
Abstract
Background
Targeting the epidermal growth factor receptor (EGFR) either alone or in combination with chemotherapy is an effective treatment for patients with RAS wild-type metastatic colorectal cancer (mCRC). However, only a small percentage of mCRC patients receive clinical benefits from anti-EGFR therapies, due to the development of resistance mechanisms. In this regard, HER2 has emerged as an actionable target in the treatment of mCRC patients with resistance to anti-EGFR therapy.
Methods
We have used SW48 and LIM1215 human colon cancer cell lines, quadruple wild-type for KRAS, NRAS, BRAF and PI3KCA genes, and their HER2–amplified (LIM1215-HER2 and SW48-HER2) derived cells to perform in vitro and in vivo studies in order to identify novel therapeutic strategies in HER2 gene amplified human colorectal cancer.
Results
LIM1215-HER2 and SW48-HER2 cells showed over-expression and activation of the HER family receptors and concomitant intracellular downstream signaling including the pro-survival PI3KCA/AKT and the mitogenic RAS/RAF/MEK/MAPK pathways. HER2-amplified cells were treated with several agents including anti-EGFR antibodies (cetuximab, SYM004 and MM151); anti-HER2 (trastuzumab, pertuzumab and lapatinib) inhibitors; anti-HER3 (duligotuzumab) inhibitors; and MEK and PI3KCA inhibitors, such as refametinib and pictilisib, as single agents and in combination. Subsequently, different in vivo experiments have been performed. MEK plus PI3KCA inhibitors treatment determined the best antitumor activity. These results were validated in vivo in HER2-amplified patient derived tumor xenografts from three metastatic colorectal cancer patients.
Conclusions
These results suggest that combined therapy with MEK and PI3KCA inhibitors could represent a novel and effective treatment option for HER2-amplified colorectal cancer.
Similar content being viewed by others


Background
In the last decade, the development of targeted anti-cancer therapies has revolutionized the treatment of metastatic colorectal cancer (mCRC) patients. In particular, monoclonal antibodies (mAbs) targeting the epidermal growth factor receptor (EGFR) like cetuximab and panitumumab were the first targeted agents to enter the clinical setting improving the survival of mCRC patients [1, 2]. However, efficacy in anunselected patient population was limited, whereas the identification of biomarkers has led to significantly improved patient selection [3]. In particular, mutations in KRAS and NRAS genes are found to predict resistance to anti-EGFR targeted therapies and are used in clinical practice to guide treatment decision [4]. Furthermore, at least one third of mCRC patients with RAS wild type tumors receiving first-line chemotherapy in combination with anti-EGFR mAbs fail to have a therapeutic response. These results indicate that additional genetic alterations in genes implicated in the EGFR signaling network can be involved in the primary resistance [5,6,7,8]. In fact, deregulation of other effectors of the EGFR signaling cascade, such as mutations in BRAF or PIK3CA genes, loss of PTEN expression, and amplification of KRAS may affect primary response to EGFR blockade [9,10,11,12]. Despite the implementation of biomarkers in clinical practice, patients who initially respond to anti-EGFR therapies almost invariably develop secondary resistance through several mechanisms. The most common molecular mechanisms that are responsible for acquired resistance are genetic alterations of KRAS, NRAS and BRAF genes [6, 13]. In the absence of alteration in RAS or its immediate downstream effectors, other mechanisms have been involved in the activation of the EGFR pathway. Genetic aberrations in receptor tyrosine kinase (RTK), such as HER2 and MET, have been shown to bypass EGFR signaling and activate the MAPK cascade and, therefore, to confer acquired resistance to anti-EGFR therapies [14,15,16]. In particular, HER2 amplification has been suggested as both an intrinsic as well as an acquired mechanism of resistance [17]. One explanation could be that pre-exiting infrequent HER2-amplificated clones might be expanded under