
Abstract
Follicular lymphoma (FL) is a B-cell non-Hodgkin lymphoma of germinal center (GC) origin with a distinctive tumor microenvironment (TME) and a unique spectrum of mutations. Despite the important therapeutic advances, FL is still incurable. During B-cell development, the GC reaction is a complex multistep process in which epigenetic regulators dynamically induce or suppress transcriptional programs. In FL, epigenetic gene mutations perturb the regulation of these programs, changing GC B-cell function and skewing differentiation towards tumor cells and altering the microenvironment interactions. FL pathogenesis and malignant transformation are promoted by epigenetic reprogramming of GC B cells that alters the immunological synapse and niche. Despite the extensive characterization of FL epigenetic signature and TME, the functional consequences of epigenetic dysregulation on TME and niche plasticity need to be better characterized. In this review, first we describe the most frequent epigenomic alterations in FL (KMT2D, CREBBP and EZH2) that affect the immunological niche, and their potential consequences on the informational transfer between tumor B cells and their microenvironment. Then, we discuss the latest progress to harness epigenetic targets for inhibiting the FL microenvironment. Finally, we highlight unexplored research areas and outstanding questions that should be considered for a successful long-term treatment of FL.
Similar content being viewed by others

Background
Follicular lymphoma (FL) is the most frequent indolent B-cell non-Hodgkin lymphoma (yearly incidence: 2.7 cases per 1000,000 inhabitants in the USA in the 2014–2018 period according to the National Cancer Institute Surveillance, Epidemiology and End Results program). It is characterized by abnormal B-cell clonal expansion and originates from the germinal center (GC). Immunotherapy has drastically improved FL outcome. Specifically, the use of anti-CD20 monoclonal antibodies (such as rituximab) has increased the patients’ overall survival to 90%. However, many patients experience relapses and develop treatment resistance, or transformation to more aggressive lymphoma types (usually diffuse large B cell lymphoma (DLBCL) in 30% of patients) [1, 2]. Hence, there is an urgent need of new clinical options for the long-term cure of FL. Approximately 90% of patients with FL harbor the t(14;18)(q32:q21) chromosomal translocation in which the immunoglobulin heavy chain (IGH) enhancer region at 14q32 and the B-cell lymphoma 2 (BCL2) gene at 18q21 are juxtaposed. This translocation is characterized by a transposition of the BCL2 oncogene to the regulatory region of the IGH gene, thus leading to BCL2 ectopic expression and constitutive activation of the anti-apoptotic program. In patients with FL, this translocation can be found also in bone marrow CD34+ CD19+ B-cell precursors [3]. However, the detection of BCL2 genetic aberrations at low frequency (40%) also in healthy individuals is one of the many evidences supporting the hypothesis that this translocation is insufficient for FL development and that other genetic and epigenetic alterations (such as BCL6) are required [4,5,6].
Epigenetic processes are implicated in the regulation of gene transcription and translation. They include DNA methylation and histone modifications through the transfer of acetyl or methyl groups. Deregulation of such processes (e.g. aberrant DNA hypermethylation) has been recognized as a central feature of hematologic malignancies [7, 8], particularly FL where epigenome alterations have been observed in almost all patients [9, 10]. This could be explained by the multistep GC reaction process in which gene expression is finely regulated by post-translational histone modifications and/or chromatin remodeling. In FL, mutations in epigenetic-related genes concern mainly histone methyltransferases (KMT2D and EZH2) and histone acetyltransferases (CREBBP and EP300), and they are mostly inactivating mutations, except in EZH2. They play a crucial role in gene expression regulation by modifying lysine 4 on histone 3 (H3K4) (KMT2D) and lysine 27 on histone 3 (H3K27) (EZH2, CREBBP and EP300) [9, 11]. Moreover, these epigenome abnormalities are common in adult FL, but not in the rare pediatric nodal FL that has a more favorable prognosis [12]. The impact of these alterations varies in function of the B-cell development stage. At early stages, these mutations are sufficient to initiate lymphomagenesis, while at later stages, additional epigenetic gene mutations might be required to support malignant transformation [13].
