):75–9.
Zhao S, et al. Propranolol induced apoptosis and autophagy via the ROS/JNK signaling pathway in human ovarian Cancer. J Cancer. 2020;11(20):5900–10.
Yang J, et al. Chlorpyrifos induces apoptosis and autophagy in common carp lymphocytes by influencing the TCR γ-dependent PI3K/AKT/JNK pathway. Fish Shellfish Immunol. 2020;99:587–93.
Zhu X, et al. Autophagy activated by the c-Jun N-terminal kinase-mediated pathway protects human prostate cancer PC3 cells from celecoxib-induced apoptosis. Exp Ther Med. 2017;13(5):2348–54.
Niki T, et al. DJBP: a novel DJ-1-binding protein, negatively regulates the androgen receptor by recruiting histone deacetylase complex, and DJ-1 antagonizes this inhibition by abrogation of this Complex1 1 Ministry of Education, science, sport, culture and Technology of Japan. 2 2 the nucleotide sequence reported in this paper has been submitted to the DDBJ/GenBank/EBI data Bank with accession number AB073862. Mol Cancer Res. 2003;1(4):247–61.
Takahashi K, et al. DJ-1 positively regulates the androgen receptor by impairing the binding of PIASxα to the receptor. J Biol Chem. 2001;276(40):37556–63.
Qin X, et al. DJ-1 inhibits autophagy activity of prostate cancer cells by repressing JNK-Bcl2-Beclin1 signaling. Cell Biol Int. 2020;44(4):937–46.
Li H, et al. SIRT7 promotes thyroid tumorigenesis through phosphorylation and activation of Akt and p70S6K1 via DBC1/SIRT1 axis. Oncogene. 2019;38(3):345–59.
Yu H, et al. Overexpression of sirt7 exhibits oncogenic property and serves as a prognostic factor in colorectal cancer. Clin Cancer Res. 2014;20(13):3434–45.
Haider R, et al. Sirtuin 7: a new marker of aggressiveness in prostate cancer. Oncotarget. 2017;8(44):77309–16.
Tang X, et al. SIRT7 antagonizes TGF-β signaling and inhibits breast cancer metastasis. Nat Commun. 2017;8(1):318.
Niture S, et al. TNFAIP8 promotes prostate cancer cell survival by inducing autophagy. Oncotarget. 2018;9(42):26884–99.
Kang HY, et al. Differential modulation of androgen receptor-mediated transactivation by Smad3 and tumor suppressor Smad4. J Biol Chem. 2002;277(46):43749–56.
Tektemur A, et al. TRPM2 mediates distruption of autophagy machinery and correlates with the grade level in prostate cancer. J Cancer Res Clin Oncol. 2019;145(5):1297–311.
Sethi G, et al. Pro-apoptotic and anti-cancer properties of diosgenin: a comprehensive and critical review. Nutrients. 2018;10(5):645.
Zhang J, et al. Diosgenin inhibits the expression of NEDD4 in prostate cancer cells. Am J Transl Res. 2019;11(6):3461–71.
Sun GC, Jan CR, Liang WZ. Exploring the impact of a naturally occurring sapogenin diosgenin on underlying mechanisms of ca (2+) movement and cytotoxicity in human prostate cancer cells. Environ Toxicol. 2020;35(3):395–403.
Nie C, et al. Diosgenin-induced autophagy and apoptosis in a human prostate cancer cell line. Mol Med Rep. 2016;14(5):4349–59.
Ma X, et al. Inhibition of autophagy improves the efficacy of Abiraterone for the treatment of prostate Cancer. Cancer Biother Radiopharm. 2019;34(3):181–8.
Riaz A, et al. Eriocalyxin B induces apoptosis in human triple negative breast cancer cells via inhibiting STAT3 activation and mitochondrial dysfunction. Pak J Pharm Sci. 2019;32(6(Supplementary)):2843–8.
Lu YM, et al. Eriocalyxin B blocks human SW1116 colon cancer cell proliferation, migration, invasion, cell cycle progression and angiogenesis via the JAK2/STAT3 signaling pathway. Mol Med Rep. 2016;13(3):2235–40.
