
Abstract
Background
Glioblastoma (GBM) is an aggressive brain tumor with universal recurrence and poor prognosis. The recurrence is largely driven by chemoradiation resistant cancer stem cells (CSCs). Epidermal growth factor receptor (EGFR) and its mutant EGFRvIII are amplified in ~ 60% and ~ 30% of GBM patients, respectively; however, therapies targeting EGFR have failed to improve disease outcome. EGFRvIII-mediated cross-activation of tyrosine kinase receptor, cMET, regulates GBM CSC maintenance and promote tumor recurrence. Here, we evaluated the efficacy of pan-EGFR inhibitor afatinib and Temozolomide (TMZ) combination on GBM in vitro and in vivo.
Methods
We analyzed the effect of afatinib and temozolomide (TMZ) combination on GBM cells U87MG and U251 engineered to express wild type (WT) EGFR, EGFRvIII or EGFRvIII dead kinase, CSCs isolated from U87 and U87EGFRvIII in vitro. The therapeutic utility of the drug combination was investigated on tumor growth and progression using intracranially injected U87EGFRvIII GBM xenografts.
Results
Afatinib and TMZ combination synergistically inhibited the proliferation, clonogenic survival, motility, invasion and induced senescence of GBM cells compared to monotherapy. Mechanistically, afatinib decreased U87EGFRvIII GBM cell proliferation and motility/invasion by inhibiting EGFRvIII/AKT, EGFRvIII/JAK2/STAT3, and focal adhesion kinase (FAK) signaling pathways respectively. Interestingly, afatinib specifically inhibited EGFRvIII-cMET crosstalk in CSCs, resulting in decreased expression of Nanog and Oct3/4, and in combination with TMZ significantly decreased their self-renewal property in vitro. More interestingly, afatinib and TMZ combination significantly decreased the xenograft growth and progression compared to single drug alone.
Conclusion
Our study demonstrated significant inhibition of GBM tumorigenicity, CSC maintenance in vitro, and delayed tumor growth and progression in vivo by combination of afatinib and TMZ. Our results warrant evaluation of this drug combination in EGFR and EGFRvIII amplified GBM patients.
Similar content being viewed by others


Background
Glioblastoma (GBM) accounts for 45.6% of malignant brain tumors and is universally fatal [1]. Despite the multimodality treatment options available to newly diagnosed GBM patients including surgical resection, radiotherapy and temozolomide (TMZ)-based concomitant and adjuvant chemotherapy [2], tumor recurrence is inevitable and results in poor median progression-free survival (PFS) (6.9 months) [3]. Scant progress has been made in the last decade to improve survival [4, 5].
Several histologic and cancer genome sequencing studies have revealed deregulation of EGFR and its downstream signaling pathways in GBM [6,7,8]. Specifically, 30–60% of primary GBM patients carry EGFR amplification [9, 10], and ~ 50–60% of GBM tumors with EGFR amplification also carry constitutively active EGFR variant III (EGFRvIII) [11,12,13]. In addition to EGFR, mutations in ERBB2/HER2 are also reported in 7–15% of GBM patients [14, 15]. EGFR family members control cell differentiation, proliferation, survival, and migration, while aberrant activation of these receptors results in persistent activation of the PI3K/AKT/mTOR and Ras/Raf/ERK signaling pathways implicated in the development and progression of several tumors including GBM [16]. In addition, EGFR/EGFRvIII signaling maintains GBM cancer stem cells (CSCs) also called side population (SP) [17,18,19] and control tumor progression, recurrence, and resistance to chemoradiation therapy (CRT) [20, 21]. Though CSCs from many tumors overexpress EGFR [22, 23], GBM CSCs show resistance to anti-EGFR therapies by compensatory upregulation of HER2 and HER3 [24]. In addition, co-activation of other receptor tyrosine kinases (RTKs) has also been implicated in limiting the efficacy of anti-EGFR therapies [25]. Recently, EGFRvIII was reported to cross-activate cMET RTK signaling [25,26,27], and result in increased growth and enrichment of GBM CSCs [28, 29]. Immunohistochemical analysis also showed increased co-expression of stem cell markers and cMET in GBM patient specimens [28], and patient-derived neurospheres [29]. Furthermore, cMET co-precipitates with EGFR in GBM patient biopsies and mouse xenografts [30]. Interestingly, the addition of either cMET or PDGFRα inhibitor along with erlotinib (a first generation EGFR inhibitor) significantly suppressed GBM cell growth compared to erlotinib alone. These findings highlight the need to develop therapies targeting both EGFR family members and co-activators in GBM cells for effective growth inhibition and prevention of recurrent tumor development. [25].
Afatinib is an FDA-approved, irreversible inhibitor [31] that blocks activation of EGFR, HER2, HER4, and EGFRvIII by irreversibly binding to their ATP binding site [32, 33]. Recent studies have shown a significant increase in the PFS of afatinib-treated non-small-cell lung cancer (NSCLC) patients with EGFR mutation compared to patients treated with pemetrexed plus cisplatin or gemcitabine plus cisplatin [34, 35]. Interestingly, afatinib also significantly improved the PFS of EGFR mutant NSCLC patients with brain metastases [36]. In addition, afatinib significantly increased overall survival (OS) and PFS in lung squamous cell carcinoma patients compared to erlotinib [37]. Though afatinib is shown to cross the blood-brain barrier (BBB) [38], a recent study showed no improvement in non-selected recurrent GBM patients [39]. Intriguingly, afatinib significantly increased OS (six fold) of a patient with recurrent GBM overexpressing EGFR and EGFRvIII [40].
TMZ is a DNA alkylating agent and the standard chemotherapeutic drug for GBM. TMZ in combination with radiation therapy (RT) and adjuvant significantly increased the OS (14.6 months vs 12.1 months) of GBM patients compared to RT alone [3]. Though previous study demonstrated no pharmacokinetic alteration of TMZ upon co-administration with afatinib [39], however, therapeutic efficacy and the molecular mechanism(s) of this combination is still unknown.
We analyzed the efficacy of afatinib and TMZ combination in EGFRvIII-amplified GBM using in vitro and in vivo models. Our study revealed that the combination of afatinib and TMZ synergistically decreased cell proliferation, clonogenicity, invasion, and motility of U87EGFRvIII and U251EGFRvIII cells in vitro and significantly inhibited the growth of U87EGFRvIII orthotopic xenografts in vivo. Our mechanistic studies revealed that afatinib reduces CSCs and tumor growth by inhibiting EGFRvIII-mediated cMET and JAK2/STAT3 pathway activation, enhancing TMZ-induced cytotoxicity.
Materials and methods
Temozolomide (TMZ) and afatinib were obtained from Sigma-Aldrich (T2577; St. Louis, MO) and Selleck chemicals (S1011; Houston, TX), respectively. The 24 well plate cell culture inserts (#3422) and BioCoat™ Matrigel® invasion chambers (#354480, Corning Incorporated, USA) were used to analyze migration and invasion respectively. All antibodies used in this study are summarized in Additional file 4: Table S1.
Cell culture
Human GBM cell lines U87MG and U87 cells transfected with either EGFR WT, EGFRvIII or EGFRvIII DK (dead kinase) were a generous gift from Dr. Webster K. Cavenee (University of California San Diego, CA, USA), and U251 cells transfected with EGFR WT or EGFRvIII under the control of tetracycline (Tet)-inducible promoter were gifted by Dr. Amyn A. Habib (University of Texas Southwestern Medical Center, Dallas, TX, USA). All cell lines were cultured as described earlier [41]. Cell line authentication was done by PCR-based STR analysis at the University of Arizona genetics core, Tucson, AZ.