At the cellular level, the multiple relapsing/remitting cycles that characterize FL are partly due to the presence of a common precursor cell (CPC) population enriched in these early epigenetic driver and recurrent mutations, particularly in histone modifiers. This “founder” population acts as a FL niche that sustains the tumor microenvironment (TME) reprogramming through molecular (genetic and epigenetic) mechanisms to promote anti-tumor immune evasion and transformation [10].
In studies on FL, genetic/epigenetic alterations and TME are often characterized separately; conversely, in this review we wanted to discuss the impact of the FL epigenome on TME functional plasticity to better understand this indolent cancer.
The FL epigenome influences its microenvironment
Gene regulation by epigenetic mechanisms involves the addition or removal of chemical groups on DNA or histones, catalyzed by enzymes called ‘writers’ and ‘erasers’. Moreover, ‘readers’ are a distinct group of proteins that are recruited to specific regions of the genome through recognition of the epigenetic marks deposited or erased by the ‘writers’ and ‘erasers’. Then, readers can modify/adjust the chromatin state and regulate the expression of target genes. DNA methylation and histone modifications are the most common epigenetic modifications. The mechanisms through which epigenetic regulators regulate gene expression have been described elsewhere [14, 15], and will not be discussed in this review. Here, we will focus on the mutations of the histone acetyltransferase CREBBP and the methyltransferases KMT2D and EZH2 that play a key role in the FL microenvironment, and have detrimental consequences on the anti-tumor immune response.
FL is a cancer addicted to epigenetic gene mutations that are detected in almost all patients (80%), and concern particularly the histone methyltransferases KMT2D (90% of patients) and EZH2 (25%) and the histone acetyltransferases CREBBP (30–60%) and EP300 (9%). They are frequently combined with other mutations, for instance in linker and core histone genes and chromatin remodeling complexes [16]. All these mutations lead to a modification of the epigenetic landscape in FL that supports tumor progression.
A specific feature of FL is its strong dependency on its microenvironment. This is explained by various mechanisms [17,18,19], but is essentially correlated to the epigenetic changes that occur in the early stage of tumorigenesis. Indeed, the epigenetic dysregulation detected in FL can affect the expression of genes that regulate quantitatively and qualitatively the TME cell composition, thus influencing antigen presentation mechanisms, immune signaling pathways, and cell recruitment to escape immune surveillance and maintain an immunosuppressive microenvironment [19,20,21]. Therefore, these epigenetic alterations substantially affect the patient outcomes by providing survival signals for cancer cells and by hindering the anti-tumor immune response (Table 1).
CREBBP mutations affect antigen presentation
In FL, the unbalance toward abnormal repression of gene transcription might be explained by the loss of histone 3 lysine 27 acetylation (H3K27ac; an active transcription mark) catalyzed by CREBBP. CREBBP, one of the most frequently mutated gene in FL (∼65%), encodes a lysine acetyltransferase that activates gene expression through acetylation of histone H3 lysine 18 (H3K18Ac), H3K27Ac, and other residues. Most CREBBP mutations lead to single–amino acid changes within the catalytic acetyltransferase domain that reduce its acetyltransferase activity [22]. FL sequencing identified CREBBP mutations as early oncogenic events, likely in the CPC founder population [23], thus partly explaining their association with poor outcome [24]. Interestingly, in FL, CREBBP mutations are correlated with reduced tumor expression of genes involved in the MHC class II antigen presentation pathway, and also with decreased B-cell receptor (BCR) and interferon signaling, accompanied by reduced proliferation of tumor-infiltrating T cells [23, 25]. Overall, as the lack of recognition and presentation of tumor antigens by immune cells contributes to tumor invasion and progression [25], these findings suggest that CREBBP mutations directly promote lymphomagenesis by controlling the antigen presentation and interferon signaling pathways (Fig. 1). Interestingly, MHC pathway deregulation is observed in the early stage of FL, and also in transformed FL and DLBCL. FL progression to a more aggressive form is associated with increased somatic hypermutation activity, leading to accumulation of mutations on several downstream targets [26, 27]. All these studies indicate that MHC deregulation is an important mechanism in FL pathogenesis and that it occurs at every FL stage.

