
Abstract
The discovery of the role of non-coding RNAs (ncRNAs) in the onset and progression of malignancies is a promising frontier of cancer genetics. It is clear that ncRNAs are candidates for therapeutic intervention, since they may act as biomarkers or key regulators of cancer gene network. Recently, profiling and sequencing of ncRNAs disclosed deep deregulation in human cancers mostly due to aberrant mechanisms of ncRNAs biogenesis, such as amplification, deletion, abnormal epigenetic or transcriptional regulation. Although dysregulated ncRNAs may promote hallmarks of cancer as oncogenes or antagonize them as tumor suppressors, the mechanisms behind these events remain to be clarified. The development of new bioinformatic tools as well as novel molecular technologies is a challenging opportunity to disclose the role of the “dark matter” of the genome. In this review, we focus on currently available platforms, computational analyses and experimental strategies to investigate ncRNAs in cancer. We highlight the differences among experimental approaches aimed to dissect miRNAs and lncRNAs, which are the most studied ncRNAs. These two classes indeed need different investigation taking into account their intrinsic characteristics, such as length, structures and also the interacting molecules. Finally, we discuss the relevance of ncRNAs in clinical practice by considering promises and challenges behind the bench to bedside translation.
Similar content being viewed by others
Background
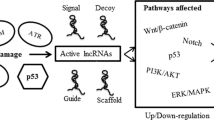
Carcinogenesis is a multistep process in which normal cells acquire genetic and epigenetic alterations that drive the onset of “hallmarks” of cancer finally resulting in development and progression of malignancies [1]. Despite most cancer studies have been focused on protein-coding genes, the evidence that about 97% of human genome consists of non protein-coding sequences led scientists to investigate this genetic “dark matter” in tumorigenesis. The untranslated transcripts, called non-coding RNAs (ncRNAs) can be classified in short (19–31 nucleotides), mid (20–200 nucleotides) and long (> 200 nucleotides) based on their length. Among them, the most extensively studied in cancer are micro-RNAs (miRNAs), which belong to short ncRNAs class (22–25 nucleotides in length) [2] and long-ncRNAs (lncRNAs), which represent the largest class of non-coding transcripts with about 55,000 genes along the human genome [3]. According to miRNA-mRNA complementarity, miRNAs can mediate post-transcriptional gene regulation by translational repression or mRNA degradation, while lncRNAs may regulate gene expression through their interaction domains for DNA, mRNAs, miRNAs and proteins. These events are dependent from both their sequence and secondary structure [4]. Specifically, ncRNAs can affect cancer cell fate and survival through a variety of different mechanisms, including transcriptional and post-transcriptional modification, chromatin remodeling and signal transduction. However, to date the exact function and mechanism of action of most of them is still unknown. As far as we know, ncRNAs create a complex network of mutual interactions [5] and act as oncogenes or tumor suppressors. They present a tissue specific expression pattern, which is highly dysregulated in cancer, and are considered promising diagnostic, prognostic and therapeutic targets [6,7,8]. Therefore, the understanding of the role of ncRNAs in tumorigenesis is a challenging goal in current biology. In this review we describe the in silico and in vitro approaches aimed to investigate the ncRNA transcriptome by providing a comprehensive overview of strategies and tools to characterize ncRNA structure and to study their contribution in cancer onset and progression (Fig. 1). Moreover we underline the promises and limits of these approaches in terms of translational relevance.

Schematic representation of the approaches discussed in this review for the investigation of the role of ncRNAs in human cancer
NcRNAs expression profiling
The aberrant expression of ncRNAs is an important feature of human cancer [9,10,11]. The ncRNAs have cell type, tissue and cancer specificity, thus RNA profiling has become a mean to identify useful biomarkers of tumor development, progression and metastasis. Although miRNAs represent the most widely investigated ncRNAs, lncRNAs are emerging as cancer key regulators [7, 12]. Arrays and next generation sequencing (NGS) are high-throughput methods to detect and quantify ncRNAs, even though several caveats are to be considered. Typically, both miRNAs and lncRNA are expressed at lower abundance if compared to mRNAs (~ 0.01% and ~ 0.1% of total RNA, respectively) [13]. In particular, miRNA profiling requires RNA isolation procedures to retain the small RNA fraction. In addition, miRNAs lack a common sequence, such as poly(A) tail, that is typically observed in mRNAs, so it is necessary to selectively detect this class of ncRNAs among different RNA species. Moreover, miRNAs of the same family may show high similarity or differ from the reference sequence due to post-transcriptional modifications (isomiRs). In contrast, lncRNAs have similarities with mRNAs such as size, RNA polymerase II transcription, 5′-capping, RNA splicing, and also about 60% of lncRNAs include a poly(A) tail. For this reason, lncRNAs can be profiled together with mRNAs while miRNAs require different approaches. However, the design of probes for many lncRNAs is difficult because most lncRNAs are located at intergenic regions with high GC content or are antisense transcripts of known protein coding genes. Here we report various methodologies to investigate differences in the pattern of gene expression between normal vs cancer cell, in order to discover ncRNAs potentially involved in tumorigenesis [14].
Microarray
Microarray is a well-established method to profile both miRNAs and lncRNAs, although it was initially designed for protein-coding mRNAs. This technology is based on nucleic acid hybridization between labeled RNA targets and their specific and complementary probes. Advantages of microarrays are the high parallel analysis coupled with relative cost and the ability to detect low levels of molecules of RNAs without the need of PCR-enrichment steps. Various platforms for miRNA profiling include different direct miRNA labelling procedures without amplification. LncRNA microarray platforms systematically profile lncRNAs together with mRNAs. Generally, lncRNA platforms include an in vitro transcription (IVT)-based amplification step and are characterized by less technical variations respect to miRNA platforms and mainly differ for the number of lncRNAs analyzed. For example, the Agilent SurePrint G3 Gene Expression v3 microarray, targeting 30,606 human lncRNA transcripts, covers all of LNCipedia 2.1, the Arraystar LncRNA microarray (release Human v5.0) profiles 39,317 lncRNAs, Clariom D human array from Thermo Fisher Scientific covers more than 55,900 lncRNA NONCODE transcripts. Limitations of microarrays for ncRNA analysis are a restricted linear range of quantification, the design of probes limited to known sequences, the need for continuous updating of annotations, the relative quantification limited to compare different status (for example, healthy versus affected). An example of microarray technology application has been reported by Zhou et al. which profiled 389 colon cancer patients identifying a signature of six lncRNAs (linc0184, AC105243.1, LOC101928168, ILF3-AS1, mir31HG, and AC006329.1) associated to risk of cancer recurrence [15], or by Liang et al., which identified through this technology, six miRNAs involved in breast cancer pathogenesis (hsa-miR-21b, hsa-miR-29b, and hsa-miR-155 as upregulated and hsa-miR-10b, hsa-miR-125 and hsa-miR-145 as downregulated) [16]. Another technique based on microarray is the Tiling array that differs because of the use of probes that may cover either specific chromosomal sequences as well as contiguous regions or even the whole genome. Bertone et al. found 10.595 transcribed sequences not detected with other methods in 2004 [17] but in the current biology, this technology has been replaced by NGS approaches.
Sage
Serial Analysis of Gene Expression (SAGE) is the first high-throughput sequencing technology developed to analyze the transcriptome in term of identification and quantification of transcripts, including ncRNAs [18]. It is based on the restriction enzymes-mediated generation of short-stretches of unbiased cDNAs sequences (9 bp SAGE tags) followed by concatenation, cloning and sequencing. This method has been implemented in the “SuperSAGE” variant that allows the profiling of 26 bp tags and provides the advantage of in tag-to gene annotation by generating more throughput data with a better quality/cost ratio [19]. Gibb et al. reported lncRNA expression profile across 26 normal and 19 tumoral tissues by analyzing 24 million SAGE tags [20].
RNA-seq
RNA sequencing (RNA-seq) allows the detection and quantification of all classes of ncRNAs through the construction of different cDNA libraries, specific for each type of ncRNA. cDNA library preparation is followed by massive parallel sequencing of transcripts of interest. The small RNA-seq is suitable for the sequencing of small ncRNAs, while total RNA-seq is recommended for lncRNA sequencing as many lncRNAs may not be polyadenilated. Compared to microarray, RNA-seq offers a more comprehensive coverage of whole transcriptomes. Importantly, RNA-seq is design-free probe allowing the detection of unknown/novel transcripts and also the detection of sequences that differ, even for a single nucleotide, such as transcripts harboring mutations or isoforms. The main limitations of RNA-seq are the complexity of data analysis and the high deep reads needed to detect low amount of the target. Using RNA-seq technology Yamada et al. identified a signature of 27 upregulated and 22 downregulated lncRNAs associated with colorectal cancer (CRC) as alternative biomarkers and/or treatment targets [21]. Yu N et al. identified tumor suppressor in lung adenocarcinoma by integrating data from miRNA-seq and RNA-seq [22]. The most advanced application of RNA-seq is the single cell transcriptomic sequencing [23]. For example, Designed Primer-based RNA-sequencing strategy (DP-seq) allows the amplification of RNA from 50 pg of sample [24], while Quartz-Seq is a single cell RNA seq method able to reveal genetic changes between single cells into the same cell type and also into the same cell-cycle phase [25].
Cage
Cap analysis gene expression (CAGE) is an NGS-based technology allowing the generation of a snapshot of the 5′ end of the mRNA. Similarly to SAGE, sequencing is preceded by cDNAs-tag generation, concatenation and cloning, but the main differences between the two approaches is the ability of CAGE to identify the exact location of the 5′ capped- transcript. Respect from RNA-seq, the advantage of CAGE consists in the identification of transcriptionally active promoter regions and RNA polymerase II transcription start sites (TSS). Horie et al. revealed a set of 49 coding and 10 non-coding genes upregulated in non-small cell lung cancer (NSCLC) due to promoter hypomethylation, by performing an integrative analysis of promoter level expression profiles generated through CAGE method [26].
In silico investigation
High-throughput ncRNAs expression profiling methods require bioinformatic contribution to analyze data generated from different platforms, including microarray and NGS technologies described above. In particular, the application of NGS is becoming predominant to explore in depth patient specific genetic background underlying intra and inter-individual variability, which acquire increasing relevance in the era of personalized medicine [27,28,29,30]. Analysis of ncRNAs data may have different aims such as discovery and annotation of novel ncRNAs, expression pattern profiling, validation and structural reconstruction of known ncRNAs and integrative analysis of their behavior and functions.