Sun HD, et al. Diterpenoids from Isodon eriocalyx var. laxiflora. Phytochemistry. 1995;38(6):1451–5.
Yu Z, Chen Y, Liang C. Eriocalyxin B induces apoptosis and autophagy involving Akt/mammalian target of rapamycin (mTOR) pathway in prostate cancer cells. Med Sci Monit. 2019;25:8534–43.
Hahm ER, Singh SV. Cytoprotective autophagy induction by withaferin a in prostate cancer cells involves GABARAPL1. Mol Carcinog. 2020;59(10):1105–15.
Waldvogel-Abramowski S, et al. Physiology of iron metabolism. Transfus Med Hemother. 2014;41(3):213–21.
Kühn LC. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics. 2015;7(2):232–43.
Zhang C, Zhang F. Iron homeostasis and tumorigenesis: molecular mechanisms and therapeutic opportunities. Protein Cell. 2015;6(2):88–100.
Ba Q, et al. Iron deprivation suppresses hepatocellular carcinoma growth in experimental studies. Clin Cancer Res. 2011;17(24):7625–33.
Pullarkat V, et al. Iron chelators induce autophagic cell death in multiple myeloma cells. Leuk Res. 2014;38(8):988–96.
Yang C, et al. Curcumin induces apoptosis and protective autophagy in castration-resistant prostate cancer cells through iron chelation. Drug Des Devel Ther. 2017;11:431–9.
Chen X, Zhang T, Zhang Y. Endoplasmic reticulum stress and autophagy in HIV-1-associated neurocognitive disorders. J Neurovirol. 2020:1–10. https://doi.org/10.1007/s13365-020-00906-4.
Ashrafizadeh M, et al. Therapeutic effects of kaempferol affecting autophagy and endoplasmic reticulum stress. Phytother Res. 2020;34(5):911–23.
Fontana F, et al. δ-Tocotrienol induces apoptosis, involving endoplasmic reticulum stress and autophagy, and paraptosis in prostate cancer cells. Cell Prolif. 2019;52(3):e12576.
Kirtonia A, Sethi G, Garg M. The multifaceted role of reactive oxygen species in tumorigenesis. Cell Mol Life Sci. 2020;77(22):4459–83.
Tochhawng L, et al. Gelsolin-cu/ZnSOD interaction alters intracellular reactive oxygen species levels to promote cancer cell invasion. Oncotarget. 2016;7(33):52832–48.
Kim C, et al. Formononetin-induced oxidative stress abrogates the activation of STAT3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett. 2018;431:123–41.
Subramaniam A, et al. An anthraquinone derivative, emodin sensitizes hepatocellular carcinoma cells to TRAIL induced apoptosis through the induction of death receptors and downregulation of cell survival proteins. Apoptosis. 2013;18(10):1175–87.
Zhang ZH, et al. ROS-mediated genotoxic stress is involved in NaAsO (2)-induced cell cycle arrest, stemness enhancement and chemoresistance of prostate cancer cells in a p53-independent manner. Ecotoxicol Environ Saf. 2021;208:111436.
Lee JH, et al. Ophiopogonin D, a steroidal glycoside abrogates STAT3 signaling cascade and exhibits anti-cancer activity by causing GSH/GSSG imbalance in lung carcinoma. Cancers (Basel). 2018;10(11):427.
Kim SM, et al. 6-Shogaol exerts anti-proliferative and pro-apoptotic effects through the modulation of STAT3 and MAPKs signaling pathways. Mol Carcinog. 2015;54(10):1132–46.
Tian Y, et al. Autophagy induced by ROS aggravates testis oxidative damage in diabetes via breaking the feedforward loop linking p62 and Nrf2. Oxidative Med Cell Longev. 2020;2020:7156579.
Kim KY, et al. Inhibition of autophagy promotes salinomycin-induced apoptosis via reactive oxygen species-Mediated PI3K/AKT/mTOR and ERK/p38 MAPK-dependent signaling in human prostate cancer cells. Int J Mol Sci. 2017;18(5):1088.