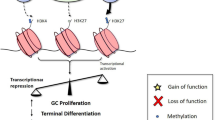
Epigenetic dysregulation in FL modifies its microenvironment. Somatic mutations occur during the early steps of FL lymphomagenesis (dashed circle, 1). Accumulation of genetic and epigenetic mutations promotes tumor progression. All these alterations accumulate during the different phases of the GC reaction, contributing to the dysfunctional B-T cell crosstalk (dashed circle, 2) that favors tumor growth, escape, and dissemination. 1- Schematic summary of the epigenetic regulators frequently mutated in FL: CREBBP, KMT2D and EZH2. These mutations occur concurrently in most FL, but it is not known whether they act alone or cooperatively in driving B cell malignancy and in shaping the FL epigenome. H3Kac, histone H3 lysine acetylation; H3K4me3, histone H3 lysine 4 trimethylation; H3K27me3, histone H3 lysine 27 trimethylation. Loss-of-function: -, gain-of-function: + . 2- Consequences of epigenetic gene mutations on the immunological synapse. Impact of CREBBP, KMT2D and EZH2 gene mutations in FL B cells on the crosstalk of tumor cells, T follicular helper (TFH) cells, and follicular dendritic cells (FDC). Alterations of this cross-talk lead to inhibition of TFH immune synapse formation and antigen presentation, and increased FDC interactions. Specifically, CREBBP gene mutations affect antigen presentation, and KMT2D and EZH2 gene mutations lead to a disrupted immune synapse. Red stars: decreased expression. Green stars: increased expression. Dashed arrows: abnormal pathway
EZH2 and KMT2D mutations disrupt the immune synapse
EZH2 mutations were the first epigenetic alterations to be reported in FL and to be investigated using murine models that showed their crucial role in early FL [28]. In FL, the unbalance toward abnormal repression of gene transcription is characterized by an increase in the repressive trimethylation of lysine 27 on histone 3 (H3K27me3) mark catalyzed by EZH2, the best characterized writer in lymphoma. EZH2 is part of the polycomb repressor 2 complex, and is normally strongly expressed in GC B cells. Indeed, EZH2 is one of the main regulators of the GC phenotype by repressing the expression of regulators of B-cell differentiation (such as PRDM1 and IRF4) through H3K27me3 deposition [28]. EZH2 mutations are the third most important recurrent gene alterations in FL, leading to increased H3K27me3 levels [29].
Recently, Beguelin et al. showed that in Ezh2 conditional knock-in mice harboring the EZH2Y641F mutation in the catalytic domain that increases H3K27me3 levels, GC B cells accumulate in the light zone because of the lack of the MYC-associated recycling signature, thus impairing cell re-entry in the dark zone and promoting terminal differentiation. An important effect of these Ezh2 mutations is the prevention of the induction of genes (e.g. Cd69, Icosl, Icam1, Icam2, Slam, Ly108, and Basp1) that are normally upregulated in GC centrocytes. This is associated with a preferential spreading of the H3K27me3 mark from gene promoters to neighboring chromatin (due to their shared localization within topologically associating domains) [9]. As these genes are usually implicated in the formation of stable immune synapses with T-follicular helper (TFH) cells, this impairment results in the loss of the crosstalk with these cells and in the reduction of the CD40/CD40L response. Importantly, GC B cells harboring Ezh2 mutations maintain GCs in the absence of CD40 signaling and TFH interaction thanks to their increased crosstalk with follicular dendritic cells (FDCs). This is possible through aberrant overexpression of genes involved in the interaction with FDCs, such as Tnfrsf13c (BAFF receptor), Ltb (FDC precursor) [9, 30,31,32]. In line with these observations, human FL with EZH2 mutations have an atypical denser FDC network within follicles. Particularly, EZH2 mutant GC B cells expand GCs until hyperplasia by enhancing centrocyte survival and by evading TFH-directed clonal selection and affinity maturation [9]. These studies suggest that EZH2 mutations lead to aberrant repression of genes normally expressed during the GC reaction, thus preventing the normal clonal selection of GC B-cells, and allowing their survival and proliferation within the LZ by depending only on survival signals from FDCs. This reflects an epigenetic reprogramming that modifies the GC B cell interactions with TFH cells and FDCs, thus promoting malignant transformation and the establishment of the specific features of the FL immunological synapse and niche (Fig. 1). As GC B cells are very T-cell dependent, all these results might help to understand FL resistance to T-cell-based therapies.