the selective pressure of anti-EGFR therapy, leading to disease progression. In this regard, HER2 amplification was found in 5% of mCRC patients with RAS wild type tumors and seem to be associated with resistance to anti-EGFR therapy [18, 19]. In a large cohort of 85 patient-derived colorectal cancer xenografts, Bertotti and colleagues identified HER2 gene amplification in some xenografts, which were resistant to cetuximab and did not harbour mutations in KRAS, NRAS or BRAF genes [17, 20, 21]. Moreover, patient-derived mCRC xenografts with HER2 amplification were treated with various HER2-targeted therapies, alone or in combination. In these preclinical models of human colorectal cancer, the combination of an anti-HER2 antibody (pertuzumab or trastuzumab) and an HER2 tyrosine kinase inhibitor (TKI) (lapatinib) induced pronounced tumor shrinkage [17]. These preclinical results were the proof of concept for clinical trials targeting HER2 genetic alterations in mCRC patients [22]. The phase II HERACLES-A trial of dual HER2-targeted therapy (trastuzumab plus lapatinib) in patients with KRAS wild-type, HER2-positive mCRC who were refractory to standard-of-care treatments, including cetuximab or panitumumab was conducted [23]. Of the 27 patients evaluated for efficacy, eight patients (30%) achieved an overall objective response, meeting the primary endpoint of the trial. None of the patients enrolled who were evaluable for response to anti-EGFR therapy had achieved an objective response with either cetuximab or panitumumab. Based on these results, several clinical trials have been conducted exploiting HER2 as a target for mCRC and also case reports of patients with HER2-positive mCRC who have achieved substantial clinical benefit with targeted anti-HER2 therapy have recently been published [19, 24, 25]. However, 40–50% of patients treated within the HERACLES-A trial did not achieve partial response or prolonged stable disease despite HER2 gene amplification [23, 26]. Notably, even in patients initially responding, acquired resistance occurred in almost all cases [23]. Understanding the mechanisms of resistance to HER2 blockade is a priority to develop more effective and additional options for therapy in this disease setting.
In order to elucidate the possible mechanism(s) of resistance to anti-HER2 treatments, in this study we have used LIM1215 and SW48 human colon cancer cell lines and their HER2-amplified derivatives (LIM1215-HER2 and SW48-HER2) to perform in vitro and in vivo studies using different xenograft models in order to identify novel therapeutic options [17, 23, 26]. Furthermore, human mCRC patient derived tumor xenografts with HER2 gene amplification were used to further validate the potential efficacy of these therapeutic strategies.
Methods
Drugs
5-Fluorouracil, oxaliplatin and irinotecan were obtained from the pharmacy of the University of Campania “Luigi Vanvitelli”. Cetuximab, panitumumab, SYM004, MM151, trastuzumab, pertuzumab and duligotuzumab antibodies were kindly provided by Merck, Amgen, Symphogen, Merrimack Pharmaceuticals, Roche and Genentech, respectively. Refametinib, a selective MEK 1/2 inhibitor was kindly provided by Bayer Italy; Pictilisib, a PI3Kα inhibitor and lapatinib were purchased from Selleckchem.
Human cancer cell lines
The human LIM1215 and SW48 colon cancer cell lines were purchased from the American Type Culture Collection (ATCC). Cells were grown in RPMI 1460 medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin and maintained in a humidified controlled atmosphere at 37 °C.
Colony formation assay
LIM1215-HER2 and SW48-HER2 cells were treated with different concentrations of trastuzumab, pertuzumab, lapatinib, cetuximab, SYM004, MM-151, duligotuzumab, refametinib and pictilisib alone and in combination (range: 0,05–10 μg/ml-μM), for 96 h. After 14 days, cells were fixed with 4% paraformaldehyde and stained with crystal violet. Finally, the plates were inspected by microscopy, photographed and the colonies area was calculated by ImageJ plugins [27]. Results represent the median of three separate experiments, each performed in triplicate.