Erkisa M, et al. A promising therapeutic combination for metastatic prostate cancer: chloroquine as autophagy inhibitor and palladium (II) barbiturate complex. Biochimie. 2020;175:159–72.
Yu Z, et al. Germacrone induces apoptosis as well as protective autophagy in human prostate cancer cells. Cancer Manag Res. 2020;12:4009–16.
Shanmugam MK, et al. Celastrol attenuates the invasion and migration and augments the anticancer effects of Bortezomib in a xenograft mouse model of multiple myeloma. Front Pharmacol. 2018;9:365.
Kuchta K, et al. Celastrol, an active constituent of the TCM plant Tripterygium wilfordii Hook.F., inhibits prostate cancer bone metastasis. Prostate Cancer Prostatic Dis. 2017;20(2):156–64.
Sanna V, et al. Nanoencapsulation of natural triterpenoid celastrol for prostate cancer treatment. Int J Nanomedicine. 2015;10:6835–46.
Wolfram J, et al. Evaluation of anticancer activity of celastrol liposomes in prostate cancer cells. J Microencapsul. 2014;31(5):501–7.
Kashyap D, et al. Natural product-based nanoformulations for cancer therapy: opportunities and challenges. Semin Cancer Biol. 2021;69:5–23.
Guo J, et al. Downregulation of miR-17-92a cluster promotes autophagy induction in response to celastrol treatment in prostate cancer cells. Biochem Biophys Res Commun. 2016;478(2):804–10.
Guo J, et al. Celastrol induces autophagy by targeting AR/miR-101 in prostate cancer cells. Plos One. 2015;10(10):e0140745.
Mortezavi A, et al. Inhibition of autophagy significantly increases the antitumor effect of Abiraterone in prostate cancer. World J Urol. 2019;37(2):351–8.
Eberli D, et al. Apalutamide in combination with autophagy inhibitors improves treatment effects in prostate cancer cells. Urol Oncol. 2020;38(8):683.e19–26.
Yang A, et al. Huaier suppresses proliferative and metastatic potential of prostate cancer PC3 cells via downregulation of Lamin B1 and induction of autophagy. Oncol Rep. 2018;39(6):3055–63.
Wang B, et al. Antitumor effect of sunitinib in human prostate cancer cells functions via autophagy. Exp Ther Med. 2017;13(4):1285–94.
Etani T, et al. NCL1, a highly selective lysine-specific demethylase 1 inhibitor, suppresses castration-resistant prostate cancer growth via regulation of apoptosis and autophagy. J Clin Med. 2019;8(4):442.
Bellisai C, et al. Reverse transcriptase inhibitors promote the remodelling of nuclear architecture and induce autophagy in prostate cancer cells. Cancer Lett. 2020;478:133–45.
Tai S, et al. Combination of Rad001 (everolimus) and propachlor synergistically induces apoptosis through enhanced autophagy in prostate cancer cells. Mol Cancer Ther. 2012;11(6):1320–31.
Chiu HW, et al. Monascuspiloin enhances the radiation sensitivity of human prostate cancer cells by stimulating endoplasmic reticulum stress and inducing autophagy. Plos One. 2012;7(7):e40462.
Peng X, et al. Inhibition of proliferation and induction of autophagy by atorvastatin in PC3 prostate cancer cells correlate with downregulation of Bcl2 and upregulation of miR-182 and p21. Plos One. 2013;8(8):e70442.
Gafar AA, et al. Lithocholic acid induces endoplasmic reticulum stress, autophagy and mitochondrial dysfunction in human prostate cancer cells. PeerJ. 2016;4:e2445.
Lin HY, et al. The anti-proliferative activity of secondary metabolite from the marine streptomyces sp. against prostate cancer cells. Life (Basel). 2021;11(12):1414.
Li X, et al. Oridonin up-regulates expression of P21 and induces autophagy and apoptosis in human prostate cancer cells. Int J Biol Sci. 2012;8(6):901–12.
Xiao D, et al. Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J Biol Chem. 2010;285(34):26558–69.
Akhtar N, et al. The pentacyclic triterpenoid, plectranthoic acid, a novel activator of AMPK induces apoptotic death in prostate cancer cells. Oncotarget. 2016;7(4):3819–31.