KMT2D encodes a SET domain-containing lysine methyltransferase and catalyzes almost all mono-methylation of lysine 4 on histone 3 (H3K4me1). KMT2D is the most recurrently mutated gene in FL, resulting in its loss of function, thus promoting lymphomagenesis [33].
Transcriptomic analysis of a mouse model in which Kmt2d was ablated showed an abnormal enrichment in genes encoding immune-related proteins, such as CD40, JAK-STAT, Toll-like receptor (TLR), IL-6, TNF, and BCR signaling molecules. Among the KMT2D target genes, there is also the TNFRSF14 (HVEM) tumor suppressor that is frequently mutated in FL (40% of patients) [13]. These mutations lead to the loss of the signaling cascade induced by HVEM interaction with its inhibitory receptor BTLA. Disruption of the HVEM/BTLA crosstalk causes hyperactivity of BCR signaling and consequently infiltration of BTLA+ TFH cells that produce high amounts of TNF-α, IL-4, and lymphotoxin a1b2 (crucial regulators of stroma differentiation and maintenance), reflecting an enrichment in stromal TME [34].
Interestingly, Haebe et al. performed single-cell analysis of FL samples from two sites/patient and found that in most patients, the two B-cell sub-clones grow separately with phenotypic divergences that correlate with TFH cell infiltration through the CD40-CD40L crosstalk, whereas the overall TME profile remains similar at both sites [35]. This discrepancy might be explained by the loss of the GC program flexibility and the ability to de-synchronize and adopt different functional circuits, independently of the original clone [36]. These studies suggest the existence of founder neoplastic clones with intrinsic characteristics to modulate the TME in favor of disease progression. Additionally, it has been reported that some gene alterations (e.g. cathepsin S, beta2 microglobulin, CIITA and RRAGC) [5, 27, 37,38,39] also support the establishment of a biased immune synapse in FL, independently of epigenome modifications. However, this is outside the scope of this review and will not be further discussed.
Epigenetic modifying agents for FL treatment?
Due to FL indolence and slow progression, this malignancy is often diagnosed at an advanced stage with high tumor burden. The classical chimeric anti-CD20 monoclonal antibody rituximab is an effective immunotherapy, as single agent or in combination with first-line chemotherapy, to achieve long-term remission. Progression-free survival is variable because of the non-responsiveness of some patients [40].
Various agents are currently under investigation to restore the anti-FL response and to reduce/inhibit its supportive TME in order to improve the anti-tumor immunity by reprogramming immune cells [41, 42] (NCT02953509). Here, we discuss strategies to target epigenetic alterations that might restore the immunological synapse in FL.
Because of the high rate of epigenetic dysregulation in FL, epigenetic regulators represent targets of choice. Ongoing clinical trials focus on CREBBP, EZH2, and HVEM with promising responses in refractory/relapsed FL. Although the most common loss-of-function KMT2D and CREBBP mutations appear early in FL pathogenesis, they are hard to target. However, as CREBBP and p300, its closely related activator, regulate regulatory T cell (Treg) differentiation, the inhibition of CREBBP/p300 loss-of function mutations in FL might have some effects also on non-tumor cells within the TME [43].
As genes silenced upon CREBBP mutation are direct targets of the BCL6/HDAC3 tumor-repressor complex, the use of selective HDAC3 inhibitors (e.g. BRD3308) allows reversing CREBBP loss-of-function effects by restoring the immune response (T-cell anti-tumoral function optimization by inducing antigen presentation by MHC class II molecules, and interferon signaling) and inhibiting lymphoma growth [44].
The EZH2 inhibitor tazemetostat is the only epigenetic modifying agent approved by the US Food and Drug Administration. An early clinical trial in non-Hodgkin lymphoma (NCT01897571) showed its effectiveness, but with no alternative treatment options for refractory patients [45]. Although EZH2 inhibitors showed promising effects on the non-tumor microenvironment of other cancers [46,47,48], their effects on the non-tumor microenvironment of FL has not been investigated yet. Importantly, as EZH2 mutations occur later in FL, they might not be the best therapeutic target for long-term cure. Moreover, EZH2 is physiologically involved in the GC reaction and in other important cellular mechanisms (e.g. T and natural killer cell development), agents that target mutated EZH2 in FL, rather than wild type EZH2, should be developed. In addition, the specific targeting of EZH2 mutations is interesting to prevent the CPC population rebound correlated with FL relapses and transformation.