MTT assay
The cytotoxicity of TMZ- and afatinib-treated GBM cells was measured by MTT assay as described earlier [42] Briefly, U87MG (3 × 103/well) and U87EGFRvIII (2 × 103/well) cells were seeded in a 96-well plate overnight and incubated with drugs or vehicle [(0.02% dimethyl sulfoxide (DMSO)] for 24–72 h at 37 °C. MTT (5 mg/ml) was added and cells were incubated for 4 h at 37 °C. Formazan crystals were dissolved in DMSO, plates were read at 570 nm using a Spectra MAX 190 plate reader (Molecular Devices, LA, USA), and viable cells were calculated [42].
Combination index (CI)
The combination effects or index (CI) of TMZ and afatinib was calculated using CompuSyn software as described earlier [43]. A CI < 1 means synergistic effect, whereas CI equal to 1 or > 1 indicate additive and antagonist effects, respectively.
Clonogenic survival and soft agar assay
Both the colony formation and soft agar assays were done as described earlier [42]. Briefly, 2000 cells/well were seeded in a six-well plate with complete media. After overnight incubation, cells were treated with either 0.02% of DMSO, 1 μM of afatinib, 25 μM of TMZ or combination for 48 h. After washing with PBS, cells were allowed to grow in 2% media for 12 days, and colonies were fixed in methanol and stained with crystal violet solution. Colonies were dissolved in 10% acetic acid [44] and absorbance was measured at 570 nm using a Spectra MAX 190 plate reader.
For soft agar assay, plates were coated with 0.5% agarose, and 5000 cells containing DMEM with 20% FBS and 0.25% agarose were seeded on the top of base agar. The next day, cells were treated with vehicle, 1 μM of afatinib, 25 μM of TMZ or combination for 30 days with fresh media change every third day. Colonies were stained with crystal violet solution (0.1% crystal violet in 20% methanol) and counted [42]. Colony sizes of ≥70 μm was considered as big colonies [45].
Cell cycle analysis and SA-β-gal staining
The effect of afatinib and TMZ on the cell cycle was analyzed by flow cytometry as described earlier [46]. Quantitative in situ senescence-associated β-galactosidase (SA-β-gal) staining was done as described earlier [47]. Images were taken using light microscopy, and multiple representative areas (n = 7) were randomly selected for the quantification of SA-β-gal positive cells.
Migration and invasion assay
The transwell migration and invasion assays were done as described earlier [42]. Briefly, after treating U87MG and U87EGFRvIII cells with drugs for 48 h, 2.5 × 104 and 5.0 × 104 cells each were re-suspended in 100 μl serum-free DMEM medium with TMZ (25 μM), afatinib (1 μM), or combination and applied on the upper chamber of non-coated and Matrigel-coated transwell chambers for motility and invasion, respectively. The bottom chamber was filled with 600 μl DMEM containing 10% FBS. After 16 h of incubation at 37 °C in a CO2 incubator, non-migrated cells in the upper chamber were removed using a cotton swab and migrated/invaded cells were stained with Diff-Quick® cell stain kit (Dade-Behring Inc., Newark, DE, USA) and counted using Image J software.
Side population assay
Flow cytometry was used to analyze the SP/CSCs as described previously [48]. Briefly, 1 × 106 cells/ml of 10% FBS containing DMEM were incubated with 5 μg/mL Hoechst 33342 (AnaSpec Inc., Fremont, CA, USA) for 60 min at 37 °C. CSCs were sorted by FACS analysis using LSR II Green (BD Biosciences). Verapamil (50 μM/ml), an ABC transporters inhibitor, was used as a control to identify CSCs.
Neurosphere assay
Cells were cultured in DMEM-F12 (Invitrogen-Life Technologies), together with basic fibroblast growth factor (bFGF) (10 ng/mL), epidermal growth factor (EGF) (20 ng/mL), leukemia inhibitory factor (LIF) (10 ng/mL) and 10% knockout serum (all from Sigma). Two-hundred μL of the medium containing 2000 cells/well were plated in 96-well low attachment culture plates. Those spheres with a diameter of ≥100 μm within each well were counted after 10 days of culture. Images were captured using a Carl Zeiss microscope.
Intracranial injection and bioluminescence imaging
All animal experiments were carried out according to protocols approved by the Institutional Animal Care and Use Committee (IACUC). Intracranial injection into four to six-week-old athymic nude mice was done as described earlier with slight modifications [49]. Briefly, mice were anesthetized by intraperitoneal (i.p.) injection of ketamine and xylazine and immobilized on a stereotactic frame (Stoelting Co, IL, USA). A Hamilton syringe with a 26-gauge needle was inserted at 1-mm dorsal and 2-mm lateral to the bregma to a depth of 3.5-mm and then pulled back 0.5-mm to allow space for tumor cells. Following this, U87EGFRvIII luciferase-transfected cells (2 × 104 in 2 μl of PBS) were injected at an injection speed of 0.5 μl/min. After 5 days, luciferase substrate was injected (i.p. 100 μl of 5 mM CycLuc1) and tumor growth was measured using bioluminescence imaging after 10 min in an IVIS spectrum, Caliper life sciences (PerkinElmer, MA, USA). Tumor volume (photon flux) of the mice was measured using Living Image® software, PerkinElmer, MA, USA. After 5 days of tumor implantation, based on tumor volume, animals were randomized into four groups and treated with vehicle, TMZ (25 mg/kg BW), afatinib (10 mg/kg BW) or combination for 5 days a week by oral gavage for 30 days (treatment start date was considered as day 1). Vehicle-treated and afatinib-treated animals were sacrificed when they were very weak. TMZ and combination group animals were euthanized after 30 days.
Statistical analysis
Each experiment was repeated at least three times and data were expressed as mean values ± SD. The student t-test and ANOVA were used to determine significant differences between the groups with p-values <0.05 considered statistically significant.
Results
Afatinib and TMZ combination differentially inhibit the proliferation and clonogenic survival of EGFR and EGFRvIII expressing GBM cells
EGFRvIII is known to increase the proliferation, survival and modulate therapeutic response of cancer cells [50]. We validated the expression of EGFR, EGFRvIII and EGFRvIII DK in U87MG and U251 cells by western blot analysis (Fig. 1a), and analyzed the proliferation rates by MTT assay. In concordance with the previous report [51], we observed significantly increased (p < 0.0001) proliferation of U87EGFRvIII cells compared to parental U87MG, EGFR and EGFRvIII DK over expressed cells (Fig. 1b). To analyze the cytotoxic effects of TMZ and afatinib on GBM, U87MG and U87EGFRvIII cells were treated with varying concentrations of TMZ (10–500 μM) or afatinib (0.25–5.0 μM) for 48–72 h and analyzed by MTT assay. A dose-dependent decrease in the viability of GBM cells was observed following TMZ and afatinib treatment. The inhibitory concentration of 25% (IC25) values was approximately 25 μM and 300 μM for TMZ, and 2 μM and 1 μM of afatinib for U87MG and U87EGFRvIII cells, respectively. Recently, a pharmacokinetic analysis on 35 GBM patients revealed that approximately 10–25 μM concentrations of TMZ reaches Cerebrospinal Fluid (CSF) [52, 53] and therefore to mimic the in vivo conditions, we used 25 μM of TMZ and 1 μM afatinib for all our in vitro experiments. Accordingly, our CI plot revealed an additional and near synergistic decrease in cell viability upon combining afatinib with TMZ in U87EGFRvIII cells (Additional file 1: Figure S1 A-F).