Western blot analysis
LIM1215-HER2 and SW48-HER2 cells were treated with refametinib and pictilisib alone and in combination for 24 h. Equal amounts of total proteins were incubated with following primary polyclonal antibodies: EGFR and phospho-EGFR, HER2 and phospho-HER2, HER3 and phospho-HER3, HER4 and phospho-HER4, AKT and phospho-AKT, MEK1/2 and phospho-MEK1/2, p44/42MAPK and phospho-p44/42 purchased from Cell Signaling. Monoclonal anti- α-tubulin antibody was provided by Sigma-Aldrich. After incubation with secondary anti-goat antibody, the membranes were developed using an enhanced chemi-luminescence (ECL) detection system (BioRad). As concerning the in vivo experiments, tumor samples were homogenized as previously described [28]. Indicated proteins were probed with the same antibodies, as described above.
Tumor xenografts in nude mice
Four- to six-week old female balb/c athymic (nu+/nu+) mice were purchased from Envigo Laboratories. Three different in vivo experiments were performed. Briefly for the first experiment, mice were injected subcutaneously in the right flank with LIM1215-HER2 and SW48-HER2 cells and were randomly assigned to one of the following groups (10 mice/group). Group 1: vehicle administrated intraperitoneally (i.p.); group 2: refametinib administrated by oral gavage (o.g.) every day (25 mg/kg); group 3: pictilisib administrated by o.g. every day (75 mg/kg); group 4: combined treatment of refametinib (25 mg/kg) and pictilisib (75 mg/kg). Treatments were continued for 4 weeks. In the second in vivo experiment, groups of 90 mice injected in the right flank with LIM1215-HER2 and SW48-HER2 cells were treated with oxaliplatin (10 mg/kg) once every 2 weeks (i.p) plus trastuzumab (10 mg/kg) twice a week (i.p). At the end of 4 weeks treatment (induction treatment), mice were randomized into one of the following nine groups: Group 1: Veichle control; group 2: Pictilisib administrated by o.g. every day (75 mg/kg); group 3: refametinib administrated by o.g. every day (25 mg/kg); group 4: trastuzumab (10 mg/kg) twice a week i.p.; group 5: refametinib plus trastuzumab; group 6: pictilisib plus trastuzumab; group 7: pictilisib plus refametinib; group 8: lapatinib plus trastuzumab; group 9: the triplet combination of pictilisib, refametinib and trastuzumab. All drugs in the combined treatment are used at same concentration as single agent. Treatments continue for 8 weeks (maintenance treatment) and afterwards animals were followed for additional 16 weeks (follow-up period). At the end of maintenance treatment, one animal per group was sacrificed and tumor sample were collected for western blot analysis. In the third in vivo experiment, mice injected in the right flank with LIM1215-HER2 and SW48-HER2 cells were divided into four groups. Group 1: Vehicle control; group 2: refametinib (25 mg/kg) plus pictilisib (75 mg/kg) for 26 weeks; group 3: lapatinib (30 mg/kg) plus trastuzumab (10 mg/kg) for 26 weeks; group 4: lapatinib (30 mg/kg) plus trastuzumab (10 mg/kg) until progression. At the time of progression to lapatinib plus trastuzumab, mice were treated in second-line with combination of refametinib (25 mg/kg) and pictilisib (75 mg/kg). Tumor volume was measured using the formula π/6 × larger diameter x (smaller diameter)2. For monitoring tumor responses to therapy, we measured volumetric changes and used an arbitrary classification method modified from clinical research methods as previously reported [28].
Tumor specimen collection and annotation
Tumor and matched normal samples were obtained from patients treated by liver metastasectomy at the Candiolo Cancer Institute (Torino, Italy), Mauriziano Umberto I, and San Giovanni Battista (Torino). All patients provided informed consent.
Patient derived xenograft (PDX) models and in vivo treatments
Tumor implantation and expansion were performed in 6-week-old NOD/SCID mice as previously described [17, 29]. Mice were randomized and treated with the following agents: trastuzumab; refametinib; pictilisib. Tumor size was measured twice a week. Results were considered interpretable when a minimum of 4 mice per treatment group reached the pre-specified endpoints (at least 3 weeks on therapy or development of tumors with volumes larger than 1500 mm3) [30].
Statistical analysis
Statistical analyses of the in vitro data were performed using a one-way analysis of variance (ANOVA). Quantitative data were reported as mean ± standard deviation (SD). Results were compared by ANOVA.