Importantly, mutations in these different epigenetic genes might interact and influence each other in FL. This would imply the concomitant use of different epigenetic modifying agents as a global epi-therapeutic strategy in FL.
Therapeutic combinations are among the best options to reduce the exceptional supporting role of the TME in FL and to improve the anti-tumor response. This could include the combination of epigenetic modifying agents with immunotherapies or other anti-cancer drugs. For instance, the response of HDAC3 inhibitors can be enhanced by combining them with anti-PDL1 immune checkpoint inhibitors to prevent interferon-induced adaptive immune suppression [49]. Recently, two CD19-directed CAR-T cell products (axicabtagene ciloleucel and lisocabtagene maraleucel) have been approved for B-cell lymphoma (DLBCL and FL at advanced stage), including in combination with an anti-CD20 monoclonal antibody and bendamustine, an alkylating agent [50,51,52].
All these preliminary results suggest that combination therapies synergistically improve FL outcomes, and that epigenetic modifying agents might be best used in association with other therapeutic agents (Table 1).
In conclusion, epigenetic modifying agents seems to be a very promising avenue for FL therapy; however, i) their complete response rate, as single agents, is still low; ii) their effects too broad; and iii) their toxicity is high due to their systemic delivery. This makes more difficult (as it is also the case with other therapeutic agents) to treat disease heterogeneity, non-responsiveness, and resistance to treatment [53]. High density lipoprotein (HDL)-based natural biomaterials could be used to concentrate their effects on the target and to minimize their toxicity, by associating the benefits of therapeutic targeting and controlled release of the active compound [54, 55].
Concluding remarks
Currently, FL management is based on immunotherapies and chemo/radiotherapies, thus undervaluing the importance of epigenetic dysregulation and its strong impact on the TME. FL is addicted to epigenetic modifications and strongly relies on its microenvironment, suggesting a mutual impact and crucial role in FL pathogenesis. Understanding FL epigenetic landscape will require detailed genetic, epigenetic and transcriptomic approaches to determine how all these factors contribute independently to FL development. Moreover, the functional characterization of FL epigenome would help to dissect how the complex interplay of these alterations contributes to modulate the TME and leads to immunosuppression, thus to FL pathogenesis. This will also help to answer other outstanding questions (Table 2). Importantly, the presence of many epigenetic alterations in the CPC population increases the interest of epigenetic therapies for eradicating this founder clone in FL and preventing transformation. Due to the flexibility and reversibility of epigenetic modifications and the particular enrichment of epigenetic gene mutations in FL, they are perfect therapeutic targets and accurate biomarkers for early diagnosis. However, the complexity of the genetic/epigenetic alteration landscape within the same tumor, as a result of the concerted epigenetic regulator crosstalk, and the substantial impact of epigenetic drugs also on the non-malignant microenvironment may make it more difficult to predict the efficacy of epigenetic modifying agents in FL. Thus, for clinical translation, their effects must be restricted to the targeted tumor population and their off-target effects strongly reduced by using natural nanomaterials (such as HDL-based nanoparticles). Moreover, although epigenetic modifying agents represent an interesting therapeutic option for FL, their complete response rates, as single agents, have been low [56, 57]. Hence, combination therapies targeting the epigenetic dysregulations (upstream), the immunological synapse (downstream), and FL metabolic pathway would help to better attack all the pro-tumor mechanisms of FL. Lastly, the development of high throughput data analysis and whole sequencing will allow addressing important questions concerning the impact of FL epigenome on its complex informational immunological synapse. Ultimately, a personalized therapy based on each patient’s malignant epigenetic signature might be the best ways to improve FL outcome.