Afatinib and TMZ combination differentially inhibit proliferation and clonogenic survival of EGFR and EGFRvIII expressing GBM cells. a, b EGFR and EGFRvIII expression differentially effect cell proliferation. a U87MG, U87EGFR WT, U87EGFRvIII, U87EGFRvIII DK, U251, U251EGFR WT (Tet-inducible system) and U251EGFRvIII (Tet-inducible system) cell lysates were analyzed for EGFR (full length), EGFRvIII and pEGFR (Tyr-1068). b The graph shows increased proliferation rate of U87EGFRvIII cells compared to U87MG, U87EGFR WT, and U87EGFRvIII DK cells. Tet - tetracycline. c, d U87MG, U87EGFR WT, U87EGFRvIII and U87EGFRvIII DK cells were seeded and treated as specified for 48 h. After washing with PBS, cells were allowed to grow for 12 days in 2% media. Colonies formed were fixed with methanol, stained with crystal violet solution, dissolved in 10% acetic acid and absorbance was measured at 570 nm. The graph shows the mean (± SD) percentage of colony formation. The experiment was repeated three times (*$ P ≤ 0.05); * significant compared to control; $ significant compared to TMZ. e, f Afatinib and TMZ combination decreases the anchorage-independent growth of TMZ-resistant EGFRvIII GBM cells. U87MG, U87 EGFR WT, U87EGFRvIII and U87EGFRvIII DK cells (5 × 103) were seeded with 0.25% agarose on the top of the 0.5% base agar. After overnight incubation, cells were incubated with TMZ (25 μM) or afatinib (1 μM) or combination of both for a month. The 0.1% crystal violet stained colonies were counted (n = 3). The graph shows mean (± SD) number of small (blue) and big colonies (orange). (* P ≤ 0.05); * number of small colonies, significant compared to control; # number of big colonies, significant compared to control
Colony formation assay revealed that both afatinib and TMZ significantly decreased the clonogenicity of GBM cells U87MG, U87EGFR WT, U87EGFRvIII, and U87EGFRvIII DK compared to vehicle-treated control cells (Fig. 1c, d). TMZ inhibited colony formation more than afatinib in U87MG, U87EGFR WT, and U87EGFRvIII DK cells, while U87EGFRvIII cells were relatively resistant (Fig. 1c, d). However, combining afatinib with TMZ abolished the colony forming ability of U87EGFRvIII cells (Fig. 1c, d). The percentage decreases in colony formation of U87MG cells were 96.3 ± 0.3, 58 ± 6.2, 98.4 ± 0.7 for TMZ (p = 0.01), afatinib (p = 0.03) and TMZ plus afatinib (p = 0.01) compared to vehicle-treated control, respectively. The percentage decreases in colony formation of U87 EGFR WT were 66.5 ± 1.8, 41.4 ± 1.9 and 80.5 ± 1.2 for TMZ, afatinib and combination group compared to control (p = 0.001), respectively. U87EGFRvIII formed bigger colonies than U87MG, U87EGFR WT, and U87EGFRvIII DK cells. Similarly, the mean percentage decreases in colonies formed by U87EGFRvIII were 89.6 ± 3.0, 56.6 ± 7.0 and 94.5 ± 0.6 for TMZ-, afatinib- and combination-treated groups compared to control, respectively. In addition, afatinib plus TMZ significantly decreased EGFRvIII colonies when compared to either TMZ (p = 0.048) or afatinib (p = 0.005) alone. The percentage decreases in colony formation of U87EGFRvIII DK were 85.0 ± 2.3, 78.3 ± 1.5 and 95.3 ± 0.3 for TMZ (p = 0.02), afatinib (p = 0.02) and combination group (p = 0.01) when compared to control, respectively (Fig. 1d). Overall, combination treatment significantly decreased U87MG, U87EGFR WT, and U87EGFRvIII colony growth when compared to TMZ alone (p = 0.01). Similar results were observed for U251 and U251EGFRvIII GBM cells (data not shown).
We analyzed whether afatinib could block the anchorage-independent growth of U87EGFRvIII cells, since EGFRvIII plays an important role in cancer cell proliferation [51], and anchorage-independent growth predicts in vivo tumorigenicity [54]. TMZ significantly decreased the colony size, as well as the number of colonies in U87MG, U87EGFR WT, and U87EGFRvIII DK, while TMZ had less effect on EGFRvIII cells. (Fig. 1e). TMZ alone or in combination with afatinib significantly decreased the number of U87MG small colonies (p = 0.03) and big colonies (p = 0.02) as well. TMZ and afatinib individually significantly decreased the number of U87EGFR WT big colonies (p = 0.05) while TMZ and afatinib combined significantly decreased both the number of small colonies (p = 0.03) and big colonies (p = 0.03). Afatinib or combination treatment significantly decreased the number of U87EGFRvIII small [(afatinib (p = 0.02); combination (p = 0.01)] and big colonies (afatinib (p = 0.02); combination (p = 0.01)). TMZ or afatinib or combination treatment significantly decreased the number of U87EGFRvIII DK number of small colonies (p = 0.03), while TMZ and combination treatment significantly decreased the number of big colonies [TMZ (p = 0.048); combination (p = 0.038)]. Overall, combination therapy with afatinib completely abrogated anchorage-independent growth of GBM cells regardless of the activation status of EGFR (Fig. 1f).
Afatinib inhibits EGFR activation in GBM cells
We examined the effect of afatinib alone or in combination with TMZ on EGFR activation by immunoblotting. Afatinib treatment significantly inhibited EGFR activation (phosphorylated EGFR) in U87MG, U87EGFR WT, U87EGFRvIII, and U87EGFRvIII DK, whereas TMZ had no effect (Fig. 2a, b). Interestingly, we observed that afatinib inhibited pEGFR longer and more potently than erlotinib (first-generation EGFR inhibitor) in EGFRvIII cells (Fig. 2b). Afatinib and erlotinib had no effect on total EGFR (Fig. 2b). Afatinib had similar inhibitory effects on EGFR activation in GBM cell lines U251 and U251EGFRvIII (Fig. 2c).

Afatinib inhibits EGFR and EGFRvIII activation and augments G2/M arrest with TMZ. a U87MG, U87EGFR WT and U87EGFRvIII DK cells were incubated with TMZ (25 μM), afatinib (1 μM) or combination of drugs for 48 h, and cell lysates were analyzed for pEGFR (Tyr-1068) by western blot analysis. β-actin serves as a loading control. b U87EGFRvIII cells were treated with TMZ, afatinib, erlotinib or combination of TMZ and afatinib and analyzed for pEGFR (Tyr-1068). c U251 and U251EGFRvIII cells were treated with afatinib, TMZ or combination for 48 h. Cell lysates were analyzed for pEGFR (Tyr-1068) and pAKT (Ser-473) by western blot analysis. d U87EGFRvIII cells were synchronized with double thymidine block and treated with TMZ (25 μM), afatinib (1 μM) or combination of drugs for 48 h. Cells were trypsinized, fixed with 70% ethanol, stained with Telford reagent and analyzed by flow cytometry. e The bar diagram shows the mean (± SD) percentage of distribution of the cells (*$# P ≤ 0.05); * significant compared to control; $ significant compared to TMZ; # significant compared to afatinib (n = 3)
Afatinib and TMZ combination induces cell cycle arrest in EGFRvIII GBM cells
TMZ is known to induce G2/M arrest and inhibit U87MG GBM cell proliferation [55]. U87EGFRvIII cells are more resistant to the cytotoxic drug cisplatin than U87MG cells [56]. We analyzed the effect of afatinib and TMZ on the cell cycle in U87EGFRvIII cells by flow cytometry. Afatinib and TMZ significantly induced G1 (p = 0.001) and G2/M arrest (p = 0.001), respectively, while decreasing the proportion of EGFRvIII cells in S-phase compared to vehicle-treated control cells (Fig. 2d, e). We further observed that the combination of afatinib and TMZ significantly reduced the percentage of S-phase cells (p = 0.001), but increased the proportion of G2/M-arrested (p = 0.001) U87EGFRvIII cells (Fig. 2d, e).