Bioinformatic analysis
In the case of data generated by array technology, bioinformatic data analysis includes: i) the identification of differentially expressed genes between two classes (such as normal versus tumor specimens, pharmacological treated versus non-treated cells, etc.), ii) clustering, which consists in building clusters of genes in term of expression level, iii) classification and/or, iv) analysis of pathways and interaction networks. Microarray raw data processing involves 4 phases: 1) pre-processing, which includes background adjustment, normalization and summarization, 2) annotation to enrich preprocessed data, 3) statistical and/or data mining analysis and 4) biological interpretation. Well-known algorithms of microarray data preprocessing are MAS4.0, MAS5.0, RMA, PLIER and GCRMA. Background correction is essential to remove noise in the optical detection system due to non-specific hybridization. Normalization, within and between arrays, is needed in order to remove systematic technical artifacts that could be due to different efficiency of reverse transcription, labeling or hybridization reactions, or other laboratory conditions. Summarization unifies signals generated from multiple probes, designed for the same transcripts, with multiple locations on the array. Once summarized, data can be annotated by adding information such as gene symbols or function. Data mining is the process by which groups of samples are compared in order to find differentially expressed genes on the basis of their expression values. Many of the methods for visualization and interpretation of gene expression data can be used for both microarray and RNA-seq experiment including clustering analysis, gene set enrichment analysis and pathway (Gene Ontology, KEGG, Ingenuity, Reactome, WikiPathways) or network analysis [31, 32].
In the case of RNA-seq, ncRNAs analysis workflow starts from raw NGS data. The first step is the filtering of low-quality reads from raw data. This process is usually performed by using tools for pre-processing of files containing short-reads encoded into FASTA-FASTQ files, which map the sequences to reference data (stored into databases). Example of such programs are the FASTX-Toolkit, Blat, SHRiMP, LastZ, MAQ and many others [33]. Once filtered, the second step is to construct transcript assembly using, for instance BowTie [34] or TopHat [35]. After the assembly, known genomic sequences, i.e. known coding genes, are filtered by tools, such as Bowtie. At the end of this step all the sequences may represent potential noncoding RNAs that have to be assessed and mapped with respect to existing ncRNA databases using assessment tools such as CPAT or Pfamscan. The NCBI NT and NR database is the preferred mapping database in this step since they include sequences for all species. NcRNAs extraction from RNA-seq and assembled transcripts processing can be instead performed by using many different approaches, many of which implement sequential filters based on features such as transcripts length, number of exons for each identified transcript or Open Reading Frame (ORF) size. Sun et al. presented a pipeline called lncRNAscan able to detect novel lncRNAs from the transcripts file generated by RNA sequencing [36]. Machine learning based algorithms and comparative sequence analysis have also been investigated in literature [37,38,39]. The above described pipeline is used in almost all the bioinformatic studies related to ncRNAs [2, 5, 40, 41] in which ncRNAs expression pattern has been correlated with clinical outcome of cancer patients. Finally the function of ncRNAs has to be investigated by analyzing existing databases (summarized in Table 1) hosting a large number of ncRNA sequences and, when available, information about biological studies. These databases can be queried to identify known ncRNAs in a given dataset but, due to the lack of conservation, many known ncRNAs are only valid for well-annotated species. For example, Song et al. [42] used the lnRNAdb to support the identification of lncRNAs with a potential role in human gastric cancer occurrence and development. Hou et al. used NONCODE database to detect coding and noncoding genes extracted from cell RNA-seq data and bulk hepatocellular carcinomas cells data [43].
Computational biology prediction
In the context of the investigation and discovery of ncRNAs involved in cancer cell biology, it is important to identify and predict such molecules by computational methods. Therefore, several algorithms have been developed or used for accurate and fast prediction of ncRNAs, with the aim to avoid expensive experimental methods [44]. Notwithstanding, the accuracy of these algorithms is reduced by increasing ncRNA nucleotide sequence length, thus lncRNAs prediction suffers from numerous limitations and approximations, while miRNAs are more easy to be modelled. The algorithms development for predicting ncRNAs requires specific models [45], among them i) Minimal Folding Energy (MFE), ii) Hidden Markov Model (HMM) and iii) Stochastic Context Free Grammar (SCFG), whose application led to the reconstruction of the 2D structure of ncRNAs (Fig. 2). MFE is an RNA structure-based model that considers base pairs and their related energy. Therefore, by applying the canonical base pairing between A-U, C-G and the unusual G-U [46, 47] together with thermodynamic laws [48], the structure showing the lowest energy is selected as the most stable. However, the MFE predictions are based on different assumptions about native RNA structures and suffer some limitations, which enable MFE to identifies ~ 70% of bp correctly for RNAs under ~ 700 nt in length, only [49]. Among MFE based algorithms RNAfold, RNAstructure and Mfold have been widely used to predict 2D structure of ncRNAs. For example, Rahimi et al. explored potential hairpin structures and differentiated real miRNA precursors from pseudo ones through the application of RNAfold, allowing the selection of hsa-miR-B43 as one of the best candidates which might have a potential metastasis-related function in breast cancer [50]. RNAfold and RNAstructure were also able to demonstrate that the free energy of miR-302c stem-loop structure was more negative in the presence of the wild-type compared to the variant allele, leading to the suggestion that rs199971565 SNP is a novel INDEL biomarker located in the seed site of miR-302c, which may have crucial roles in the susceptibility to gastric cancer [51]. HMM is a probabilistic model and belongs to methods able to find similarity between sequences. BLAST and FASTA algorithms are considered the easiest methods, which can determine sequences similarity between homologous families [52]. HMM is to be preferred in the case where sequence similarity between distant families is required. Thus, it is useful to identify the most likely positions containing ncRNAs sequences. In particular, the similarity between query sequence and the consensus sequences is established through the alignment employing the constructed scoring matrix. Vorozheykin et al. applied this model in a web server for prediction of pre-miRNAs, miRNAs, and their binding sites [53]. Voss et al. developed RNAlishapes [54]: an ab initio algorithm able to predict ncRNA genes applying HMM method. In particular, it predicts ncRNAs through the identification of transcription start points or other unique positions in a genome. Finally, SCFG is a statistical method capable of modeling interactions between base pairs in the structure of RNA and is used to predict structure and sequence of ncRNAs. As HMM extends regular grammars, SCFGs extends context-free grammars in which each production is attached to a probability. Simply, ncRNA secondary structures are assimilated to symbols and the comparison of similarity between target sequences and such symbols can predict most likely sequences as ncRNAs. In this model, the dynamic programming algorithm detects secondary structures with the maximum score for their functions. Two important algorithms, that use this model, are Rfam and tRNAscan [55].

Three of the most important models required for the development of algorithms predicting 2D ncRNAs: Minimal Folding Energy (MFE), Hidden Markov Model (HMM) and Stochastic Context Free Grammar (SCFG)
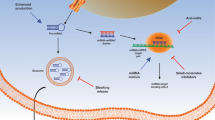
Computational techniques are also useful in the identification of ligands interacting with lncRNA. In this field, a useful software is Inforna. It is able to predict motifs (secondary structures) within the target and drives sequence-based design of small molecules (SMs) targeting structured RNAs [56]. Recent studies also demonstrated the suitability of classical computational methods, such as docking and molecular dynamics, by working on lncRNA 3D structures, thanks to the target of discrete binding pockets in nucleic acids [57, 58] (Fig. 3). Unfortunately, the number of available lncRNA crystallographic structures is still very small, thus making more difficult the identification or the design of specific inhibitors. Currently, the literature reported successful drug discovery studies on three particular lncRNAs: TERRA, MALAT-1 and HOTAIR [59,60,61].

Possible application of computational biology prediction starting from lncRNAs 3D structure
Validation of ncRNAs expression
Data obtained from microarray or RNA-seq, as well as from the in silico predictions need hortogonal validation. The evaluation of ncRNAs expression level can be performed through different approaches, such as northern blot (NB), reverse transcription quantitative PCR (RT-qPCR), in situ hybridization (ISH) or fluorescence in situ hybridization (FISH).
Northern blot analysis
NB analysis is the earliest technique used to analyze gene expression splicing variants of a given ncRNA. The procedure involves the use of gel electrophoresis to separate RNA samples followed by RNA transfer onto a nylon membrane, RNA-probe hybridization and finally RNA-probe detection. An example of the application of this approach has been reported from Vanas et al., which used NB to measure miRNA-21 in osteosarcoma cell lines, by demonstrating that its expression is involved in cell proliferation and regulation of cisplatin activity [62], while Liu J et al. described lncRNA PANDAR as new prognostic and therapeutic target in gastric cancer, on the bases of expression levels [63]. The main limitation of this method is the low sensitivity and the high time consumption. A large amount of total RNA for samples is required and this is very problematic for low-abundant miRNAs or limited cell or tissue source of RNA samples. Moreover, the use of isotope labeling in the classical protocol is hazardous and thus restricted by many institutions. In recent years, several improvements have been made to the classical method using non-radioactive labeling such as digoxigenin (DIG)-labeled modified probes [64]. Probes have been modified with locked nucleic acid (LNA) structure or biotin to increase affinity and sensitivity [65].
Reverse transcription quantitative PCR
The RT-qPCR is one of the most used method of detection and quantification of ncRNAs as easily to be incorporated in laboratory workflow. This method is often used to validate data obtained from microarray, as reported i.e. by He et al., which analyzed the differential expression profile of miRNAs in peripheral blood of lung cancer patients [66]. RT-qPCR technique include both TaqMan and SYBR green assays. The reverse transcription step varies on the basis of ncRNA of interest. In TaqMan assay, miRNAs are reverse transcribed using a specific stem loop RT primer, while SYBR green protocol includes the addition of a poly-A tail to miRNA sequence to allow primer binding. For lncRNA reverse transcription is performed using random primers or a specific RT primer followed by qPCR with real-time monitoring of reaction product accumulation using both TaqMan or SYBR green chemistry. Commercially available customizable plates and microfluidic cards can be designed either to examine a small set of ncRNAs or to provide more comprehensive coverage. Using RT-qPCR as a quantification method, it has been possible to demonstrate that the lncRNA HOTAIR is an independent predictor of metastatic spread and death in breast cancer patients [67].
In situ hybridization and fluorescence in situ hybridization
In recent years, advances in probe technologies and detection methods have improved ncRNA visualization by the application of ISH and FISH methods, based on the use of fluorescent probes binding the nucleic acid sequence presenting the highest degree of complementarity. FISH and ISH provide information regarding the spatial-temporal expression of ncRNAs and their subcellular localization providing novel information on ncRNA biological function. For example, confocal microscopy for FISH demonstrated that lncRNA NKILA exerts its critical function in cellular cytoplasm preventing NF-κB activation through stabilization of NF-κB/IκB complex by playing an essential role in turning off cancer-associated inflammation [68]; while ISH method applied to miRNAs allowed the identification of miR-375 downregulation as prognostic factor of esophageal squamous cell carcinoma [69]. The use of fluorophore-labeled DNA or RNA probes methods, is highly challenging due to the short length and the presence of repetitive sequence. Examples are the application of fluorophore-labeled multiple oligo probe sets [70], LNA probes [71] and branched-DNA probes [72]. The use of modified oligonucleotides, such as LNA or 2′-O-methyl (2OMe) [73] has significantly increased specificity and affinity to RNA targets. In particular, the use of hapten-labeled LNA oligos has been found to be highly advantageous in the detection of miRNAs in experimental and clinical tissue samples [74] whereas only a few reports are published for lncRNAs detection [75]. To overcome the limitations of these methods to detect low abundance of ncRNAs, researchers have developed and applied single-molecule RNA FISH based on hybridization of multiple short fluorescently labeled oligonucleotide of a single cell [76]. The use of a single oligo probe, optimally designed and with minimum cross-binding to other RNAs, reduces the risk of off-target probe hybridization.