Teiten MH, et al. Anti-proliferative potential of curcumin in androgen-dependent prostate cancer cells occurs through modulation of the wingless signaling pathway. Int J Oncol. 2011;38(3):603–11.
Lin JF, et al. Zoledronic acid induces autophagic cell death in human prostate cancer cells. J Urol. 2011;185(4):1490–6.
Wang M, et al. A small molecule inhibitor of isoprenylcysteine carboxymethyltransferase induces autophagic cell death in PC3 prostate cancer cells. J Biol Chem. 2008;283(27):18678–84.
Jiang H, et al. Marchantin M: a novel inhibitor of proteasome induces autophagic cell death in prostate cancer cells. Cell Death Dis. 2013;4(8):e761.
Gioti K, et al. Glycyrrhiza glabra-enhanced extract and Adriamycin Antiproliferative effect on PC-3 prostate Cancer cells. Nutr Cancer. 2020;72(2):320–32.
Luty M, et al. Fenofibrate augments the sensitivity of drug-resistant prostate cancer cells to docetaxel. Cancers (Basel). 2019;11(1):77.
Lee MS, et al. Anti-prostate cancer potential of gossypetin via inducing apoptotic and autophagic cell death. Mol Carcinog. 2017;56(12):2578–92.
Inamura SO, et al. Low-dose docetaxel enhanced the anticancer effect of Temsirolimus by overcoming autophagy in prostate cancer cells. Anticancer Res. 2019;39(10):5417–25.
Luo C, et al. Hydroxytyrosol promotes superoxide production and defects in autophagy leading to anti-proliferation and apoptosis on human prostate cancer cells. Curr Cancer Drug Targets. 2013;13(6):625–39.
Lin HJ, et al. Cytolethal distending toxin enhances radiosensitivity in prostate cancer cells by regulating autophagy. Front Cell Infect Microbiol. 2017;7:223.
Chang L, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014;5(10):e1437.
Brohée L, et al. Propranolol sensitizes prostate cancer cells to glucose metabolism inhibition and prevents cancer progression. Sci Rep. 2018;8(1):7050.
Bosseboeuf A, et al. A potential antineoplastic peptide of human prostate cancer cells derived from the lesser spotted dogfish (Scyliorhinus canicula L.). Mar Drugs. 2019;17(10):585.
Hsieh CL, et al. A novel Salicylanilide derivative induces autophagy cell death in castration-resistant prostate cancer via ER stress-activated PERK signaling pathway. Mol Cancer Ther. 2020;19(1):101–11.
Cheng K, et al. α-Viniferin activates autophagic apoptosis and cell death by reducing glucocorticoid receptor expression in castration-resistant prostate cancer cells. Med Oncol. 2018;35(7):105.
Selvaraj S, et al. Resveratrol activates autophagic cell death in prostate cancer cells via downregulation of STIM1 and the mTOR pathway. Mol Carcinog. 2016;55(5):818–31.
Di Giacomo V, et al. MRJF4, a novel histone deacetylase inhibitor, induces p21 mediated autophagy in PC3 prostate cancer cells. Cell Mol Biol (Noisy-le-grand). 2015;61(3):17–23.
Wang Y, et al. Hispidulin inhibits proliferation, migration, and invasion by promoting autophagy via regulation of PPARγ activation in prostate cancer cells and xenograft models. Biosci Biotechnol Biochem. 2021;85(4):786–97.
Li S, et al. Potent antitumour of the mTORC1/2 dual inhibitor AZD2014 in docetaxel-sensitive and docetaxel-resistant castration-resistant prostate cancer cells. J Cell Mol Med. 2021;25(5):2436–49.
Xu H, et al. Enoxacin exerts anti-tumor effects against prostate Cancer through inducing apoptosis. Technol Cancer Res Treat. 2021;20:1533033821995284.
Nazim UM, Yin H, Park SY. Neferine treatment enhances the TRAIL-induced apoptosis of human prostate cancer cells via autophagic flux and the JNK pathway. Int J Oncol. 2020;56(5):1152–61.