The G2/M phase arrest may either allow cells to repair damaged DNA and proliferate or induce cell death by apoptosis, senescence, or mitotic catastrophe [57]. As we did not observe EGFRvIII cells in sub-G0 (apoptosis) after drug treatment, we analyzed senescence-associated β-galactosidase (SA-β-gal) activity, as a marker for cellular senescence. TMZ and afatinib combination treatment led to significantly increased SA-β-gal-stained EGFRvIII cells (p = 0.0001) (24 ± 4.5%) as compared with TMZ, afatinib or treatment control cells (6.6 ± 1.6%, 10 ± 2%, and 2 ± 1%, respectively) (Additional file 2: Figure S2 A-B).
Afatinib and TMZ combination decreases GBM cell migration and invasion
GBM is locally aggressive and invades the perivascular regions [58]. We analyzed the effect of afatinib and TMZ on U87MG and U87EGFRvIII GBM cell migration and invasion. Afatinib alone or in combination with TMZ significantly decreased the migration of U87EGFRvIII cells compared to U87 cells (Fig. 3a, b). Afatinib alone had no significant effect on U87MG cells (Fig. 3 a, b). The number of U87MG-migrated cells decreased significantly in the TMZ (p = 0.02) and combination (p = 0.001) groups; TMZ vs TMZ plus afatinib (p = 0.04)). Similarly, TMZ and combination treatment significantly decreased the migration of U87EGFRvIII (control vs any treatment (p = 0.02); TMZ vs TMZ plus afatinib (p = 0.02)). TMZ (p = 0.03) and combination (p = 0.02) treatments significantly decreased the number of invasive U87MG cells when compared to control (Fig. 3c, d). The number of invasive U87EGFRvIII cells decreased significantly in treated cells when compared to control [TMZ (p = 0.005); afatinib (p = 0.01)]; combination (p = 0.0002)). Further, both in U87MG and U87EGFRvIII, combination treatment significantly decreased the number of invasive cells when compared to TMZ alone [U87MG (p = 0.001); U87EGFRvIII (p = 0.03)].

Afatinib and TMZ combination decreases migration and invasion of U87MG and U87 EGFRvIII cells. a, b U87MG and U87EGFRvIII cells (2.5 × 104) were incubated with TMZ (25 μM), afatinib (1 μM) or combination of drugs for 48 h. Non-migrated cells in the upper chamber were removed with a cotton swab, and the migrated cells were stained and counted. Representative images are shown (10X magnification). The bar graph shows the mean (± SD) percentage of migrated cells. Experiments were repeated three times and 5 random fields were chosen for quantification (*$ P ≤ 0.05); * significant compared to control; $ significant compared to TMZ. c, d Afatinib alone or in combination with TMZ decreases the invasion of U87EGFRvIII cells. U87MG and U87 EGFRvIII cells (5.0 × 104) were incubated with TMZ (25 μM), afatinib (1 μM) or combination of drugs for 48 h. Non-invaded cells in the upper chamber was removed with a cotton swab, and the invaded cells were stained and counted. The bar graph shows the mean (± SD) percentage of invaded U87MG and U87EGFRvIII cells. Experiments were repeated three times (*$ P ≤ 0.05); *significant compared to control; $ significant compared to TMZ. e, f Afatinib inhibits EGFRvIII-mediated JAK2/STAT3 and FAK signaling. U87EGFRvIII cells were treated with TMZ (25 μM), afatinib (1 μM) or combination for 48 h, and lysates were analyzed for pEGFR (Tyr-1068), pJAK2 (Tyr-1007/1008), pSTAT3 (Tyr-705), pFAK (Tyr-925) and pAKT (Ser-473) by western blot analysis. g EGFRvIII kinase domain mediates FAK (Tyr-925) activation. U87, U87 EGFR WT, U87EGFRvIII and U87EGFRvIII DK cell lysates were analyzed for pFAK (Tyr-925), pFAK (Tyr-576/577) and pFAK (Tyr-397)
EGFRvIII is known to activate JAK2/STAT3 signaling, and its inhibition has been shown to decrease invasion both in vitro and in vivo [59]. STAT3 activation is found to be higher in GBM than in low-grade astrocytoma, and it is co-expressed with EGFR in GBM [60]. Inhibition of JAK2/STAT3 signaling sensitized U87EGFR WT and U87EGFRvIII cells to the anti-EGFR drug gefitinib [60]. Interestingly, our results showed that TMZ and afatinib together significantly decreased STAT3 signaling as well as cell survival AKT signaling (Fig. 3f). In addition, EGFR also mediates FAK phosphorylation (Y925) through Src, resulting in cytoskeletal reorganization and increased cell motility/invasion [59, 61,62,63]. To investigate whether the kinase activity of EGFRvIII mediates FAK Y925 phosphorylation, we analyzed FAK activation (pFAK-Y925) in U87MG, U87EGFR WT, U87EGFRvIII, U87EGFRvIII DK and U251EGFRvIII cells. As shown in Fig. 3g and (Additional file 3: Figure S3), pFAK (Y925) signaling was specific to EGFRvIII, as no activation was observed in U87MG and U87EGFRvIII DK cells. Importantly, afatinib treatment resulted in complete downregulation of both pJAK2 and pFAK in U87EGFRvIII cells (Fig. 3e), suggesting their involvement in EGFRvIII-mediated GBM cell invasion. No changes in other FAK phosphorylation sites (Y397 and Y576/577) were observed in EGFRvIII cells (Fig. 3g).
Afatinib reduces SP/CSCs by inhibiting EGFRvIII-cMET cross-activation
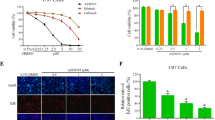
Cancer stem cells are highly resistant to CRT and play a major role in tumor recurrence [20, 21]. We observed a significantly higher proportion of CSCs in U87EGFRvIII cells compared to U87MG cells (p = 0.03). While afatinib significantly decreased the percentage of CSCs in both U87MG and U87EGFRvIII cells (p = 0.02) (Fig. 4a, b), TMZ decreased CSCs in only U87MG cells, (Fig. 4a, b). The average percentages of CSCs in U87EGFRvIII GBM cells were 1.03 ± 0.2, 1.0 ± 0.2, 0.13 ± 0.05 and 0.2 ± 0 in control, TMZ, afatinib and combination groups, respectively.