Investigation of ncRNAs interactome
Non-coding RNAs exert their functions by direct interaction with other partners, which could be RNA in the case of miRNAs, and RNA, DNA and/or proteins in the case of lncRNAs.
In a previous manuscript we discussed about the integration of multi-omics data from different molecular levels in order to underline the complexity of the biological interactions. In that context we mentioned integrative analyses performed between transcriptomic data (e.g. miRNA and mRNA expression) together with genomics and epigenomics data (e.g. methylation profiling) to highlight the functional interactions between coding and non coding genes [77]. Here, we afford to point out the networks involving lncRNAs, miRNAs and coding genes in terms of cooperation and reciprocal regulation in the biological pathways which have a driver role in human cancer. The molecular mechanisms behind these interactions have been described, even tough technological advances allow the continuous updating and refinement of the understanding of these molecular events [78, 79]. MiRNAs work as negative regulators of coding transcripts by direct binding to mRNA. On the other hands, lncRNAs modulate the biologicial pathways through various mechanism at genomic, trascriptional and post-trascriptional level such as i) chromatin remodeling through histone modifications, ii) recruitment of transcription factors, iii) RNA polymerase II binding, iv) alternative splicing, v) mRNA stability, vi) recruitment of polysomes, vii) gene expression regulation in neighbor cells through extracellular vesicles and viii) miRNA interaction [80]. For what concerns the lncRNA-miRNA direct post-trascriptional interaction, 4 different mechanisms have been proposed: i) miRNA-triggered lncRNA decay, in which lncRNA degradation is induced by miRNA binding ii) lncRNA acting as miRNA sponge/decay, in which lncRNAs sequestrate miRNAs by removing their negative control on target mRNA iii) lncRNA-miRNA competition for mRNAregulation and iv) lncRNA generating miRNAs by alternative splicing [81].
Several studies focusing on non coding-coding genes interactions in human cancer, have been aimed to identify lncRNA-miRNA-mRNA axes which may promote tumor growth. For example Yu Lian et al. identified the role of the oncogenic lncRNA AFAP-AS1 to promote nasopharyngeal carcinoma metastasis by binding miR-423-5p and modulating the RAB11B and LASP1 coding genes involved in the Rho/Rac signaling pathway [82]. Han Li et al. demonstrated a feedback loop in the regulation of the malignant behaviors of glioma cells in which are involved the lncRNA SNHG1, microRNA-154-5p or miR-376b-3p and the coding gene FOXP2. This axis leads to the enhanced expression of KDM5B, which is an RNA-binding protein able increase the stability of SNHG1 [83]. A transcriptomic analysis of mRNA-lncRNA and miRNA interaction, performed by Xia Tang et al., revealed their synergistic network in hepatocellular carcinoma by highlighting the interaction between 16 miRNAs, 3 lncRNAs and 253 mRNAs [84]. Other functional network involving non coding and coding RNAs have been reported e.g. in breast cancer [85, 86], CRC [87], gastric cancer [88] and NSCLC [89].
Here we describe the most relevant methods developed to investigate the ncRNAs interactome, for example, dCHIRP (domain specific chromatin isolation by RNA purification) is a method for simultaneous mapping of RNA-RNA, RNA-DNA and RNA-protein interactions at single domain level [90]. We classified these methods on the basis of the interactors and on the technical approach.
Detection of RNA-RNA interaction
The interaction between two RNAs (inter-molecular) or between different regions of the same RNA molecule (intra-molecular) are one of mechanisms of the regulatory action of ncRNAs. Since computational methods can provide just a prediction of RNA-RNA interaction (RRI), different low and high- throughput methods have been developed to directly solve these molecular events [91].
Low-throughput techniques
RRI may be directly investigated through low-throughput biophysical and biochemical methods, such as electrophoretic mobility shift assay (EMSA), surface plasmon resonance (SPR) or single molecule forster resonance energy transfer (FRET). In EMSA, RNA fragments are extracted from cells and RRI is evaluated through electrophoresis based on molecular mass (larger in the case of interaction) [92]. In SPR, RRI is detected in real time through the immobilization of one RNA fragment on a sensor by streptavidin-biotin [93]. In FRET, the fragment is immobilized on quartz surface and the real time monitoring is based on the interaction of two fluorescent dyes in a closed space. These methods are not able to identify the precise region of the interaction. An example of the application of RRI techniques has been provided by Tianyou Liu et al., which characterized lncRNA DLEU1 in the context of CRC progression, and found by EMSA that DLEU1 directly binds SMARCA1 [94].
High-throughput targeted techniques
NGS technologies have been applied to investigate RRI at transcriptomic level. Among them, we mention i) crosslinking, ligation and sequencing hybrid (CLASH) and ii) hybrid and individual-nucleotide resolution ultraviolet cross-linking and Immunoprecipitation (HiCLIP RNA), which are able to identify duplex of two ligated RNAs, iii) RNA interactome analysis followed by deep sequencing (RIA-seq) and iv) RNA antisense purification and sequencing (RAP-seq) that explore the interactome for a target RNA. For example, Helwak et al. mapped the human miRNA interactome by CLASH and revealed non-canonical binding sites [95].
Transcriptome-wide techniques
The last frontier for RRI detection techniques is based on sequencing-based methods at transcriptome-wide level. i) psoralen analysis of RNA interactions and structures (PARIS) [96], ii) sequencing of psoralen crosslinked, ligated, and selected hybrids (SPLASH) [97] and iii) ligation of interacting RNA followed by high-throughput sequencing (LIGR-seq) [98] are three methods which differs in the isolation and enrichment of RNA-RNA duplex but all rely on cross-linking of RNAs, ligation of duplexes and high-throughput sequencing. These techniques allow the identification of all types of RRI, included unknown interactors and unexplored regions that can be mapped at high resolution.
Study of RNA- chromatin interaction: hybridization-based methods
To investigate lncRNAs binding sites on chromatin i) Chromatin isolation by RNA purification (ChIRP) ii) RNA antisense purification (RAP) and iii) capture hybridization analysis of RNA targets (CHART) are the most common experimental approaches [99]. i) ChIRP is a technology that enables the analysis of lncRNA-DNA complexes by the extraction of chromatin from cross-linked cultured cells, sonication, hybridization with biotinylated oligos and separation with magnetic streptavidin beads [100]. The output of the analysis depends from the method used, from the wet lab techniques, such as real-time PCR to the more recent and high-throughput methodologies, such as ChIRP-seq. ii) RAP differs from ChIRP for the use of longer antisense RNA probes with enhanced affinity to the target lncRNA. The products isolated with this method could undergo NGS analysis. iii) CHART similarly to ChIRP and RAP involves the purification of cross-linked RNA, DNA and proteins complexes, but differs for the use of short affinity-tagged oligonucleotides targeted to the region of predicted lncRNAs open binding sites [101]. An example of the application of hybridization-based approaches has been provided by Megan E. Forrest et al. who demonstrated by ChIRP-Seq the direct association of the colon cancer-upregulated lincDUSP with genes implicated in the replication-associated DNA damage response and in cell-cycle control [102].
Analysis of RNA-protein interaction
lncRNAs may interact with RNA-binding proteins (RBPs) to play their regulatory roles. Immunoprecipitation and affinity-based approaches have been developed to identify proteins involved in the functional complexes.
Immunoprecipitation-based methods
RNA Immunoprecipitation (RIP) is the most frequently used method to study lncRNA-protein interaction and is based on the immunoprecipitation of the complex by the use of an antibody directed to a target protein. After purification, lncRNAs can be analyzed through PCR, microarray (RIP-Chip) or NGS (RIP-seq) [103]. Subsequently, the method has been improved to map the precise binding sites, for example cross-linking and Immunoprecipitation (CLIP) differs from RIP in the use of UV radiation to cross-link RNA and binding proteins allowing stringent purification condition. In the last years, CLIP has been combined with other techniques such as NGS (CLIP-Seq) [104] and further modified to improve cross-linking efficiency and sequence read resolution (Photoactivatable ribonucleotide-enhanced cross-linking and immunoprecipitation: PAR-CLIP) [105]. Krell J et al. combined RIP-seq and PAR-CLIP-Seq to identify the precise binding site between AGO2-bound miRNAs and their mRNA targets, by determining the control of AGO2 loading by TP53 as a novel miRNA-mediated mechanism in cancer development [106].
However, CLIP or RIP are applicable only if an antibody against a specific protein is available. To overcome this limit, different approaches have been developed, such as RNA-tagging [107], or Targets of RNA-binding proteins Identified By Editing (TRIBE-seq) [108], which are able to detect and analyze protein-RNA interaction in vivo independently of a specific antibody, by using fusion proteins which bind the target RNAs.
Affinity-based methods
To move towards the identification of RNA interactors at the proteomic level, the best approach is Biotinylated RNA-protein pull-down followed by liquid chromatography-mass spectrometry/mass spectrometry (LC–MS/MS). In this method, target RNA is synthesized, labeled with biotinylated uridines, incubated with cellular lysates, pulled-down with streptavidin beads and finally, the RNA-binding protein complexes are separated through Sodium Dodecyl Sulphate - PolyAcrylamide Gel Electrophoresis (SDS-PAGE) and analyzed through MS. Anbang Wang et al. demonstrated through this technique that the lncRNA EGFR-AS1 interact with HuR, which affects mRNA stability of EGFR by promoting cell growth and metastasis in renal cancer [109]. However, in the last years, many approaches have been described to increase specificity or sensitivity such as ChIRP-MS [110].