Gong C, et al. Co-delivery of autophagy inhibitor ATG7 siRNA and docetaxel for breast cancer treatment. J Control Release. 2017;266:272–86.
Wang Q, et al. Induction of autophagy-dependent apoptosis by the survivin suppressant YM155 in prostate cancer cells. Cancer Lett. 2011;302(1):29–36.
Kolluru V, et al. Induction of endoplasmic reticulum stress might be responsible for defective autophagy in cadmium-induced prostate carcinogenesis. Toxicol Appl Pharmacol. 2019;373:62–8.
Kim RH, et al. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res. 2009;69(2):700–8.
Mouratidis PX, et al. Differential role of apoptosis and autophagy associated with anticancer effect of lupulone (hop β-acid) derivatives on prostate cancer cells. Anti Cancer Agents Med Chem. 2014;14(8):1169–78.
Kumano M, et al. Cotargeting stress-activated Hsp27 and autophagy as a combinatorial strategy to amplify endoplasmic reticular stress in prostate cancer. Mol Cancer Ther. 2012;11(8):1661–71.
Morell C, et al. Up-regulated expression of LAMP2 and autophagy activity during neuroendocrine differentiation of prostate Cancer LNCaP cells. Plos One. 2016;11(9):e0162977.
Kim RH, Bold RJ, Kung HJ. ADI, autophagy and apoptosis: metabolic stress as a therapeutic option for prostate cancer. Autophagy. 2009;5(4):567–8.
Zhang J, et al. shRNA-armed conditionally replicative adenoviruses: a promising approach for cancer therapy. Oncotarget. 2016;7(20):29824.
Ofer P, et al. Both IGF1R and INSR knockdown exert antitumorigenic effects in prostate cancer in vitro and in vivo. Mol Endocrinol. 2015;29(12):1694–707.
Ashrafizadeh M, et al. Polychemotherapy with curcumin and doxorubicin via biological Nanoplatforms: enhancing antitumor activity. Pharmaceutics. 2020;12(11):1084.
Ashrafizade M, et al. Biomedical application of Chitosan-based nanoscale delivery systems: potential usefulness in siRNA delivery for cancer therapy. Carbohydr Polym. 2021;260:117809.
Delfi M, et al. Self-assembled peptide and protein nanostructures for anti-cancer therapy: targeted delivery, stimuli-responsive devices and immunotherapy. Nano Today. 2021;38:101119.
Chen J, et al. Mesoporous silica nanoparticles combined with AKR1C3 siRNA inhibited the growth of castration-resistant prostate cancer by suppressing androgen synthesis in vitro and in vivo. Biochem Biophys Res Commun. 2021;540:83–9.
Dai L, et al. PSMA-targeted melanin-like nanoparticles as a multifunctional nanoplatform for prostate cancer theranostics. J Mater Chem B. 2021;9(4):1151–61.
Liu N, et al. Improving radio-chemotherapy efficacy of prostate cancer by co-deliverying docetaxel and dbait with biodegradable nanoparticles. Artif Cells Nanomed Biotechnol. 2020;48(1):305–14.
Tanaudommongkon I, et al. Curcumin nanoparticles and their cytotoxicity in docetaxel-resistant castration-resistant Prostate cancer cells. Biomedicines. 2020;8(8):253.
Xiao F, et al. The targeted inhibition of prostate cancer by iron-based nanoparticles based on bioinformatics. J Biomater Appl. 2020;36:885328220975249.
Chen Y, et al. Anti prostate cancer therapy: aptamer-functionalized, curcumin and cabazitaxel co-delivered, tumor targeted lipid-polymer hybrid nanoparticles. Biomed Pharmacother. 2020;127:110181.
Chen Y, et al. Silver nanoparticles regulate autophagy through lysosome injury and cell hypoxia in prostate cancer cells. J Biochem Mol Toxicol. 2020;34(5):e22474.
Becker AL, et al. Redox-active polymer microcapsules for the delivery of a survivin-specific siRNA in prostate cancer cells. ACS Nano. 2011;5(2):1335–44.