Availability of data and materials
Not applicable
Abbreviations
- FL:
-
Follicular lymphoma
- GC:
-
Germinal center
- TME:
-
Tumor microenvironment
- IGH:
-
Immunoglobulin heavy chain
- DLBCL:
-
Diffuse large B cell lymphoma
- BCL2:
-
B-cell lymphoma 2
- CPC:
-
Common precursor cell
- H3K18Ac and H3K27Ac:
-
Histone 3 lysine 18 and 27 acetylation
- BCR:
-
B-cell receptor
- H3K27me3:
-
Histone 3 lysine 27 trimethylation
- TFH:
-
T-follicular helper
- FDC:
-
Follicular dendritic cells
- H3K4me1:
-
Lysine 4 on histone 3 monomethylation
- TLR:
-
Toll-like receptor
- HDL:
-
High density lipoproteins
References
Arber DA, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Kridel R, Sehn LH, Gascoyne RD. Pathogenesis of follicular lymphoma. J Clin Invest. 2012;122(10):3424–31.
Macintyre EA, et al. Detection of clonal CD34+19+ progenitors in bone marrow of BCL2-IgH-positive follicular lymphoma patients. Blood. 1995;86(12):4691–8.
Holmes AB, et al. Single-cell analysis of germinal-center B cells informs on lymphoma cell of origin and outcome. J Exp Med. 2020;217(10):e20200483.
Ochando J, Braza MS. T follicular helper cells: a potential therapeutic target in follicular lymphoma. Oncotarget. 2017;8(67):112116–31.
Yang H, Green MR. Epigenetic Programing of B-Cell Lymphoma by BCL6 and Its Genetic Deregulation. Front Cell Dev Biol. 2019;7:272.
Dufva O, et al. Immunogenomic Landscape of Hematological Malignancies. Cancer Cell. 2020;38(3):380-399e13.
Sermer D, et al. Emerging epigenetic-modulating therapies in lymphoma. Nat Rev Clin Oncol. 2019;16(8):494–507.
Beguelin W, et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell. 2020;37(5):655-673e11.
Okosun J, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46(2):176–81.
Yang H, Green MR. Harnessing lymphoma epigenetics to improve therapies. Blood. 2020;136(21):2386–91.
Korfi K, et al. Follicular lymphoma, a B cell malignancy addicted to epigenetic mutations. Epigenetics. 2017;12(5):370–7.
Ortega-Molina A, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199–208.
Plass C, et al. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. 2013;14(11):765–80.
Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153(1):38–55.
Li H, et al. Mutations in linker histone genes HIST1H1 B, C, D, and E; OCT2 (POU2F2); IRF8; and ARID1A underlying the pathogenesis of follicular lymphoma. Blood. 2014;123(10):1487–98.
Kiaii S, et al. Follicular lymphoma cells induce changes in T-cell gene expression and function: potential impact on survival and risk of transformation. J Clin Oncol. 2013;31(21):2654–61.
Blaker YN, et al. The tumour microenvironment influences survival and time to transformation in follicular lymphoma in the rituximab era. Br J Haematol. 2016;175(1):102–14.
Sugimoto T, Watanabe T. Follicular Lymphoma: The Role of the Tumor Microenvironment in Prognosis. J Clin Exp Hematop. 2016;56(1):1–19.
Dave SS, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351(21):2159–69.
Palumbo GA, et al. Monocytic Myeloid Derived Suppressor Cells in Hematological Malignancies. Int J Mol Sci. 2019;20(21):5459.
Pasqualucci L, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471(7337):189–95.
Meyer SN, et al. Unique and Shared Epigenetic Programs of the CREBBP and EP300 Acetyltransferases in Germinal Center B Cells Reveal Targetable Dependencies in Lymphoma. Immunity. 2019;51(3):535-547e9.
Pastore A, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16(9):1111–22.
Green MR, et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc Natl Acad Sci U S A. 2015;112(10):E1116–25.
Devaiah BN, Singer DS. CIITA and Its Dual Roles in MHC Gene Transcription. Front Immunol. 2013;4:476.
Pasqualucci L, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6(1):130–40.
Beguelin W, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23(5):677–92.
Morin RD, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5.
Ise W, et al. T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity. 2018;48(4):702-715e4.
Liu D, et al. T-B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nature. 2015;517(7533):214–8.
Zaretsky I, et al. ICAMs support B cell interactions with T follicular helper cells and promote clonal selection. J Exp Med. 2017;214(11):3435–48.