Promising approaches for therapeutic intervention
The deregulation of ncRNAs in cancer cells, in term of expression profiling, interactome, as well as other intrinsic changes promoting tumor formation, offers the rational to consider them as a class of potential therapeutic targets. Given the diversity in their potential mode of action, several types of genomic and functional approaches have been developed to directly or indirectly target ncRNAs depending on whether they are oncogenes to be inhibited or tumor suppressors to be replaced. Among them we discuss about i) post-transcriptional RNA degradation using small interfering RNA (siRNAs) or synthetic antisense oligonucleotides (ASOs); ii) modulation of ncRNA genes by using genome-editing techniques; iii) replacement of ncRNAs; iv) inhibition of RNA–protein interactions or preventing secondary structure formation by using small molecules. We report several references of preclinical studies which highlight the power of these techniques in functional investigation. However, despite all these approaches are promising as therapeutics interventions, many barriers, for example in delivery systems, need to be overcome in the vision of their clinical translation.
ncRNAs targeting: ASOs
ASOs are synthetic nucleic acids sequences that binds, via Watson-Crick base pairing, to complementary RNA substrates. The two mechanisms of action of ASOs are the recruitment of RNase H to the DNA–RNA heteroduplex to degrade RNA [111] or the inhibition of biogenesis or translation [112]. Through chemical alterations of the natural nucleotides, ASOs have been designed to retain drug-like properties. The phosphorothioate modification of the linkage leads to ASOs protection from degradation by nucleases and to increase half-life in serum, while still supporting RNase H activities. These so-called first-generation ASOs composed solely of deoxy residues were limited in clinics. Second generation ASOs contain a central region of about 10 phosphorothioate DNA nucleotides flanked by nucleotides modified at the sugar (“gapmer” design). Third generation ASOs are instead composed from LNA modified antisense oligonucleotides gapmers which are enriched with LNA in the flanking regions and DNA in a LNA-free central gap. LNAs are nucleic acid analogs in which the ribose ring is “locked” by a methylene bridge between the 2′ oxygen and the 4′ carbon. A representativstudy about the use of ASOs for functional ncRNAs validation has been reported from Amodio et al., who demonstrated that the inhibition of lncRNA MALAT1 by a LNA-gapmeR antisense oligonucleotide, antagonizes cell proliferation and triggers apoptosis both in multiple myeloma cell lines and in a murine xenograft model [7]. The anti-multiple myeloma activity of miRNA-221 has been instead demonstrated through its inhibition in vitro and in vivo mediated from a specific LNA-i-miR [113, 114].
ncRNAs targeting: RNAi
RNA interference (RNAi) is an endogenous and well-conserved post-transcriptional modulation mechanism, which works through paring of endogenous or exogenous dsRNA with a target mRNA. Specifically, a dsRNA is firstly cleaved in a 21-RNA sequence, called siRNA, by Dicer and then is loaded in RISC (RNA induced silencing complex), which is located in cytosol. Here the passenger strand is discarded, the guide strand is paired with target mRNA and, depending on complementarity, silencing is induced through degradation or translational repression [115, 116]. This physiological mechanism has been frequently applied experimentally for therapeutic task in molecular oncology and then modified to perform high throughput screening by using pools of siRNAs. As matter of fact, several libraries targeting miRNAs and lncRNAs have been developed and led to the identification of ncRNAs affecting drug response or cancer pathways [116]. For example, the use of genome wide miRNA libraries allowed i) the discovery of miR 195 synergic role in microtubule targeting agent response in lung cancer [117] ii) the identification of several miRNAs relevant in trastuzumab resistance in HER2 positive cells [118] iii) the discovery of miRNAs determining navitoclax susceptibility in CRC cell lines [119]. The application of high-throughput siRNA-based screening targeting lncRNAs clarified the oncogenic role of linc0015226 and an unprecedented reported role of DRAIC in autophagy regulation in breast cancer cells [120]. On the other hands, synthetic siRNAs has been also used as therapeutic tools inducing ncRNA degradation. For example, a siRNA-mediated HOTAIR degradation suggested a therapeutic role of HOTAIR inhibition, since its negative regulation reduced tumor cells dissemination in an in vitro breast cancer model [67]. Moreover, a siRNA-based inhibition of MALAT1 suggested its critical role in temozolomide resistance in glioblastoma multiforme, since its inhibition restored drug sensitivity attenuating cancer stem cells stemness and proliferation [121].
CRISPR-Cas9 ncRNAs genomic editing
In the past 10 years, several methods for genome editing have been developed such as Zinc-finger nucleases (ZFNs), transcription activator like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeats (CRISPR)-associated nuclease 9 (CRISPR/Cas9) [122]. Here, we focus on CRISPR/Cas9 system, which represents the last revolution in biological research, especially for ncRNAs study. This system works as a molecular “scissor” and has been developed by modifying the adaptive prokaryotic immune system in order to induce a well-defined genetic change in eukaryotic cells through a “guide RNA” and Cas9 protein. The guide RNA (gRNA) is 20 nucleotides in length and is homologous to a specific region of the target DNA flanking a 3 DNA base pair protospacer adjacent motif (PAM)-sequence recognized from the Cas9, which is an endonuclease able to induce a double stranded break (DSB). The Cas9-mediated DSB may be repaired by non-homologous end joining (NHEJ) by inducing non-in frame small insertion or deletions that disrupts the targeted locus (knock-out (KO) approach), or by homology directed repair (HDR) in the case of a donor DNA is supplied to insert a desired sequence (knock-in) [123]. Several validation studies have been performed through this strategy to investigate the function of selected lncRNAs or miRNAs in solid and hematological malignancies [124,125,126]. For example, through the CRISPR-Cas9 system, the roles of i) LncRoR as activator of MAPK/ERK pathway [127], ii) LncAK023948 as positive regulator in Akt pathway [128] and iii) LncBC200 as promoter of cell growth have been demonstrated in breast cancer [129]. CRISPR-Cas9 approach has been also used to reduce the expression of miRNAs up to 96% in vitro and in vivo by targeting the miRNAs biogenesis site. This KO approach resulted more robust, precise and stable respect than other techniques available for loss of function studies (such as antisense inhibitors) [130]. Different applications of CRISPR-Cas9 system also provided the possibility to disclose the oncosuppressive role of miR-210 in renal cell carcinoma cell lines [131] and of miRNA182-5p in chronic myeloid leukemia [132]. Furthermore, several studies focused on the possible delivery strategies for the use of CRISPR-Cas9 system as technology for miRNA therapeutics [133, 134].
Considering the high impact of this technology on molecular studies [135], CRISPR-Cas9 system have been further modified to induce genetic changes increasingly precise and sophisticated, up to base editing level. At this aim, Cas9 protein has been fused to specific domains in order to work as stimulator or suppressor of genetic transcription (CRISPR-activation or CRISPR-interference system, respectively) [136,137,138,139] and to induce transient loss of function (LOF) or gain of function (GOF).
Moreover, the last progress in this context is represented from the use of CRISPR-Cas9 pooled library, which consist in thousands of plasmids encoding for multiple barcoded gRNAs targeting different genes simultaneously, with a strong reduction of time and costs related to functional validation experiments [140]. A representative study of CRISPR-interference based screening has been performed by Liu et al. in 7 transformed cell lines by targeting ~ 16.000 lncRNAs. They identified 499 lncRNA loci involved in cellular growth and tissue specific transcriptional regulation [141]. Kurata et al. identified cell fitness-associated miRNAs with a miRNA-based CRISPR-Cas9 pooled library targeting ~ 1600 annotated human miRNA stem-loops [142]. By using a genome wide CRISPR-Cas9 LOF screen, Wallace et al. identified miRNAs involved in myeloid leukemia cell growth, of which miR-155 was the top candidate [143]. Moreover, CRISPR/Cas9-based synergistic activation mediator (SAM) system revealed the role of lncRNA AK023948 as positive regulator of AKT in breast cancer [128]. A genome-scale deletion screen of ~ 12.000 lncRNAs through a paired-guide RNA pooled library allowed the identification of 51 lncRNAs involved in a positive or negative regulation of tumor growth [144]. The major limit of this technology is represented by off-target effects, even though several approaches are going to be applied to overcome or at least reduce this important issue [145, 146]. For what concerns clinical translation, CRISPR-Cas9-based approaches are still in its infancy, especially because of the eventuality of adverse immune response due to bacterial Cas9, generally delivered by viral vectors, and because of ethical issues intrinsic to genome editing applications in human [123].
ncRNAs replacement
Many relevant ncRNAs in cancer are genomically deleted or downregulated acting as tumor suppressors, whereby their reactivation may have anticancer activity. Replacement strategies are widely applied to restore the functionality of tumor suppressor miRNAs using double-stranded RNA of 22-mer oligonucleotides sharing the same sequence of mature miRNA or its precursor, and for this reason they are known as mimics. A well known example is the replacement of miR-34a, a tumor suppressor that is lost or expressed at reduced levels in a broad range of tumor types [147]. The exogenous introduction of miR-34a mimics in vitro showed inhibition of cell proliferation, migration and invasion, alone or in combo with anticancer therapies [148]. These results have lead to the first clinical application of a liposomal formulation of miR-34a mimic (MIRX34) in clinics [149]. Another approach is the use of synthetic RNA molecules able to mimic hairpin structures of lncRNAs. An example is GAS5, a lncRNA that acts as a decoy for the glucocorticoid receptor (GCR) blocking the transcription of target genes [150]. To overcome GAS5 loss of function due to acquired mutations in the GCR response element sequence, Pickard et al. reported the generation of an oligonucleotide that mimic the mutated region sequence on breast cancer cells showing pro-apoptotic activity similar to wild-type GAS5 [151].
Limitation of RNA-based therapies
The reactivation of tumor suppressor ncRNAs as well as the use of nucleic acids-based methods requires the availability of efficient in vivo delivery systems to overcome biological drawbacks associated with such strategies. A first barrier is the transport across the cell membranes limited to diffusion of small and relatively hydrophobic compounds. In addition, RNA molecules show short half-life in vivo environments due to the highly risk of degradation by cellular nuclease such exonuclease or endonuclease. An important issue is also the activation of innate immune response to foreign RNAs through toll-like receptor and retinoic acid inducible gene I protein pathways. This results in the production of type 1 interferon and subsequent release of inflammation associated cytokines. Finally, it is important to avoid, or at least predict and recognize, off-target effects and to reduce the toxicity. At this aim siRNA and ASOs as well as ncRNAs can be encapsulated inside lipid-based nanoparticles in order to ensure its survival against biological agents and delivered into cancer cells, also in a target specific manner. In addition, the development of N-acetylgalactosamide (GalNAc) conjugated to siRNAs has enhanced hepatic uptake [152]. To extend delivery to other tissues, alternative conjugation methods including lipids such as cholesterol, peptide nucleic acids (PNAs), and antibodies have been used.