Results
In vitro generation and characterization of HER2-amplified human colorectal cancer cell lines
The human CRC cell lines SW48 and LIM1215 harbor the wild type (WT) KRAS, NRAS, BRAF, and PIK3CA genes and are considerate an optimal preclinical model of EGFR therapeutic blockade. Since both cell lines have no or little expression of HER2 protein, they were transfected with HER2 gene in order to generate their HER2-amplified derivatives cells (SW48-HER2 and LIM1215-HER2). Transfection of HER2 gene results in cell stably overexpressing the HER2 protein (Additional file 1: Figure S1).To characterize the phenotype of these cells, we have studied the HER2 protein function on cell mobility. As illustrated in Additional file 1: Figure S2 A-B, parental SW48 and LIM1215 cells exhibited significant migratory capability. On the contrary their HER2-amplified derivatives cells demonstrated no ability in migration. These findings suggest that the amplification of HER2 gene is responsible of epithelial-mesenchymal-transition (EMT) loss. To further support this hypothesis, the expressions of epithelial- and mesenchymal-related proteins were measured. As depicted in Additional file 1: Figure S2 C-D, protein expression of different epithelial and mesenchymal markers changes between parental and HER2-amplified derivatives cells. In particular, the expressions of vimentin and slug, two mesenchymal markers, were decreased or suppressed in SW48-HER2 and LIM1215-HER2 cells, as compared to parental cells, respectively. On the contrary, E-cadherin, a common epithelial marker, strongly increased in both HER2-amplified derivatives cells as compared to parental cells. Collectively, these results suggest that colon cancer cell lines with HER2 amplification have lost an invasive behavior and have acquired an epithelial phenotype. In this scenario, we have performed additional western blot analyses to evaluate differential expression of other HER family receptors and their downstream effectors among parental cell lines SW48 and LIM1215 and their derived, HER2-amplified cancer cells. Western blot analysis showed an increased expression and phosphorylation of EGFR, HER3 and HER4 in HER2-amplified derivatives, that could lead to a complex intracellular signaling which includes the activation of the pro-survival PI3KCA/AKT pathway and of the mitogenic MAPK pathway [31, 32]. Activation of MAPK, MEK and AKT with an increase in their phosphorylated forms was observed in both HER2-amplified cells as compared to their parental counterpart (Additional file 1: Figure S3 A-B and Additional file 2: Table S1).
It has been demonstrated that HER2 is the only member of the HER family receptors that has no ligand and, therefore, it is activated mostly via hetero-dimerization with other HER family receptors [24, 33]. Therefore, we have analyzed whether the activation of AKT and MAPK effectors in HER2-amplified colon cancer cells could be due to the interaction of HER2 with other HER family receptors. For this purpose, SW48, SW48-HER2, LIM1215, LIM1215-HER2 protein extracts were immune-precipitated with a specific anti-HER2 antibody and then assayed by western blotting with a specific anti-HER3, anti-HER4 and anti-EGFR antibodies. As shown in Additional file 1: Figure S3-C, HER2 immuno-precipitated together with HER3 and EGFR forming HER2/HER3 and HER2/EGFR complexes only in SW48-HER2 and LIM1215-HER2 cells, but not in SW48 and LIM1215 cells. No immune-complexs were formed between HER2 and HER4 (data not shown). These data suggested the activation of HER2 signaling and this effect leads to activation of the pro-survival PI3K/AKT pathway and the mitogenic MAPK pathway through HER2/HER3 and HER2/EGFR heterodimerization.