Zhang J, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21(10):1190–8.
Boice M, et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell. 2016;167(2):405-418e13.
Haebe S, et al. Single-cell analysis can define distinct evolution of tumor sites in follicular lymphoma. Blood. 2021;137(21):2869–80.
Milpied P, et al. Human germinal center transcriptional programs are de-synchronized in B cell lymphoma. Nat Immunol. 2018;19(9):1013–24.
Bararia D, et al. Cathepsin S Alterations Induce a Tumor-Promoting Immune Microenvironment in Follicular Lymphoma. Cell Rep. 2020;31(5):107522.
Dheilly E, et al. Cathepsin S Regulates Antigen Processing and T Cell Activity in Non-Hodgkin Lymphoma. Cancer Cell. 2020;37(5):674-689e12.
Ortega-Molina A, et al. Oncogenic Rag GTPase signaling enhances B cell activation and drives follicular lymphoma sensitive to pharmacological inhibition of mTOR. Nat Metab. 2019;1(8):775–89.
Subramanian J, et al. Rituximab in the treatment of follicular lymphoma: the future of biosimilars in the evolving therapeutic landscape. Cancer Manag Res. 2017;9:131–40.
Advani R, et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N Engl J Med. 2018;379(18):1711–21.
Valero JG, et al. The receptor of the colony-stimulating factor-1 (CSF-1R) is a novel prognostic factor and therapeutic target in follicular lymphoma. Leukemia. 2021;35(9):2635–49.
Castillo J, et al. CBP/p300 Drives the Differentiation of Regulatory T Cells through Transcriptional and Non-Transcriptional Mechanisms. Cancer Res. 2019;79(15):3916–27.
Mondello P, et al. Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma. Cancer Discov. 2020;10(3):440–59.
Morschhauser F, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21(11):1433–42.
Bugide S, Green MR, Wajapeyee N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci U S A. 2018;115(15):E3509–18.
Peng D, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527(7577):249–53.
Zingg D, et al. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017;20(4):854–67.
Ogura M, et al. A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. Br J Haematol. 2014;165(6):768–76.
Abramson JS, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–52.
Halford Z, Anderson MK, Bennett LL. Axicabtagene Ciloleucel: Clinical Data for the Use of CAR T-cell Therapy in Relapsed and Refractory Large B-cell Lymphoma. Ann Pharmacother. 2021;55(3):390–405.
Neelapu SS, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377(26):2531–44.
Wallin EF, et al. Human T-follicular helper and T-follicular regulatory cell maintenance is independent of germinal centers. Blood. 2014;124(17):2666–74.
Ochando J, Braza MS. Nanoparticle-Based Modulation and Monitoring of Antigen-Presenting Cells in Organ Transplantation. Front Immunol. 2017;8:1888.
Fay F, et al. Investigating the Cellular Specificity in Tumors of a Surface-Converting Nanoparticle by Multimodal Imaging. Bioconjug Chem. 2017;28(5):1413–21.
Evens AM, et al. A Phase I/II Multicenter, Open-Label Study of the Oral Histone Deacetylase Inhibitor Abexinostat in Relapsed/Refractory Lymphoma. Clin Cancer Res. 2016;22(5):1059–66.
Burkhardt B, et al. Treatment and Outcome Analysis of 639 Relapsed Non-Hodgkin Lymphomas in Children and Adolescents and Resulting Treatment Recommendations. Cancers (Basel). 2021;13(9):2075.
Acknowledgements
Some panels of the figure are adapted from Servier Medical Art (http://smart.servier. com) and used under a Creative Commons Attribution 3.0 Unported License (CC BY 3.0) current version 4.0 published on November 2013.
Funding
This work was supported by Ikerbasque foundation for science.
Author information
Authors and Affiliations
Contributions
MSB conceived the original idea, wrote, corrected, revised and finalized the manuscript. RA wrote, revised and complemented the manuscript, provided ideas and critical feedback. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
None
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Amin, R., Braza, M.S. The follicular lymphoma epigenome regulates its microenvironment. J Exp Clin Cancer Res 41, 21 (2022). https://doi.org/10.1186/s13046-021-02234-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-021-02234-9