Small molecules
Although the RNA therapeutics research is mainly focused on oligonucleotides, the application of SMs to target specific ncRNAs have emerged as a feasible and efficient strategy and may in part overcome limitation of RNA approaches based on Watson and Crick hybridization. Computational biology can allow the identification, prediction of docking sites and design of these SMs, as we mentioned above. Moreover, the advantages of SMs are their chemical nature that make them suitable for conventional drug development. In contrast, limitations are poor specificity and complex design compared with sequence-specific methods. SMs exert their therapeutic effect on ncRNAs by specific binding to secondary or tertiary structures as miRNA hairpin precursors or structural elements of lncRNAs such as the triple-helical structure of MALAT1 and NEAT1. In this way, SMs can destabilize the transcript or allosterically interfere with the interaction between the RNA and its protein partners. An additional mechanism of action of SMs is the binding to the Dicer or Drosha nuclease processing sites, which could affect the biogenesis of miRNAs. The development of bioinformatics tools allowed to perform high-throughput screening of ncRNA libraries against datasets of small molecules identifying strong interactions. Using this strategy Li et al. tested the ability of targaprimir-96, a bleomycin A5 conjugate, to target pri-miR-96. This compound directly block pri-miR-96 maturation via Drosha leading to the upregulation of miR-96 target FOXO1 and the induction of apoptosis in breast cancer cells [153]. Similarly, Haga et al. showed that inhibition of Dicer cleavage sites in pre-miR-544 resulting in sensitization of breast cancer cells to hypoxic stress [154].
From bench to bedside: ncRNAs in clinical practice, promise or challenge?
As we discussed above, numerous preclinical studies are focusing on ncRNAs characterization with the aim to clarify their role in tumorigenesis and to disclose their contribution for diagnostic, prognostic and therapeutic purposes. We reported several strategies, which emphasized the promising use of ncRNAs for cancer treatment. Now, we discuss about the bench to bedside translation of the ncRNAs therapeutics in the ongoing clinical trials. MiRNAs are the most extensively studied as both therapeutic candidates or targets [155, 156], followed by lncRNAs, which are emerging in the clinical setting [157,158,159,160,161].
For example, it is noteworthy that ncRNAs may play a crucial role in chemo and radio resistance, which is the major challenge of current anticancer treatments [162, 163].
On https://clinicaltrials.gov website are reported 304 studies involving miRNAs in clinical applications, of which 101 are interventional studies at different phases. Among them, we mention the phase I clinical trial NCT02369198 based on the administration of TargomiRs as 2nd or 3rd line treatment for patients with recurrent malignant pleural mesothelioma and NSCLC. This drug consists in a miR-16-based microRNA mimic, nanoparticles for delivery and an anti-EGFR bispecific antibody. Moreover 13 studies are reported concerning lncRNAs, of which 11 are observational and 3 interventional (NCT02641847 phase I/II, NCT02221999 phase II/III and NCT03000764 -phase not applicable-). However, in the majority of cases, miRNAs and lncRNAs are evaluated only in term of expression profiling to validate them as biomarkers, while RNA-based therapeutics or SMs are not yet clinical interventions under widespread investigation. This points the need to work hard to translate the large and promising preclinical studies in early clinical trials, This scenario strongly indicates the need of highly multidisciplinary efforts to make “dark matter” a major mean in the fight against cancer in the next future.
Conclusion
Here we have reviewed research strategies aimed to investigate the role of miRNAs and lncRNAs in cancer. The availability of new powerful sequencing and molecular technologies allowed the overcoming of several potential caveats, such as the low abundance of ncRNAs, the subcellular spatial localization and their instability. Improvements in wet laboratory techniques together with in silico tools significantly improved the knowledge of the “dark matter” of the genome in terms of discovery, annotation and functional validation. Apart from the most widely adopted methods that we have described, other strategies have emerged to improve the global characterization of ncRNAs in the last 10 years (Table 2) and the optimization of these methods is still ongoing. Notwithstanding, in some cases, it is not possible to completely clarify the function of non-coding transcripts out of a physiological context, especially because are poorly conserved between species, making the in vivo experiments not easily translatable for applications in humans and because, if compared to coding genes, are more difficult to be explored. A lot of novel ncRNAs are completely uncharacterized by making more complex the understanding of their role. In addition, for the majority of lncRNAs, crystallographic structures leading the design of SMs are still unknown. Moreover, despite the rapid evolution of ncRNAs targeting methods (ASOs, SMs, etc) provides an exciting rationale for clinical applications, several obstacles still stand in the way, such as delivery strategies, stability, specificity and toxicity of the treatments. Further advances in the next future are expected to better clarify the regulatory network behind ncRNAs perturbations, and mostly to move experimental results from bench to bedside.
Availability of data and materials
Not applicable.
Abbreviations
- ncRNAs:
-
Non-Coding RNAs
- miRNAs:
-
MicroRNAs
- lncRNAs:
-
Long Non Coding RNAs
- NGS:
-
Next Generation Sequencing
- IVT:
-
InVitro Transcription
- SAGE:
-
Serial Analysis of Gene Expression
- RNA-seq:
-
RNA Sequencing
- CRC:
-
Colorectal Cancer
- DP-seq:
-
Designed Primer-based Sequencing
- CAGE:
-
Cap Analysis Gene Expression
- TSS:
-
Transcription Start Sites
- NSCLC:
-
Non-Small Cell Lung Cancer
- MFE:
-
Minimal Folding Energy
- HMM:
-
Hidden Markov Model
- SCFG:
-
Stochastic Context Free Grammar
- SMs:
-
Small Molecules
- NB:
-
Northern Blot
- RT-qPCR:
-
Reverse Transcription Quantitative PCR
- ISH:
-
In Situ Hybridization
- FISH:
-
Fluorescence In Situ Hybridization
- DIG:
-
Digoxigenin
- LNA:
-
Locked Nucleic Acid
- RRI:
-
RNA-RNA Interaction
- EMSA:
-
Electrophoretic Mobility Shift Assay
- SPR:
-
Surface Plasmon Resonance
- FRET:
-
Forster Resonance Energy Transfer
- CLASH:
-
Crosslinking, Ligation and Sequencing Hybrid
- HiCLIP:
-
Hybrid and Individual-Nucleotide Resolution Ultraviolet Cross-Linking and Immunoprecipitation
- RIA-seq:
-
RNA Interactome Analysis Followed by Sequencing
- RAP-seq:
-
RNA Antisense Purification and Sequencing
- PARIS:
-
Psoralen Analysis of RNA Interactions and Structures
- SPLASH:
-
Sequencing of Psoralen Crosslinked, Ligated, and Selected Hybrids
- LIGR-seq:
-
Ligation of Interacting RNA followed by Sequencing
- ChIRP:
-
Chromatin Isolation by RNA Purification
- RAP:
-
RNA Antisense Purification
- CHART:
-
Capture Hybridization Analysis of RNA targets
- RBPs:
-
RNA-Binding Proteins
- RIP:
-
RNA Immunoprecipitation
- CLIP:
-
Cross-Linking and Immunoprecipitation
- PAR-CLIP:
-
Photoactivatable Ribonucleotide-Enhanced Cross-Linking and Immunoprecipitation
- TRIBE-seq:
-
Targets of RNA-Binding Proteins Identified By Editing followed by Sequencing
- LC–MS/MS:
-
Liquid chromatography-Mass Spectrometry/Mass Spectrometry
- SDS-PAGE:
-
Sodium Dodecyl Sulphate - PolyAcrylamide Gel Electrophoresis
- SiRNAs:
-
Small Interfering RNA
- ASOs:
-
Synthetic Antisense Oligonucleotides
- RNAi:
-
RNA Interference
- RISC:
-
RNA Induced Silencing Complex
- ZFNs:
-
Zinc-Finger Nucleases
- TALENs:
-
Transcription Activator Like Effector Nucleases
- CRISPR:
-
Clustered Regularly Interspaced Short Palindromic Repeats
- gRNA:
-
Guide RNA
- PAM:
-
Protospacer Adjacent Motif
- DSB:
-
Double Stranded Break
- NHEJ:
-
Non-Homologous End Joining
- KO:
-
Knock-Out
- HDR:
-
Homology Directed Repair
- LOF:
-
Loss Of Function
- GOF:
-
Gain Of Function
- SAM:
-
Synergistic Activation Mediator
- GCR:
-
Glucocorticoid Receptor
- GalNAc:
-
N-Acetylgalactosamide
- PNAs:
-
Peptide Nucleic Acids
- PA:
-
Public Available
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–74.
Arun G, Diermeier SD, Spector DL. Therapeutic targeting of long non-coding RNAs in Cancer. Trends Mol Med. 2018;24(3):257–77.
Chandra Gupta S, Nandan TY. Potential of long non-coding RNAs in cancer patients: from biomarkers to therapeutic targets. Int J Cancer. 2017;140(9):1955–67.
Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18(1):5–18.
Stamato MA, Juli G, Romeo E, Ronchetti D, Arbitrio M, Caracciolo D, et al. Inhibition of EZH2 triggers the tumor suppressive miR-29b network in multiple myeloma. Oncotarget. 2017;8(63):106527–37.
Amodio N, Stamato MA, Juli G, Morelli E, Fulciniti M, Manzoni M, et al. Drugging the lncRNA MALAT1 via LNA gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia. 2018;32(9):1948–57.
Amodio N, Bellizzi D, Leotta M, Raimondi L, Biamonte L, D'Aquila P, et al. miR-29b induces SOCS-1 expression by promoter demethylation and negatively regulates migration of multiple myeloma and endothelial cells. Cell Cycle. 2013;12(23):3650–62.
Tokgun PE, Tokgun O, Kurt S, Tomatir AG, Akca H. MYC-driven regulation of long non-coding RNA profiles in breast cancer cells. Gene. 2019;714:143955.
Adriaens C, Marine JC. NEAT1-containing paraspeckles: central hubs in stress response and tumor formation. Cell Cycle. 2017;16(2):137–8.
Caracciolo D, Di Martino MT, Amodio N, Morelli E, Montesano M, Botta C, et al. miR-22 suppresses DNA ligase III addiction in multiple myeloma. Leukemia. 2019;33(2):487–98.
Tang Q, Zheng F, Liu Z, Wu J, Chai X, He C, et al. Novel reciprocal interaction of lncRNA HOTAIR and miR-214-3p contribute to the solamargine-inhibited PDPK1 gene expression in human lung cancer. J Cell Mol Med. 2019.
Pritchard CC, Cheng HH, Tewari M. MicroRNA profiling: approaches and considerations. Nat Rev Genet. 2012;13(5):358–69.
Wong NK, Huang CL, Islam R, Yip SP. Long non-coding RNAs in hematological malignancies: translating basic techniques into diagnostic and therapeutic strategies. J Hematol Oncol. 2018;11(1):131.
Zhou M, Hu L, Zhang Z, Wu N, Sun J, Su J. Recurrence-associated long non-coding RNA signature for determining the risk of recurrence in patients with Colon Cancer. Mol Ther Nucleic Acids. 2018;12:518–29.
Liang F, Yang M, Tong N, Fang J, Pan Y, Li J, et al. Identification of six key miRNAs associated with breast cancer through screening large-scale microarray data. Oncol Lett. 2018;16(4):4159–68.
Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, et al. Global identification of human transcribed sequences with genome tiling arrays. Science. 2004;306(5705):2242–6.
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270(5235):484–7.
Matsumura H, Kruger DH, Kahl G, Terauchi R. SuperSAGE: a modern platform for genome-wide quantitative transcript profiling. Curr Pharm Biotechnol. 2008;9(5):368–74.
Gibb EA, Vucic EA, Enfield KS, Stewart GL, Lonergan KM, Kennett JY, et al. Human cancer long non-coding RNA transcriptomes. PLoS One. 2011;6(10):e25915.
Yamada A, Yu P, Lin W, Okugawa Y, Boland CR, Goel A. A RNA-sequencing approach for the identification of novel long non-coding RNA biomarkers in colorectal cancer. Sci Rep. 2018;8(1):575.
Yu N, Yong S, Kim HK, Choi YL, Jung Y, Kim D, et al. Identification of tumor suppressor miRNAs by integrative miRNA and mRNA sequencing of matched tumor-normal samples in lung adenocarcinoma. Mol Oncol. 2019;13(6):1356–68.
Gawronski KAB, Kim J. Single cell transcriptomics of noncoding RNAs and their cell-specificity. Wiley Interdiscip Rev RNA. 2017;8:6.
Bhargava V, Ko P, Willems E, Mercola M, Subramaniam S. Quantitative transcriptomics using designed primer-based amplification. Sci Rep. 2013;3:1740.
Sasagawa Y, Nikaido I, Hayashi T, Danno H, Uno KD, Imai T, et al. Quartz-Seq: a highly reproducible and sensitive single-cell RNA sequencing method, reveals non-genetic gene-expression heterogeneity. Genome Biol. 2013;14(4):R31.
Horie M, Kaczkowski B, Ohshima M, Matsuzaki H, Noguchi S, Mikami Y, et al. Integrative CAGE and DNA methylation profiling identify epigenetically regulated genes in NSCLC. Mol Cancer Res. 2017;15(10):1354–65.
Rossi M, Di Martino MT, Morelli E, Leotta M, Rizzo A, Grimaldi A, et al. Molecular targets for the treatment of multiple myeloma. Curr Cancer Drug Targets. 2012;12(7):757–67.
Arbitrio M, Di Martino MT, Barbieri V, Agapito G, Guzzi PH, Botta C, et al. Identification of polymorphic variants associated with erlotinib-related skin toxicity in advanced non-small cell lung cancer patients by DMET microarray analysis. Cancer Chemother Pharmacol. 2016;77(1):205–9.
Scionti F, Di Martino MT, Sestito S, Nicoletti A, Falvo F, Roppa K, et al. Genetic variants associated with Fabry disease progression despite enzyme replacement therapy. Oncotarget. 2017;8(64):107558–64.
Di Martino MT, Scionti F, Sestito S, Nicoletti A, Arbitrio M, Hiram Guzzi P, et al. Genetic variants associated with gastrointestinal symptoms in Fabry disease. Oncotarget. 2016;7(52):85895–904.
Olson NE. The microarray data analysis process: from raw data to biological significance. NeuroRx. 2006;3(3):373–83.
Pirim H, Eksioglu B, Perkins A, Yuceer C. Clustering of high throughput gene expression data. Comput Oper Res. 2012;39(12):3046–61.
Liao P, Li S, Cui X, Zheng Y. A comprehensive review of web-based resources of non-coding RNAs for plant science research. Int J Biol Sci. 2018;14(8):819–32.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Brueffer C, Saal LH. TopHat-recondition: a post-processor for TopHat unmapped reads. BMC Bioinformatics. 2016;17(1):199.
Sun L, Zhang Z, Bailey TL, Perkins AC, Tallack MR, Xu Z, et al. Prediction of novel long non-coding RNAs based on RNA-Seq data of mouse Klf1 knockout study. BMC Bioinformatics. 2012;13:331.
Achawanantakun R, Chen J, Sun Y, Zhang Y. LncRNA-ID: long non-coding RNA IDentification using balanced random forests. Bioinformatics. 2015;31(24):3897–905.
Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. CPAT: coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013;41(6):e74.
Agapito G, Guzzi PH, Cannataro M. DMET-miner: efficient discovery of association rules from pharmacogenomic data. J Biomed Inform. 2015;56:273–83.
Malek E, Jagannathan S, Driscoll JJ. Correlation of long non-coding RNA expression with metastasis, drug resistance and clinical outcome in cancer. Oncotarget. 2014;5(18):8027–38.
Zhou M, Sun Y, Sun Y, Xu W, Zhang Z, Zhao H, et al. Comprehensive analysis of lncRNA expression profiles reveals a novel lncRNA signature to discriminate nonequivalent outcomes in patients with ovarian cancer. Oncotarget. 2016;7(22):32433–48.
Song H, Sun W, Ye G, Ding X, Liu Z, Zhang S, et al. Long non-coding RNA expression profile in human gastric cancer and its clinical significances. J Transl Med. 2013;11:225.
Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016;26(3):304–19.
Emamjomeh A, Zahiri J, Asadian M, Behmanesh M, Fakheri BA, Mahdevar G. Identification, prediction and data analysis of noncoding RNAs: a review. Med Chem. 2019;15(3):216–30.
Panwar B, Arora A, Raghava GP. Prediction and classification of ncRNAs using structural information. BMC Genomics. 2014;15:127.
Hermann T, Patel DJ. RNA bulges as architectural and recognition motifs. Structure. 2000;8(3):R47–54.
Lee JC, Gutell RR. Diversity of base-pair conformations and their occurrence in rRNA structure and RNA structural motifs. J Mol Biol. 2004;344(5):1225–49.
Clote P, Ferre F, Kranakis E, Krizanc D. Structural RNA has lower folding energy than random RNA of the same dinucleotide frequency. RNA. 2005;11(5):578–91.
Mathews DH, Moss WN, Turner DH. Folding and finding RNA secondary structure. Cold Spring Harb Perspect Biol. 2010;2(12):a003665.
Rahimi A, Sedighi R, Emadi-Baygi M, Honardoost MA, Mowla SJ, Khanahmad H, et al. Bioinformatics prediction and experimental validation of a novel microRNA: hsa-miR-B43 within human CDH4 gene with a potential metastasis-related function in breast cancer. J Cell Biochem. 2019.
Raad M, Bayat A, Sharafshah A, Amiri AZ, Zohour MM, Ahmadvand M. Association and in silico investigations of miR-302c insertion/deletion variant as a novel biomarker with susceptibility to gastric cancer. J Cell Biochem. 2019;120(11):18946–55.
Yoon BJ. Hidden Markov models and their applications in biological sequence analysis. Curr Genomics. 2009;10(6):402–15.
Vorozheykin PS, Titov, II. [Web server for prediction of miRNAs and their precursors and binding sites]. Mol Biol (Mosk). 2015;49(5):846–53.
Voss B. Structural analysis of aligned RNAs. Nucleic Acids Res. 2006;34(19):5471–81.
Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33(Database issue):D121–4.
Disney MD, Winkelsas AM, Velagapudi SP, Southern M, Fallahi M, Childs-Disney JL. Inforna 2.0: a platform for the sequence-based Design of Small Molecules Targeting Structured RNAs. ACS Chem Biol. 2016;11(6):1720–8.
Abulwerdi FA, Xu W, Ageeli AA, Yonkunas MJ, Arun G, Nam H, et al. Selective small-molecule targeting of a triple Helix encoded by the long noncoding RNA, MALAT1. ACS Chem Biol. 2019;14(2):223–35.
Rocca R, Talarico C, Moraca F, Costa G, Romeo I, Ortuso F, et al. Molecular recognition of a carboxy pyridostatin toward G-quadruplex structures: why does it prefer RNA? Chem Biol Drug Des. 2017;90(5):919–25.
Rocca R, Moraca F, Costa G, Nadai M, Scalabrin M, Talarico C, et al. Identification of G-quadruplex DNA/RNA binders: Structure-based virtual screening and biophysical characterization. Biochim Biophys Acta Gen Subj. 2017;1861(5 Pt B):1329–40.
Donlic A, Morgan BS, Xu JL, Liu A, Roble C Jr, Hargrove AE. Discovery of small molecule ligands for MALAT1 by tuning an RNA-binding scaffold. Angew Chem Int Ed Engl. 2018;57(40):13242–7.
Ren Y, Wang YF, Zhang J, Wang QX, Han L, Mei M, et al. Targeted design and identification of AC1NOD4Q to block activity of HOTAIR by abrogating the scaffold interaction with EZH2. Clin Epigenetics. 2019;11(1):29.
Vanas V, Haigl B, Stockhammer V, Sutterluty-Fall H. MicroRNA-21 increases proliferation and Cisplatin sensitivity of osteosarcoma-derived cells. PLoS One. 2016;11(8):e0161023.
Liu J, Ben Q, Lu E, He X, Yang X, Ma J, et al. Long noncoding RNA PANDAR blocks CDKN1A gene transcription by competitive interaction with p53 protein in gastric cancer. Cell Death Dis. 2018;9(2):168.
Huang Q, Mao Z, Li S, Hu J, Zhu Y. A non-radioactive method for small RNA detection by northern blotting. Rice (N Y). 2014;7(1):26.
Gao Z, Peng Y. A highly sensitive and specific biosensor for ligation- and PCR-free detection of microRNAs. Biosens Bioelectron. 2011;26(9):3768–73.
He Q, Fang Y, Lu F, Pan J, Wang L, Gong W, et al. Analysis of differential expression profile of miRNA in peripheral blood of patients with lung cancer. J Clin Lab Anal. 2019:e23003.
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–6.
Liu B, Sun L, Liu Q, Gong C, Yao Y, Lv X, et al. A cytoplasmic NF-kappaB interacting long noncoding RNA blocks IkappaB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015;27(3):370–81.
Li J, Li X, Li Y, Yang H, Wang L, Qin Y, et al. Cell-specific detection of miR-375 downregulation for predicting the prognosis of esophageal squamous cell carcinoma by miRNA in situ hybridization. PLoS One. 2013;8(1):e53582.
Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5(10):877–9.
Thomsen R, Nielsen PS, Jensen TH. Dramatically improved RNA in situ hybridization signals using LNA-modified probes. RNA. 2005;11(11):1745–8.
Battich N, Stoeger T, Pelkmans L. Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat Methods. 2013;10(11):1127–33.
Molenaar C, Marras SA, Slats JC, Truffert JC, Lemaitre M, Raap AK, et al. Linear 2′ O-methyl RNA probes for the visualization of RNA in living cells. Nucleic Acids Res. 2001;29(17):E89.
Jorgensen S, Baker A, Moller S, Nielsen BS. Robust one-day in situ hybridization protocol for detection of microRNAs in paraffin samples using LNA probes. Methods. 2010;52(4):375–81.