Afatinib inhibits EGFRvIII-cMET signaling crosstalk in SP/CSCs cells. a, b Afatinib reduces TMZ-resistant U87EGFRvIII SP/CSCs. U87MG and U87EGFRvIII cells treated with either TMZ (25 μM), afatinib (1 μM) alone or combination for 48 h were trypsinized, stained with Hoechst 33342 (5 μg/ml) and analyzed for CSCs and NSP cells using flow cytometry. The bar diagram shows the mean (± SD) percentage of SP/CSCs from U87MG and U87EGFRvIII (n = 3). c, d Afatinib decreases U87EGFRvIII-mediated self-renewal properties of CSCs. U87MG and U87EGFRvIII cells treated with TMZ (25 μM), afatinib (1 μM), or combination for 48 h were plated (2 × 103 cells/well in 100 μl of stem cell medium) on a 96-well ultra-low attachment plate. Neurosphere/tumor spheres formed after 10 days were quantified and photographed (X20 magnification). The graph shows the mean (± SD) number of tumor spheres formed by U87MG and U87EGFRvIII cells (n = 4). (*$ P ≤ 0.05); * significant compared to control; $ significant compared to TMZ. e U87, U87EGFR WT, U87EGFRvIII and U87EGFRvIII DK cell lysates were analyzed for pcMET (Tyr-1234/1235) by western blot analysis. f U87EGFRvIII cells were treated with either TMZ (25 μM), afatinib (1 μM), or combination for 48 h and analyzed for pcMET (Tyr-1234/1235). g Expression of Nanog, Oct3/4 (self-renewal marker) and pcMET (Tyr-1234/1235) were analyzed in SP/CSCs and NSP cells by western blot analysis. h U87EGFRvIII SP cells were treated with TMZ (25 μM), afatinib (1 μM), or combination for 48 h and analyzed for pcMET (Tyr-1234/1235) and stemness markers Nanog and Oct3/4 by western blot analysis. i U87EGFRvIII cells were treated with varying concentrations of the cMET specific inhibitor, SU-11274 (1 – 20 μM) or afatinib (1 μM) for 48 h and lysates were analyzed for pcMET (Tyr-1234/1235), Nanog and Oct3/4
We further validated our results using an in vitro clonogenic (neurosphere) assay. We observed a significantly higher (p = 0.01) number of neurospheres formed by U87EGFRvIII CSCs cells (29 ± 7) than U87MG CSC cells (13 ± 2) (Fig. 4c, d). TMZ significantly decreased the self-renewal properties of U87MG CSCs (p = 0.003). We also observed that U87EGFRvIII CSCs were relatively resistant to TMZ compared to U87MG CSCs; however, combined afatinib and TMZ significantly decreased the number of neurospheres in both cell lines (p = 0.001) (Fig. 4c, d). The average number of U87EGFRvIII CSC neurospheres per field was 29 ± 7, 18 ± 2, 7 ± 3 and 3 ± 1 in control, TMZ, afatinib, and combination treatment groups, respectively (Fig. 4d).
cMET signaling promotes and enriches GBM CSCs [28, 29]. cMET activation was seen in stem cells of GBM patient specimens [28] as well as in several GBM patient-derived neurospheres [29]. We observed increased activation of cMET in U87EGFRvIII cells compared to U87MG (Fig. 4e). Furthermore, afatinib significantly diminished cMET activation in U87EGFRvIII cells (Fig. 4f). To determine if EGFRvIII mediates CSC maintenance through cMET activation in GBM, we analyzed the expression of various stemness markers in CSCs and non-side population (NSP) cells isolated from U87EGFRvIII cells. We observed enrichment of stemness markers Nanog and Oct3/4 and increased cMET activation in CSCs compared with NSP cells. Surprisingly, CSCs expressed lower SOX9 and CD15 levels, and no change in expression of nestin was observed (Fig. 4g). Interestingly, afatinib treatment significantly decreased cMET activation as well as stemness of U87EGFRvIII CSCs, while TMZ only showed no effect (Fig. 4h). To further validate that cMET signaling conserves stemness in GBM, we treated U87EGFRvIII cells with cMET inhibitor SU11274. Our results showed a dose dependent decrease in the expression of phosphorylated/active cMET (pcMET-Y1234/1235) and decreased expression of stemness marker Nanog and Oct3/4 (Fig. 4i).
Afatinib and TMZ combination prevents tumor growth in vivo
To analyze the effect of afatinib and TMZ in vivo, U87EGFRvIII luciferase cells were intracranially injected into athymic mice, and treatments were administered once tumors developed (Fig. 5a). We observed that afatinib as a monotherapy neither inhibited tumor growth significantly nor improved the OS of the animal. Only one of eight mice survived until the end of the study (30 days). (Figs. 5C & S 2C). Although TMZ treatment alone initially decreased tumor growth and increased animal survival, tumors progressed in 60% of the mice (Fig. 5b). By contrast, the combination of afatinib and TMZ significantly inhibited tumor growth (p = 0.03) after day 30 of treatment compared to TMZ alone. Furthermore, none of the animals exhibited tumor burden in the combination group (Fig. 5a-c), suggesting the superior efficacy of this therapy in GBM in vivo.

Afatinib and TMZ combination reduces tumor burden in vivo. a, b U87EGFRvIII luciferase transfected cells (2 × 104 in 2 μl of PBS) were intracranially injected into 4–6 week’s old mice using a stereotactic frame. After 5 days, mice were randomized into 4 groups and treated with vehicle (n = 8), TMZ (25 mg/kg BW) (n = 7), afatinib (10 mg/kg BW) (n = 8) or combination (n = 5) for 5 days a week by oral gavage. Animals treated with TMZ alone or in combination with afatinib survived longer and were sacrificed after 30 days. The tumor volume (total photon count) was measured using IVIS imaging on days 0, 7, 9, 11, 20 and 30. ($, # P ≤ 0.03); $ significant compared to TMZ; # significant compared to vehicle control (c) Kaplan Meier survival curve analysis showing effect of control, afatinib, TMZ or combination on OS of EGFRvIII orthograft mice. d, e Confocal microscopy showing expression of (d) pEGFR (Tyr-1068) (red staining), Oct3/4 (green staining) and (e) pcMET (Tyr-1234/1235) (red) and Nanog (green) images in U87EGFRvIII tumor xenografts
Using immunofluorescence, we also analyzed the expression of pEGFR (Tyr-1068), pcMET (Tyr-1234/1235), Oct3/4 and Nanog in EGFRvIII tumor xenografts. We observed low expression of these markers in afatinib-treated animal tissues compared to a very high expression in control animal tumors (Fig. 5 D-E). Although TMZ decreased tumor growth, it enriched the expression of pcMET and Nanog (stem cell markers) (Fig. 5 d, e). Overall, the in vivo studies corroborate in vitro observations and reinforce the importance of EGFRvIII/cMET activation in GBM tumorigenesis (Fig. 6).

Possible mechanism of afatinib-TMZ combination therapy in GBM. TMZ targets the differentiated proliferating GBM cells but fails to eradicate slow-growing CSCs and result in tumor progression. Afatinib decreases proliferation of U87EGFRvIII cells by inhibiting EGFRvIII/AKT signaling, cell migration and invasion by inhibiting EGFRvIII/JAK2/STAT and FAK (Tyr-925) signaling pathways. Afatinib by inhibiting EGFRvIII-cMET cross-activation decreases CSC stemness possibly by downregulating stemness transcription factors Oct3/4 and Nanog. Dotted line indicates possible effects
Discussion
Despite aggressive therapeutic interventions including surgery, RT and TMZ, the survival of GBM patients has not improved substantially [3]. Most GBM patients develop recurrence that is associated with universal fatality, suggesting an urgent need to understand the disease and develop novel therapies for improving patient survival.
Recent high-throughput data analyses have revealed genetic, epigenetic and mutational features of GBM that have paved the way for personalized medicine. Many studies have established that EGFR and mutant EGFRvIII are amplified/ overexpressed in the majority of GBM patients and are thus important therapeutic targets [6,7,8,9,10,11,12,13]. While strategies that successfully targeted EGFR in other cancers have been tested in GBM, these clinical trials were largely disappointing, possibly due to the compensatory activation of other EGFR family members and RTKs [25]. Phosphatase and tensin homolog (PTEN), a negative regulator of PI3K signaling has also been shown to impact EGFR-targeted therapy outcomes [25]. GBM SP/CSCs show resistance to anti-EGFR therapies by compensatory upregulation of HER2 and HER3. Interestingly, the addition of lapatinib (which inhibits both EGFR and HER2) decreased the SP/CSCs [24]. Therefore, novel therapies targeting concurrent signaling pathways are needed for improved GBM patient outcomes. Aligned with this concept, recently a combination of erlotinib and HGF scavenging antibody L2G7 was shown to significantly decrease tumor growth and improve the OS in EGFRvIII/cMET+/HGF+/PTEN−/− glioma models compared to single agents [64]. Herein, we investigated the efficacy of afatinib and TMZ combination in GBM models overexpressing EGFRvIII in vitro and in vivo. Our studies revealed that together these drugs inhibited proliferation, anchorage-dependent and -independent growth by inducing G2/M arrest and senescence of EGFRvIII-expressing GBM cells in vitro. In addition, afatinib with TMZ synergistically inhibited invasion and motility, possibly through inhibiting JAK2/STAT3 and FAK signaling in EGFRvIII-expressing GBM cells. This combination also inhibited cross-activation of cMET and reduced GBM SP/CSCs. Remarkably, our study showed that afatinib and TMZ together not only decreased the tumor burden but also inhibited tumor growth in orthotopic intracranial models.