Gaur S, et al. Chitosan nanoparticle-mediated delivery of miRNA-34a decreases prostate tumor growth in the bone and its expression induces non-canonical autophagy. Oncotarget. 2015;6(30):29161–77.
Dong W, et al. Carbon Nanospheres exert antitumor effects associated with downregulation of 4E-BP1 expression on prostate Cancer. Int J Nanomedicine. 2020;15:5545–59.
Freytag SO, et al. Prospective randomized phase 2 trial of intensity modulated radiation therapy with or without oncolytic adenovirus-mediated cytotoxic gene therapy in intermediate-risk prostate cancer. Int J Radiat Oncol Biol Phys. 2014;89(2):268–76.
Small EJ, et al. A phase I trial of intravenous CG7870, a replication-selective, prostate-specific antigen-targeted oncolytic adenovirus, for the treatment of hormone-refractory, metastatic prostate cancer. Mol Ther. 2006;14(1):107–17.
Botta G, et al. Inhibition of autophagy enhances the effects of E1A-defective oncolytic adenovirus dl922-947 against glioma cells in vitro and in vivo. Hum Gene Ther. 2012;23(6):623–34.
Radhakrishnan S, et al. Efficacy of oncolytic mutants targeting pRb and p53 pathways is synergistically enhanced when combined with cytotoxic drugs in prostate cancer cells and tumor xenografts. Hum Gene Ther. 2010;21(10):1311–25.
Raki M, et al. Oncolytic adenovirus Ad5/3-delta24 and chemotherapy for treatment of orthotopic ovarian cancer. Gynecol Oncol. 2008;108(1):166–72.
Siurala M, et al. Oncolytic adenovirus and doxorubicin-based chemotherapy results in synergistic antitumor activity against soft-tissue sarcoma. Int J Cancer. 2015;136(4):945–54.
Aguirre-Hernández C, et al. Sensitisation to mitoxantrone-induced apoptosis by the oncolytic adenovirus ad∆∆ through Bcl-2-dependent attenuation of autophagy. Oncogenesis. 2018;7(1):6.
Pradeepkiran JA, Reddy PH. Defective mitophagy in alzheimer’s disease. Ageing Res Rev. 2020;64:101191.
Evans CS, Holzbaur EL. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. Elife. 2020;9:e50260.
Liu L, et al. Mitophagy and its contribution to metabolic and aging-associated disorders. Antioxid Redox Signal. 2020;32(12):906–27.
Liu YJ, et al. Mitochondrial fission and fusion: a dynamic role in aging and potential target for age-related disease. Mech Ageing Dev. 2020;186:111212.
Cai Q, Jeong YY. Mitophagy in Alzheimer’s disease and other age-related neurodegenerative diseases. Cells. 2020;9(1):150.
Panigrahi DP, et al. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin Cancer Biol. 2020;66:45–58.
Deng R, et al. MAPK1/3 kinase-dependent ULK1 degradation attenuates mitophagy and promotes breast cancer bone metastasis. Autophagy. 2020:1–19. https://doi.org/10.1080/15548627.2020.1850609.
Han J, et al. Abiraterone and MDV3100 inhibits the proliferation and promotes the apoptosis of prostate cancer cells through mitophagy. Cancer Cell Int. 2019;19:332.
Vykoukal J, et al. Caveolin-1-mediated sphingolipid oncometabolism underlies a metabolic vulnerability of prostate cancer. Nat Commun. 2020;11(1):4279.
Nollet EA, et al. Androgen receptor-induced integrin α6β1 and Bnip3 promote survival and resistance to PI3K inhibitors in castration-resistant prostate cancer. Oncogene. 2020;39(31):5390–404.
Liu YQ, et al. Retigeric acid B-induced mitophagy by oxidative stress attenuates cell death against prostate cancer cells in vitro. Acta Pharmacol Sin. 2013;34(9):1183–91.
Singh R, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–5.
Schulze RJ, Sathyanarayan A, Mashek DG. Breaking fat: the regulation and mechanisms of lipophagy. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(10):1178–87.
Panda PK, et al. Deacetylation of LAMP1 drives lipophagy-dependent generation of free fatty acids by Abrus agglutinin to promote senescence in prostate cancer. J Cell Physiol. 2020;235(3):2776–91.