Zhang A, Zhao JC, Kim J, Fong KW, Yang YA, Chakravarti D, et al. LncRNA HOTAIR enhances the androgen-receptor-mediated transcriptional program and drives castration-resistant prostate Cancer. Cell Rep. 2015;13(1):209–21.
Kocks C, Boltengagen A, Piwecka M, Rybak-Wolf A, Rajewsky N. Single-molecule fluorescence in situ hybridization (FISH) of circular RNA CDR1as. Methods Mol Biol. 1724;2018:77–96.
Gallo Cantafio ME, Grillone K, Caracciolo D, Scionti F, Arbitrio M, Barbieri V, et al. From Single Level Analysis to Multi-Omics Integrative Approaches: A Powerful Strategy towards the Precision Oncology. High Throughput. 2018;7:4.
Sun B, Liu C, Li H, Zhang L, Luo G, Liang S, et al. Research progress on the interactions between long non-coding RNAs and microRNAs in human cancer. Oncol Lett. 2020;19(1):595–605.
Chan JJ, Tay Y. Noncoding RNA:RNA Regulatory Networks in Cancer. Int J Mol Sci. 2018;19:5.
Fernandes JCR, Acuna SM, Aoki JI, Floeter-Winter LM, Muxel SM. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Noncoding RNA. 2019;5:1.
Yoon JH, Abdelmohsen K, Gorospe M. Functional interactions among microRNAs and long noncoding RNAs. Semin Cell Dev Biol. 2014;34:9–14.
Lian Y, Xiong F, Yang L, Bo H, Gong Z, Wang Y, et al. Long noncoding RNA AFAP1-AS1 acts AS a competing endogenous RNA of miR-423-5p to facilitate nasopharyngeal carcinoma metastasis through regulating the rho/Rac pathway. J Exp Clin Cancer Res. 2018;37(1):253.
Li H, Xue Y, Ma J, Shao L, Wang D, Zheng J, et al. SNHG1 promotes malignant biological behaviors of glioma cells via microRNA-154-5p/miR-376b-3p- FOXP2- KDM5B participating positive feedback loop. J Exp Clin Cancer Res. 2019;38(1):59.
Tang X, Feng D, Li M, Zhou J, Li X, Zhao D, et al. Transcriptomic analysis of mRNA-lncRNA-miRNA interactions in hepatocellular carcinoma. Sci Rep. 2019;9(1):16096.
Crudele F, Bianchi N, Reali E, Galasso M, Agnoletto C, Volinia S. The network of non-coding RNAs and their molecular targets in breast cancer. Mol Cancer. 2020;19(1):61.
Zhang Y, Li Y, Wang Q, Zhang X, Wang D, Tang HC, et al. Identification of an lncRNAmiRNAmRNA interaction mechanism in breast cancer based on bioinformatic analysis. Mol Med Rep. 2017;16(4):5113–20.
Liang Y, Zhang C, Ma MH, Dai DQ. Identification and prediction of novel non-coding and coding RNA-associated competing endogenous RNA networks in colorectal cancer. World J Gastroenterol. 2018;24(46):5259–70.
Mao Y, Liu R, Zhou H, Yin S, Zhao Q, Ding X, et al. Transcriptome analysis of miRNA-lncRNA-mRNA interactions in the malignant transformation process of gastric cancer initiation. Cancer Gene Ther. 2017;24(6):267–75.
Li DY, Chen WJ, Luo L, Wang YK, Shang J, Zhang Y, et al. Prospective lncRNA-miRNA-mRNA regulatory network of long non-coding RNA LINC00968 in non-small cell lung cancer A549 cells: a miRNA microarray and bioinformatics investigation. Int J Mol Med. 2017;40(6):1895–906.
Quinn JJ, Ilik IA, Qu K, Georgiev P, Chu C, Akhtar A, et al. Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nat Biotechnol. 2014;32(9):933–40.
Gong J, Ju Y, Shao D, Zhang QC. Advances and challenges towards the study of RNA-RNA interactions in a transcriptome-wide scale. Quantitative Biology. 2018;6(3):239–52.
Bak G, Han K, Kim KS, Lee Y. Electrophoretic mobility shift assay of RNA-RNA complexes. Methods Mol Biol. 2015;1240:153–63.
Di Primo C, Dausse E, Toulme JJ. Surface plasmon resonance investigation of RNA aptamer-RNA ligand interactions. Methods Mol Biol. 2011;764:279–300.
Liu T, Han Z, Li H, Zhu Y, Sun Z, Zhu A. LncRNA DLEU1 contributes to colorectal cancer progression via activation of KPNA3. Mol Cancer. 2018;17(1):118.
Helwak A, Kudla G, Dudnakova T, Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153(3):654–65.
Lu Z, Gong J, Zhang QC. PARIS: Psoralen analysis of RNA interactions and structures with high throughput and resolution. Methods Mol Biol. 1649;2018:59–84.
Aw JGA, Shen Y, Nagarajan N, Wan Y. Mapping RNA-RNA interactions globally using Biotinylated Psoralen. J Vis Exp. 2017;123.
Sharma E, Sterne-Weiler T, O'Hanlon D, Blencowe BJ. Global mapping of human RNA-RNA interactions. Mol Cell. 2016;62(4):618–26.
Kashi K, Henderson L, Bonetti A, Carninci P. Discovery and functional analysis of lncRNAs: methodologies to investigate an uncharacterized transcriptome. Biochim Biophys Acta. 2016;1859(1):3–15.
Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44(4):667–78.
Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, et al. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci U S A. 2011;108(51):20497–502.
Forrest ME, Saiakhova A, Beard L, Buchner DA, Scacheri PC, LaFramboise T, et al. Colon Cancer-Upregulated long non-coding RNA lincDUSP regulates cell cycle genes and potentiates resistance to apoptosis. Sci Rep. 2018;8(1):7324.
Gagliardi M, Matarazzo MR. RIP: RNA Immunoprecipitation. Methods Mol Biol. 2016;1480:73–86.
Bottini S, Pratella D, Grandjean V, Repetto E, Trabucchi M. Recent computational developments on CLIP-seq data analysis and microRNA targeting implications. Brief Bioinform. 2018;19(6):1290–301.
Garzia A, Morozov P, Sajek M, Meyer C, Tuschl T. PAR-CLIP for discovering target sites of RNA-binding proteins. Methods Mol Biol. 1720;2018:55–75.
Krell J, Stebbing J, Frampton AE, Carissimi C, Harding V, De Giorgio A, et al. The role of TP53 in miRNA loading onto AGO2 and in remodelling the miRNA-mRNA interaction network. Lancet. 2015;385 Suppl 1:S15.
Lapointe CP, Wilinski D, Saunders HA, Wickens M. Protein-RNA networks revealed through covalent RNA marks. Nat Methods. 2015;12(12):1163–70.
McMahon AC, Rahman R, Jin H, Shen JL, Fieldsend A, Luo W, et al. TRIBE: hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell. 2016;165(3):742–53.
Wang A, Bao Y, Wu Z, Zhao T, Wang D, Shi J, et al. Long noncoding RNA EGFR-AS1 promotes cell growth and metastasis via affecting HuR mediated mRNA stability of EGFR in renal cancer. Cell Death Dis. 2019;10(3):154.
Chu C, Chang HY. ChIRP-MS: RNA-directed proteomic discovery. Methods Mol Biol. 1861;2018:37–45.
Wu H, Lima WF, Zhang H, Fan A, Sun H, Crooke ST. Determination of the role of the human RNase H1 in the pharmacology of DNA-like antisense drugs. J Biol Chem. 2004;279(17):17181–9.
Rigo F, Seth PP, Bennett CF. Antisense oligonucleotide-based therapies for diseases caused by pre-mRNA processing defects. Adv Exp Med Biol. 2014;825:303–52.
Gulla A, Di Martino MT, Gallo Cantafio ME, Morelli E, Amodio N, Botta C, et al. A 13 mer LNA-i-miR-221 inhibitor restores drug sensitivity in Melphalan-refractory multiple myeloma cells. Clin Cancer Res. 2016;22(5):1222–33.
Gallo Cantafio ME, Nielsen BS, Mignogna C, Arbitrio M, Botta C, Frandsen NM, et al. Pharmacokinetics and Pharmacodynamics of a 13-mer LNA-inhibitor-miR-221 in Mice and Non-human Primates. Mol Ther Nucleic Acids. 2016;5:6.
Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431(7006):343–9.
Mohr SE, Smith JA, Shamu CE, Neumuller RA, Perrimon N. RNAi screening comes of age: improved techniques and complementary approaches. Nat Rev Mol Cell Biol. 2014;15(9):591–600.
Yu X, Zhang Y, Ma X, Pertsemlidis A. miR-195 potentiates the efficacy of microtubule-targeting agents in non-small cell lung cancer. Cancer Lett. 2018;427:85–93.
Leivonen SK, Sahlberg KK, Makela R, Due EU, Kallioniemi O, Borresen-Dale AL, et al. High-throughput screens identify microRNAs essential for HER2 positive breast cancer cell growth. Mol Oncol. 2014;8(1):93–104.
Notzold L, Frank L, Gandhi M, Polycarpou-Schwarz M, Gross M, Gunkel M, et al. The long non-coding RNA LINC00152 is essential for cell cycle progression through mitosis in HeLa cells. Sci Rep. 2017;7(1):2265.
Tiessen I, Abildgaard MH, Lubas M, Gylling HM, Steinhauer C, Pietras EJ, et al. A high-throughput screen identifies the long non-coding RNA DRAIC as a regulator of autophagy. Oncogene. 2019;38(26):5127–41.
Kim SS, Harford JB, Moghe M, Rait A, Pirollo KF, Chang EH. Targeted nanocomplex carrying siRNA against MALAT1 sensitizes glioblastoma to temozolomide. Nucleic Acids Res. 2018;46(3):1424–40.
Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31(7):397–405.
Yang J, Meng X, Pan J, Jiang N, Zhou C, Wu Z, et al. CRISPR/Cas9-mediated noncoding RNA editing in human cancers. RNA Biol. 2018;15(1):35–43.
Zare K, Shademan M, Ghahramani Seno MM, Dehghani H. CRISPR/Cas9 knockout strategies to ablate CCAT1 lncRNA gene in Cancer cells. Biol Proced Online. 2018;20:21.
Peng L, Pan P, Chen J, Yu X, Wu J, Chen Y. A tetracycline-inducible CRISPR/Cas9 system, targeting two long non-coding RNAs, suppresses the malignant behavior of bladder cancer cells. Oncol Lett. 2018;16(4):4309–16.