GBM is highly aggressive and frequently infiltrates/invades surrounding normal brain tissues, hampering complete surgical resection [58] and resulting in progression. It has been previously demonstrated that EGFRvIII signaling can activate JAK2/STAT3 signaling, and inhibiting JAK2 with either a JAK2 inhibitor or siRNA decreased the invasive nature of EGFRvIII cells both in vitro and in vivo [59]. Harada et al. showed that JAK2 signaling plays an important role in developing acquired resistance to erlotinib in lung cancer cells with EGFR-activating mutations [65]. Our study revealed afatinib completely inhibited EGFRvIII-mediated JAK2 signaling. Importantly, afatinib demonstrated prolonged inhibition of EGFRvIII signaling compared to erlotinib, suggesting superior efficacy of afatinib in preventing JAK2-mediated tumorigenic signaling. In addition to JAK2 activation, EGFR-mediated FAK phosphorylation (Y925) and activation also promote migration and invasion of cancer cells [61,62,63]. Consistent with these results, our study showed U87EGFRvIII cells have elevated pFAK (Y925) levels and greater invasion potential than U87 cells. Remarkably, afatinib treatment completely abrogated EGFRvIII-mediated FAK (Y925) activation/phosphorylation, GBM cell migration, and invasion.
Cancer stem cells, through intrinsic and acquired resistance to CRT, are involved in tumor progression [20, 21]. Many signaling pathways including EGFR are involved in the maintenance of CSCs. Recently, EGFRvIII was shown to cross-activate cMET RTKs [25,26,27] and enrich GBM CSCs [28, 29]. Using the breast cancer cell line MDA-MB-453 and GBM cell line U-373, treatment with cMET receptor ligand hepatocyte growth factor/scatter factor was shown to impart resistance to doxorubicin/adriamycin and cisplatin [66, 67]. Furthermore, inhibition of cMET by siRNA or the pharmacological inhibitor SU11274 decreased tumorsphere formation in several GBM CSCs in vitro [28]. In addition, liposome-conjugated cMET siRNA also decreased GBM tumor growth in an orthotopic mouse model [28]. In concordance with these and our previous results in head and neck squamous cell carcinoma [57], we observed a significant reduction of CSCs with afatinib. Here we conclusively established that afatinib decreases CSCs by abolishing EGFRvIII-cMET signaling.
A recent study showed that the combination of the cMET inhibitor crizotinib with erlotinib significantly decreased stem cell marker expression, neurosphere growth and in vivo tumor growth of human GBM xenografts [68]. While this combination decreased growth in subcutaneous xenograft tumors, the non-permeability of crizotinib through the BBB limited the efficacy in both preclinical and clinical models of brain tumors [68, 69]. Studies have shown that the BBB restricts the availability of not only crizotinib but also most chemotherapeutic drugs to brain tumors and limits their therapeutic efficacy. However, a recent prospective multicenter study of patients with NSCLC and leptomeningeal carcinomatosis showed significant benefits of afatinib, even though only 2.45 ± 2.91% of afatinib penetrated to CSF from blood [70]. Our studies showed afatinib alone has no effects on tumor growth and survival in U87EGFRvIII orthograft-bearing mice. This reduced efficacy may be due to the low dose of afatinib used in our study as opposed to the higher doses used in an NSCLC brain metastases model, which led to tumor regression [71]. Although TMZ reduced growth and overall tumor burden in this model, 60% (4/7) of the animals experienced tumor re-growth, suggesting its limitations as a monotherapy. In contrast, afatinib and TMZ together significantly reduced tumor growth and completely prevented the development of tumor re-growth (5/5). Several studies have shown that chemotherapeutic drugs kill the bulk of differentiating tumor cells, but enrich SP/CSCs, resulting in tumor re-growth. Our results align with these reports as EGFRvIII tumor xenografts showed significant upregulation of CSC markers upon TMZ treatment, but significant downregulation of these markers in mice treated with combined afatinib and TMZ (Fig. 6).
Conclusion
In summary, our studies demonstrated that the addition of afatinib to TMZ significantly reduced proliferation, clonogenic survival and invasion of U87EGFRvIII GBM cells in vitro and significantly inhibited tumor growth in pre-clinical orthotopic models. Though afatinib was disappointing as a monotherapy in a clinical trial of unselected recurrent GBM patients, it significantly reduced tumor burden when combined with TMZ in U87EGFRvIII xenografts in our pre-clinical mouse model. This work warrants further evaluation of this treatment combination in GBM patients with EGFR amplification or mutant EGFRvIII expression.
Availability of data and materials
Not applicable.
Abbreviations
- BBB:
-
Blood brain barrier
- bFGF:
-
Basic fibroblast growth factor
- CI:
-
Combination index
- CRT:
-
Chemoradiotherapy
- CSCs:
-
Cancer stem cells
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- FAK:
-
Focal adhesion kinase
- GBM:
-
Glioblastoma
- LIF:
-
Leukemia inhibitory factor
- NSCLC:
-
Non-small-cell lung cancer
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- PTEN:
-
Phosphatase and tensin homolog
- RT:
-
Radiation therapy
- RTKs:
-
Receptor tyrosine kinases
- SA-β-gal:
-
Senescence-associated β-galactosidase
- SP:
-
Side population
- TMZ:
-
Temozolomide
References
Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2008-2012. Neuro-Oncology. 2015;17(Suppl 4):iv1–iv62.
Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31(32):4085–91.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96.
Johnson DR, O'Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. J Neuro-Oncol. 2012;107(2):359–64.
Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro-Oncology. 2013;15(1):4–27.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12.
Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8.
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–710.
Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci U S A. 1992;89(10):4309–13.
Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64(19):6892–9.
Yang RY, Yang KS, Pike LJ, Marshall GR. Targeting the dimerization of epidermal growth factor receptors with small-molecule inhibitors. Chem Biol Drug Des. 2010;76(1):1–9.
Frederick L, Wang XY, Eley G, James CD. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60(5):1383–7.
Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990;87(21):8602–6.
Koka V, Potti A, Forseen SE, Pervez H, Fraiman GN, Koch M, et al. Role of Her-2/neu overexpression and clinical determinants of early mortality in glioblastoma multiforme. Am J Clin Oncol. 2003;26(4):332–5.
Yan M, Parker BA, Schwab R, Kurzrock R. HER2 aberrations in cancer: implications for therapy. Cancer Treat Rev. 2014;40(6):770–80.
Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer. 2012;12(8):553–63.
Kim EJ, Kim SO, Jin X, Ham SW, Kim J, Park JB, et al. Epidermal growth factor receptor variant III renders glioma cancer cells less differentiated by JAGGED1. Tumour Biol. 2015;36(4):2921–8.