Kaini RR, et al. Autophagy regulates lipolysis and cell survival through lipid droplet degradation in androgen-sensitive prostate cancer cells. Prostate. 2012;72(13):1412–22.
Ahdoot M, et al. MRI-targeted, systematic, and combined biopsy for prostate Cancer diagnosis. N Engl J Med. 2020;382(10):917–28.
Kasivisvanathan V, et al. MRI-targeted or standard biopsy for prostate-Cancer diagnosis. N Engl J Med. 2018;378(19):1767–77.
Cheng Y, et al. Autophagy-related genes are potential diagnostic and prognostic biomarkers in prostate cancer. Transl Androl Urol. 2020;9(6):2616–28.
Hu D, et al. Development of an autophagy-related gene expression signature for prognosis prediction in prostate cancer patients. J Transl Med. 2020;18(1):160.
Jiang X, et al. Autophagy defects suggested by low levels of autophagy activator MAP 1S and high levels of autophagy inhibitor LRPPRC predict poor prognosis of prostate cancer patients. Mol Carcinog. 2015;54(10):1194–204.
Jiang X, et al. Elevated levels of mitochondrion-associated autophagy inhibitor LRPPRC are associated with poor prognosis in patients with prostate cancer. Cancer. 2014;120(8):1228–36.
Huang CY, et al. Genetic variants of the autophagy pathway as prognostic indicators for prostate cancer. Sci Rep. 2015;5:14045.
Ashrafizadeh M, et al. PD-1/PD-L1 axis regulation in cancer therapy: the role of long non-coding RNAs and microRNAs. Life Sci. 2020;256:117899.
Wu L, et al. Identification of a novel six autophagy-related genes signature for the prognostic and a miRNA-related autophagy predictor for anti-PD-1 therapy responses in prostate cancer. BMC Cancer. 2021;21(1):15.
Lu J, et al. Autophagy induced by overexpression of DCTPP1 promotes tumor progression and predicts poor clinical outcome in prostate cancer. Int J Biol Macromol. 2018;118(Pt A):599–609.
Burdelski C, et al. Cytoplasmic accumulation of Sequestosome 1 (p62) is a predictor of biochemical recurrence, rapid tumor cell Proliferation, and genomic instability in prostate Cancer. Clin Cancer Res. 2015;21(15):3471–9.
Acknowledgments
The authors would like to thank Dr. Pooyan Makvandi and Dr. Navid Rabiee for providing us with their valuable comments during revision to improve article quality.
Funding
This research was supported by the Canadian Institutes of Health Research (#141635, #144159, #153081, #173338), Terry Fox Research Institute (#1062) - (awarded to YW); National Institutes of Health (GM131919) - (awarded to DJK); Singapore Ministry of Education (MOE-T2EP30120–0016) and the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Center of Excellence initiative to Cancer Science Institute of Singapore, National University of Singapore - (awarded to APK).
Author information
Authors and Affiliations
Contributions
Milad Ashrafizadeh, conceptualization, writing first-draft and revision; Mahshid Deldar Abad Paskeh, writing first-draft and revision; Speideh Mirzaei, writing first-draft and revision; Mohammad Hossein Gholami, writing first-draft and revision; Ali Zarrabi, software and drawing figures; Farid Hashemi, writing first draft and revision; Kiavash Hushmandi, revisions on first draft; Noushin Nabavi, revisions on first draft; Francesco Crea, English editing; Jun Ren, English editing; Daniel J, Klionsky, English editing; Alan Prem Kumar, conceptualization, supervision and editing final manuscript; Yuzhuo Wang, conceptualization, supervision and editing final manuscript. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
We confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship but are not listed. We further confirm that the order of authors listed in the manuscript has been approved by all.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ashrafizadeh, M., Paskeh, M.D.A., Mirzaei, S. et al. Targeting autophagy in prostate cancer: preclinical and clinical evidence for therapeutic response. J Exp Clin Cancer Res 41, 105 (2022). https://doi.org/10.1186/s13046-022-02293-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-022-02293-6