Zhen S, Hua L, Liu YH, Sun XM, Jiang MM, Chen W, et al. Inhibition of long non-coding RNA UCA1 by CRISPR/Cas9 attenuated malignant phenotypes of bladder cancer. Oncotarget. 2017;8(6):9634–46.
Peng WX, Huang JG, Yang L, Gong AH, Mo YY. Linc-RoR promotes MAPK/ERK signaling and confers estrogen-independent growth of breast cancer. Mol Cancer. 2017;16(1):161.
Koirala P, Huang J, Ho TT, Wu F, Ding X, Mo YY. LncRNA AK023948 is a positive regulator of AKT. Nat Commun. 2017;8:14422.
Singh R, Gupta SC, Peng WX, Zhou N, Pochampally R, Atfi A, et al. Regulation of alternative splicing of Bcl-x by BC200 contributes to breast cancer pathogenesis. Cell Death Dis. 2016;7(6):e2262.
Chang H, Yi B, Ma R, Zhang X, Zhao H, Xi Y. CRISPR/cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci Rep. 2016;6:22312.
Yoshino H, Yonemori M, Miyamoto K, Tatarano S, Kofuji S, Nohata N, et al. microRNA-210-3p depletion by CRISPR/Cas9 promoted tumorigenesis through revival of TWIST1 in renal cell carcinoma. Oncotarget. 2017;8(13):20881–94.
Arya D, Sachithanandan SP, Ross C, Palakodeti D, Li S, Krishna S. MiRNA182 regulates percentage of myeloid and erythroid cells in chronic myeloid leukemia. Cell Death Dis. 2017;8(1):e2547.
Zhen S, Takahashi Y, Narita S, Yang YC, Li X. Targeted delivery of CRISPR/Cas9 to prostate cancer by modified gRNA using a flexible aptamer-cationic liposome. Oncotarget. 2017;8(6):9375–87.
Mout R, Ray M, Yesilbag Tonga G, Lee YW, Tay T, Sasaki K, et al. Direct cytosolic delivery of CRISPR/Cas9-Ribonucleoprotein for efficient gene editing. ACS Nano. 2017;11(3):2452–8.
Aquino-Jarquin G. Emerging role of CRISPR/Cas9 technology for MicroRNAs editing in Cancer research. Cancer Res. 2017;77(24):6812–7.
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154(2):442–51.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173–83.
La Russa MF, Qi LS. The new state of the art: Cas9 for gene activation and repression. Mol Cell Biol. 2015;35(22):3800–9.
Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517(7536):583–8.
Esposito R, Bosch N, Lanzos A, Polidori T, Pulido-Quetglas C, Johnson R. Hacking the Cancer genome: profiling therapeutically actionable long non-coding RNAs using CRISPR-Cas9 screening. Cancer Cell. 2019;35(4):545–57.
Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D, et al. CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science. 2017;355:6320.
Kurata JS, Lin RJ. MicroRNA-focused CRISPR-Cas9 library screen reveals fitness-associated miRNAs. RNA. 2018;24(7):966–81.
Wallace J, Hu R, Mosbruger TL, Dahlem TJ, Stephens WZ, Rao DS, et al. Genome-wide CRISPR-Cas9 screen identifies MicroRNAs that regulate myeloid leukemia cell growth. PLoS One. 2016;11(4):e0153689.
Zhu S, Li W, Liu J, Chen CH, Liao Q, Xu P, et al. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat Biotechnol. 2016;34(12):1279–86.
Kimberland ML, Hou W, Alfonso-Pecchio A, Wilson S, Rao Y, Zhang S, et al. Strategies for controlling CRISPR/Cas9 off-target effects and biological variations in mammalian genome editing experiments. J Biotechnol. 2018;284:91–101.
Tycko J, Wainberg M, Marinov GK, Ursu O, Hess GT, Ego BK, et al. Mitigation of off-target toxicity in CRISPR-Cas9 screens for essential non-coding elements. Nat Commun. 2019;10(1):4063.
Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17(2):193–9.
Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR, et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: in vitro and in vivo evidence. Clin Cancer Res. 2012;18(22):6260–70.
Hong DS, Kang YK, Borad M, Sachdev J, Ejadi S, Lim HY, et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br J Cancer. 2020.
Pickard MR, Williams GT. Molecular and cellular mechanisms of action of tumour suppressor GAS5 LncRNA. Genes (Basel). 2015;6(3):484–99.
Pickard MR, Williams GT. The hormone response element mimic sequence of GAS5 lncRNA is sufficient to induce apoptosis in breast cancer cells. Oncotarget. 2016;7(9):10104–16.
Bajan S, Hutvagner G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells. 2020;9:1.
Li Y, Disney MD. Precise small molecule degradation of a noncoding RNA identifies cellular binding sites and modulates an oncogenic phenotype. ACS Chem Biol. 2018;13(11):3065–71.
Haga CL, Velagapudi SP, Childs-Disney JL, Strivelli J, Disney MD, Phinney DG. Rapid generation of miRNA inhibitor leads by bioinformatics and efficient high-throughput screening methods. Methods Mol Biol. 2017;1517:179–98.
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16(3):203–22.
Smolle MA, Calin HN, Pichler M, Calin GA. Noncoding RNAs and immune checkpoints-clinical implications as cancer therapeutics. FEBS J. 2017;284(13):1952–66.
Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. 2017;16(3):167–79.
Tang Q, Hann SS. HOTAIR: an oncogenic long non-coding RNA in human Cancer. Cell Physiol Biochem. 2018;47(3):893–913.
Meng J, Li P, Zhang Q, Yang Z, Fu S. A four-long non-coding RNA signature in predicting breast cancer survival. J Exp Clin Cancer Res. 2014;33:84.
Zhou M, Zhao H, Wang Z, Cheng L, Yang L, Shi H, et al. Identification and validation of potential prognostic lncRNA biomarkers for predicting survival in patients with multiple myeloma. J Exp Clin Cancer Res. 2015;34:102.
Jariwala N, Sarkar D. Emerging role of lncRNA in cancer: a potential avenue in molecular medicine. Ann Transl Med. 2016;4(15):286.
Wang WT, Han C, Sun YM, Chen TQ, Chen YQ. Noncoding RNAs in cancer therapy resistance and targeted drug development. J Hematol Oncol. 2019;12(1):55.
Corra F, Agnoletto C, Minotti L, Baldassari F, Volinia S. The network of non-coding RNAs in Cancer drug resistance. Front Oncol. 2018;8:327.
Wilkinson KA, Merino EJ, Weeks KM. Selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE): quantitative RNA structure analysis at single nucleotide resolution. Nat Protoc. 2006;1(3):1610–6.
Owens MC, Clark SC, Yankey A, Somarowthu S. Identifying Structural Domains and Conserved Regions in the Long Non-Coding RNA lncTCF7. Int J Mol Sci. 2019;20:19.
Schmidt K, Weidmann CA, Hilimire TA, Yee E, Hatfield BM, Schneekloth JS Jr, et al. Targeting the oncogenic long non-coding RNA SLNCR1 by blocking its sequence-specific binding to the androgen receptor. Cell Rep. 2020;30(2):541–54 e5.
Saus E, Willis JR, Pryszcz LP, Hafez A, Llorens C, Himmelbauer H, et al. nextPARS: parallel probing of RNA structures in Illumina. RNA. 2018;24(4):609–19.
Bartonicek N, Maag JL, Dinger ME. Long noncoding RNAs in cancer: mechanisms of action and technological advancements. Mol Cancer. 2016;15(1):43.
Fang Y, Fullwood MJ. Roles, functions, and mechanisms of long non-coding RNAs in Cancer. Genomics Proteomics Bioinformatics. 2016;14(1):42–54.
Uzilov AV, Underwood JG. High-throughput nuclease probing of RNA structures using FragSeq. Methods Mol Biol. 2016;1490:105–34.
Zampetaki A, Albrecht A, Steinhofel K. Long non-coding RNA structure and function: is there a link? Front Physiol. 2018;9:1201.
Sakurai M, Yano T, Kawabata H, Ueda H, Suzuki T. Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat Chem Biol. 2010;6(10):733–40.
Barbieri I. Kouzarides T. Nat Rev Cancer: Role of RNA modifications in cancer; 2020.
Imamachi N, Tani H, Mizutani R, Imamura K, Irie T, Suzuki Y, et al. BRIC-seq: a genome-wide approach for determining RNA stability in mammalian cells. Methods. 2014;67(1):55–63.
Tani H, Numajiri A, Aoki M, Umemura T, Nakazato T. Short-lived long noncoding RNAs as surrogate indicators for chemical stress in HepG2 cells and their degradation by nuclear RNases. Sci Rep. 2019;9(1):20299.
Tani H, Mizutani R, Salam KA, Tano K, Ijiri K, Wakamatsu A, et al. Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res. 2012;22(5):947–56.
Zhu S, Qing T, Zheng Y, Jin L, Shi L. Advances in single-cell RNA sequencing and its applications in cancer research. Oncotarget. 2017;8(32):53763–79.
Lee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343(6177):1360–3.
Ashouri A, Sayin VI, Van den Eynden J, Singh SX, Papagiannakopoulos T, Larsson E. Pan-cancer transcriptomic analysis associates long non-coding RNAs with key mutational driver events. Nat Commun. 2016;7:13197.
Sun M, Gadad SS, Kim DS, Kraus WL. Discovery, annotation, and functional analysis of long noncoding RNAs controlling cell-cycle gene expression and proliferation in breast Cancer cells. Mol Cell. 2015;59(4):698–711.
Acknowledgements
Not applicable.
Funding
This work has been partially supported by Italian Association for Cancer Research (AIRC) Investigator Grant “Small molecule-based targeting of lncRNAs 3D structure: a translational platform for treatment of multiple myeloma” Project N. 21588; AIRC Special Program for Molecular Clinical Oncology-5 per mille 2010–15 and its Extension Program 2016–18 N. 9980: “Phase I studies of LNA-i-miR-221 and LNA Gapmer 17-92 inhibitor in refractory multiple myeloma patients”; CARICAL foundation “microRNA-based immunotherapeutic strategies for Multiple Myeloma and Chronic Lymphocytic Leukemia” Project N. 16695.
Author information
Authors and Affiliations
Contributions
KG contributed to conception, design and revision of the manuscript sections, wrote sections of the manuscript, designed Fig.1 and created Table 1. CR, FS, RR, GT, PHG, SA, MTDM wrote sections of the manuscript. RR designed Fig.2 and Fig.3. CR created Table 2. PT and PT supervised the manuscript by providing critical feedback and revision. The authors read and approved the final manuscript.
Authors’ information
(optional)
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Grillone, K., Riillo, C., Scionti, F. et al. Non-coding RNAs in cancer: platforms and strategies for investigating the genomic “dark matter”. J Exp Clin Cancer Res 39, 117 (2020). https://doi.org/10.1186/s13046-020-01622-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-020-01622-x