Stockhausen MT, Kristoffersen K, Stobbe L, Poulsen HS. Differentiation of glioblastoma multiforme stem-like cells leads to downregulation of EGFR and EGFRvIII and decreased tumorigenic and stem-like cell potential. Cancer Biol Ther. 2014;15(2):216–24.
Emlet DR, Gupta P, Holgado-Madruga M, Del Vecchio CA, Mitra SS, Han SY, et al. Targeting a glioblastoma cancer stem-cell population defined by EGF receptor variant III. Cancer Res. 2014;74(4):1238–49.
Carruthers R, Ahmed SU, Strathdee K, Gomez-Roman N, Amoah-Buahin E, Watts C, et al. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol Oncol. 2015;9(1):192–203.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60.
Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27(47):6120–30.
Ma L, Zhang G, Miao XB, Deng XB, Wu Y, Liu Y, et al. Cancer stem-like cell properties are regulated by EGFR/AKT/beta-catenin signaling and preferentially inhibited by gefitinib in nasopharyngeal carcinoma. FEBS J. 2013;280(9):2027–41.
Clark PA, Iida M, Treisman DM, Kalluri H, Ezhilan S, Zorniak M, et al. Activation of multiple ERBB family receptors mediates glioblastoma cancer stem-like cell resistance to EGFR-targeted inhibition. Neoplasia. 2012;14(5):420–8.
Stommel JM, Kimmelman AC, Ying H, Nabioullin R, Ponugoti AH, Wiedemeyer R, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–90.
Huang PH, Mukasa A, Bonavia R, Flynn RA, Brewer ZE, Cavenee WK, et al. Quantitative analysis of EGFRvIII cellular signaling networks reveals a combinatorial therapeutic strategy for glioblastoma. Proc Natl Acad Sci U S A. 2007;104(31):12867–72.
Pillay V, Allaf L, Wilding AL, Donoghue JF, Court NW, Greenall SA, et al. The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia. 2009;11(5):448–58 2 p following 58.
Joo KM, Jin J, Kim E, Ho Kim K, Kim Y, Gu Kang B, et al. MET signaling regulates glioblastoma stem cells. Cancer Res. 2012;72(15):3828–38.
Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, et al. C-met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc Natl Acad Sci U S A. 2011;108(24):9951–6.
Velpula KK, Dasari VR, Asuthkar S, Gorantla B, Tsung AJ. EGFR and c-met cross talk in glioblastoma and its regulation by human cord blood stem cells. Transl Oncol. 2012;5(5):379–92.
Dungo RT, Keating GM. Afatinib: first global approval. Drugs. 2013;73(13):1503–15.
Solca F, Dahl G, Zoephel A, Bader G, Sanderson M, Klein C, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343(2):342–50.
Li D, Ambrogio L, Shimamura T, Kubo S, Takahashi M, Chirieac LR, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–11.
Sequist LV, Yang JC, Yamamoto N, O'Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–34.
Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15(2):213–22.
Schuler M, Wu YL, Hirsh V, O'Byrne K, Yamamoto N, Mok T, et al. First-line Afatinib versus chemotherapy in patients with non-small cell lung Cancer and common epidermal growth factor receptor gene mutations and brain metastases. J Thorac Oncol. 2016;11(3):380–90.
Soria JC, Felip E, Cobo M, Lu S, Syrigos K, Lee KH, et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol. 2015;16(8):897–907.
Hoffknecht P, Tufman A, Wehler T, Pelzer T, Wiewrodt R, Schutz M, et al. Efficacy of the irreversible ErbB family blocker afatinib in epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI)-pretreated non-small-cell lung cancer patients with brain metastases or leptomeningeal disease. J Thorac Oncol. 2015;10(1):156–63.
Reardon DA, Nabors LB, Mason WP, Perry JR, Shapiro W, Kavan P, et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro-Oncology. 2015;17(3):430–9.
Alshami J, Guiot MC, Owen S, Kavan P, Gibson N, Solca F, et al. Afatinib, an irreversible ErbB family blocker, with protracted temozolomide in recurrent glioblastoma: a case report. Oncotarget. 2015;6(32):34030–7.
Narita Y, Nagane M, Mishima K, Huang HJ, Furnari FB, Cavenee WK. Mutant epidermal growth factor receptor signaling down-regulates p27 through activation of the phosphatidylinositol 3-kinase/Akt pathway in glioblastomas. Cancer Res. 2002;62(22):6764–9.
Macha MA, Rachagani S, Gupta S, Pai P, Ponnusamy MP, Batra SK, et al. Guggulsterone decreases proliferation and metastatic behavior of pancreatic cancer cells by modulating JAK/STAT and Src/FAK signaling. Cancer Lett. 2013;341(2):166–77.
Chou TC MN. CompuSyn for drug combinations: PC Software and User's Guide: a computer program for quantitation of synergism and antagonism in drug combinations, and the determination of IC50 and ED50 and LD50 values. Paramus (NJ): ComboSyn 2005. Available from: : http://www.combosyn.com/ for video demonstration.
Groenendijk FH, Mellema WW, van der Burg E, Schut E, Hauptmann M, Horlings HM, et al. Sorafenib synergizes with metformin in NSCLC through AMPK pathway activation. Int J Cancer. 2015;136(6):1434–44.
Horibata S, Vo TV, Subramanian V, Thompson PR, Coonrod SA. Utilization of the soft agar Colony formation assay to identify inhibitors of Tumorigenicity in breast Cancer cells. J Vis Exp. 2015(99):e52727.
Lakshmanan I, Ponnusamy MP, Das S, Chakraborty S, Haridas D, Mukhopadhyay P, et al. MUC16 induced rapid G2/M transition via interactions with JAK2 for increased proliferation and anti-apoptosis in breast cancer cells. Oncogene. 2012;31(7):805–17.
Macha MA, Rachagani S, Pai P, Gupta S, Lydiatt WM, Smith RB, et al. MUC4 regulates cellular senescence in head and neck squamous cell carcinoma through p16/Rb pathway. Oncogene. 2015;34(13):1698–708.
Karmakar S, Seshacharyulu P, Lakshmanan I, Vaz AP, Chugh S, Sheinin YM, et al. hPaf1/PD2 interacts with OCT3/4 to promote self-renewal of ovarian cancer stem cells. Oncotarget. 2017;8(9):14806–20.
Dinca EB, Sarkaria JN, Schroeder MA, Carlson BL, Voicu R, Gupta N, et al. Bioluminescence monitoring of intracranial glioblastoma xenograft: response to primary and salvage temozolomide therapy. J Neurosurg. 2007;107(3):610–6.
Zhang X, Peng L, Liang Z, Kou Z, Chen Y, Shi G, et al. Effects of aptamer to U87-EGFRvIII cells on the proliferation, Radiosensitivity, and radiotherapy of glioblastoma cells. Mol Ther Nucleic Acids. 2018;10:438–49.
Jones JE, Esler WP, Patel R, Lanba A, Vera NB, Pfefferkorn JA, et al. Inhibition of acetyl-CoA carboxylase 1 (ACC1) and 2 (ACC2) reduces proliferation and De novo lipogenesis of EGFRvIII human glioblastoma cells. PLoS One. 2017;12(1):e0169566.
Bobola MS, Kolstoe DD, Blank A, Silber JR. Minimally cytotoxic doses of temozolomide produce radiosensitization in human glioblastoma cells regardless of MGMT expression. Mol Cancer Ther. 2010;9(5):1208–18.
Ostermann S, Csajka C, Buclin T, Leyvraz S, Lejeune F, Decosterd LA, et al. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin Cancer Res. 2004;10(11):3728–36.
Gao CF, Xie Q, Su YL, Koeman J, Khoo SK, Gustafson M, et al. Proliferation and invasion: plasticity in tumor cells. Proc Natl Acad Sci U S A. 2005;102(30):10528–33.
Wang HH, Chang TY, Lin WC, Wei KC, Shin JW. GADD45A plays a protective role against temozolomide treatment in glioblastoma cells. Sci Rep. 2017;7(1):8814.
Nagane M, Levitzki A, Gazit A, Cavenee WK, Huang HJ. Drug resistance of human glioblastoma cells conferred by a tumor-specific mutant epidermal growth factor receptor through modulation of Bcl-XL and caspase-3-like proteases. Proc Natl Acad Sci U S A. 1998;95(10):5724–9.
Macha MA, Rachagani S, Qazi AK, Jahan R, Gupta S, Patel A, et al. Afatinib radiosensitizes head and neck squamous cell carcinoma cells by targeting cancer stem cells. Oncotarget. 2017;8(13):20961–73.
Paw I, Carpenter RC, Watabe K, Debinski W, Lo HW. Mechanisms regulating glioma invasion. Cancer Lett. 2015;362(1):1–7.
Zheng Q, Han L, Dong Y, Tian J, Huang W, Liu Z, et al. JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII-expressing glioblastoma. Neuro-Oncology. 2014;16(9):1229–43.
Lo HW, Cao X, Zhu H, Ali-Osman F. Constitutively activated STAT3 frequently coexpresses with epidermal growth factor receptor in high-grade gliomas and targeting STAT3 sensitizes them to Iressa and alkylators. Clin Cancer Res. 2008;14(19):6042–54.
Deramaudt TB, Dujardin D, Hamadi A, Noulet F, Kolli K, De Mey J, et al. FAK phosphorylation at Tyr-925 regulates cross-talk between focal adhesion turnover and cell protrusion. Mol Biol Cell. 2011;22(7):964–75.
Chen HC, Chan PC, Tang MJ, Cheng CH, Chang TJ. Tyrosine phosphorylation of focal adhesion kinase stimulated by hepatocyte growth factor leads to mitogen-activated protein kinase activation. J Biol Chem. 1998;273(40):25777–82.
McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5(7):505–15.
Lal B, Goodwin CR, Sang Y, Foss CA, Cornet K, Muzamil S, et al. EGFRvIII and c-met pathway inhibitors synergize against PTEN-null/EGFRvIII+ glioblastoma xenografts. Mol Cancer Ther. 2009;8(7):1751–60.
Harada D, Takigawa N, Ochi N, Ninomiya T, Yasugi M, Kubo T, et al. JAK2-related pathway induces acquired erlotinib resistance in lung cancer cells harboring an epidermal growth factor receptor-activating mutation. Cancer Sci. 2012;103(10):1795–802.
Fan S, Ma YX, Wang JA, Yuan RQ, Meng Q, Cao Y, et al. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3′ kinase. Oncogene. 2000;19(18):2212–23.
Bowers DC, Fan S, Walter KA, Abounader R, Williams JA, Rosen EM, et al. Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase- and AKT-dependent pathways. Cancer Res. 2000;60(15):4277–83.
Goodwin CR, Rath P, Oyinlade O, Lopez H, Mughal S, Xia S, et al. Crizotinib and erlotinib inhibits growth of c-met(+)/EGFRvIII(+) primary human glioblastoma xenografts. Clin Neurol Neurosurg. 2018;171:26–33.
Costa DB, Kobayashi S, Pandya SS, Yeo WL, Shen Z, Tan W, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29(15):e443–5.
Tamiya A, Tamiya M, Nishihara T, Shiroyama T, Nakao K, Tsuji T, et al. Cerebrospinal fluid penetration rate and efficacy of Afatinib in patients with EGFR mutation-positive non-small cell lung Cancer with leptomeningeal Carcinomatosis: a multicenter prospective study. Anticancer Res. 2017;37(8):4177–82.
Zhang SR, Zhu LC, Jiang YP, Zhang J, Xu RJ, Xu YS, et al. Efficacy of afatinib, an irreversible ErbB family blocker, in the treatment of intracerebral metastases of non-small cell lung cancer in mice. Acta Pharmacol Sin. 2017;38(2):233–40.
Acknowledgments
We thank Dr. Webster K. Cavenee (University of California San Diego, CA, USA) for providing human GBM cell lines U87MG and U87 cells transfected with either EGFR WT, EGFRvIII or EGFRvIII DK. We also thank Dr. Amyn A. Habib (University of Texas Southwestern Medical Center, Dallas, TX, USA) for giving us U251 and U251 cells transfected with EGFR WT or EGFRvIII. We also grateful to Flow cytometry Research Facility and confocal core facility at the University of Nebraska Medical Center. We would like to thank Dr. Jessica Mercer for editing this manuscript.
Funding
National Institutes of Health (P01 CA217798, P30 CA036727; R01 CA183459, R01 CA210637, U01185148); Fred and Pamela Buffett Cancer Center, University of Nebraska Medical Center, Omaha, NE, USA. Dr. Raghupathy Vengoji is supported by the Fred and Pamela Buffett Cancer Center, University of Nebraska Medical Center, Omaha, NE, and William Langford Creative Mind Fund.
Author information
Authors and Affiliations
Contributions
Study concept and design: SKB, RV, MAM, MPP and NS; acquisition of data: RV and RKN; analysis and interpretation of data: RV, MPP, MAM and SKB; drafting of the manuscript: RV.; critical revision of the manuscript: MAM, MJ, MPP, NS, and SKB; technical and material support: SR, JAS., SG and KM; study supervision: SKB All authors read and accept the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
SKB is one of the co-founder of Sanguine Diagnostics and Therapeutics, Inc. The other authors disclosed no potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. TMZ and afatinib synergistically inhibit U87EGFRvIII proliferation (A-D). U87MG (3 × 103 cells/well) and U87EGFRvIII (2 × 103 cells/well) were seeded in a 96-well plate and treated with different concentrations of TMZ and afatinib for 48–72 h; viable cells were measured by MTT assay. (E-F) Combination treatment significantly decreased the proliferation rate of U87EGFRvIII cells. U87MG and U87EGFRvIII cells were treated with TMZ (25 μM), afatinib (1 μM) or combination for 48 h, and viable cells were measured by MTT assay. Combination index (CI) was calculated using CompuSyn software. (E) CI values for non-constant combination: T + A. (F) Logarithmic CI graph shows that additional and near synergistic effects of TMZ and afatinib in U87EGFRvIII cells. T - TMZ; A - afatinib. (JPG 215 kb)
Additional file 2:
Figure S2. Combination of afatinib and TMZ treatment decreases the proliferation of U87EGFRvIII cells by inducing cellular senescence. (A) Representative image shows SA-β-galactosidase-positive staining in drug-treated EGFRvIII cells. (B) The bar graph shows the mean (±SD) number of senescent cells (*$ P ≤ 0.05); * significant compared to control; $ significant compared to TMZ. (C) U87EGFRvIII luciferase cells were injected intracranially and treated with afatinib 10 mg/kg/BW (5 days a week p.o.); tumor growth was measured by IVIS imaging at indicated time points. (JPG 139 kb)
Additional file 3:
Figure S3. U251EGFRvIII brings FAK (Y925) activation. U251EGFRvIII (Tet-inducible system) cells were cultured in the presence and absence of tetracycline and lysates were analyzed for pFAK (Tyr-925), pFAK (Tyr-576/577) and pFAK (Tyr-397). (JPG 44 kb)
Additional file 4:
Table S1. Antibodies list (DOCX 12 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Vengoji, R., Macha, M.A., Nimmakayala, R.K. et al. Afatinib and Temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. J Exp Clin Cancer Res 38, 266 (2019). https://doi.org/10.1186/s13046-019-1264-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-019-1264-2