Sensitivity to chemotherapeutic agents and to anti-EGFR monoclonal antibodiesin parental and in HER2-amplified human colon cancer cell lines
We first tested in vitro the activity of different chemotherapeutic agents in both parental and HER2–amplified colon cancer cell lines. All cancer cells we treated with 5-fluorouracil, oxaliplatin and irinotecan for 96 h. As shown in Additional file 1: Figure S4-A, there was no differential sensitivity to chemotherapies among all cancer cells. We next evaluated the sensitivity to the cell growth inhibiting effects of different anti-EGFR mAbs, including cetuximab, panitumumab, SYM004, and MM-151. As shown in Additional file 1: Figure S4-B, there was a differential sensitivity to anti-EGFR mAbs-induced cell growth inhibition. In fact, parental SW48 and LIM1215 cancer cells were significantly sensitive to cetuximab, panitumumab, SYM004 and MM-151 antibodies inducing growth inhibition, as expected being “quadruple wild type” for KRAS, BRAF, NRAS and PIK3CA genes. On the contrary, HER2-amplified colon cancer cells were resistant to all anti-EGFR inhibitors, expanding and confirming the results from previous studies regarding the role of HER2 amplification in either primary and acquired resistance to EGFR targeted therapies [17, 34, 35].
Effects of anti-HER2 inhibitors alone and/or in combination with anti-EGFR and anti-HER3 monoclonal antibodies on HER2-amplified human colon cancer cells
In previous studies, it has been demonstrated that HER2 amplification represents not only a biomarker of resistance to EGFR inhibition, but also a positive predictor of response to HER2 targeting agents [17]. In fact, mCRC patient derived xenografts with HER2 amplification were sensitive to HER2-blockade with trastuzumab in combination with lapatinib, but not to either agent alone. In HER2-amplified colon cancer cells monotherapy with either HER2 tyrosine kinase inhibitor or anti-HER2 antibodies was almost ineffective, whereas only the combination of a monoclonal antibody, trastuzumab, and a tyrosine kinase inhibitor, lapatinib, induced a significant anti-proliferative activity (Fig. 1a-e). Our data showed that, whereas pertuzumab, trastuzumab and lapatinib treatments had no or little effects as single agents on cell growth in LIM1215-HER2 and SW48-HER2 cells, a dual blockade of HER2 with trastuzumab and lapatinib had the most anti-proliferative effect compared to other treatments (Fig. 1 a-e). Moreover, this effect was not ameliorated by the addition of the other two anti-HER2 mAbs, as trastuzumab and pertuzumab. Furthermore, in HER2-amplified LIM1215-HER2 and SW48-HER2 colon cancer cells HER2 activation was accompanied by the interaction of HER2 with both EGFR and HER3, respectively, by forming HER2/EGFR and HER2/HER3 complexes. Therefore, we investigated whether a dual blockade of HER2 by using lapatinib and trastuzumab, that is considered the most active therapeutic strategy, in combination with anti-EGFR or anti-HER3 inhibitors could be a more effective treatment in HER2-amplified cells. As depicted in Fig. 1 a-e, the addiction of anti-EGFR mAbs to lapatinib plus trastuzumab combined treatment antagonized the anti-proliferative activity of the dual anti-HER2 blocked. In particular, in LIM1215-HER2 cells the IC50 for the triplet combination with trastuzumab, lapatinib and cetuximab was 3 μg/ml, 60 times higher than the IC50 of dual anti-HER2 blockade. Similar results were obtained by using triplet combinations with the anti-HER3 antibody duligotuzumab (Fig. 1 a-e).

Effects of dual block of anti-HER2 inhibitors alone and/or in combination with anti-EGFR and anti-HER3 mAbs on HER2-amplified (SW48-HER2 and LIM1215-HER2) human colon cancer cells. a-e Cells sensitivity to different drugs was evaluated by clonogenic assay in LIM1215-HER2 and SW48-HER2 cells. Data were represented as cell colony area calculated by ImageJ software after staining with crystal violet, as described in Materials and Methods. e Table with IC50 value for each drug, used as single agent or in combination
Effects of refametinib and pictilisib on cell proliferation and on EGFR-dependent intracellular pathways in parental SW48 and LIM1215 and in HER2-amplifiedhuman colon cancer cell lines
Given the observed concomitant activation of MAPK and PI3KCA-driven intracellular signals in HER2-amplified colon cancer cells, we next evaluated the cell growth inhibiting effects of a selective MEK1/2 inhibitor, refametinib, and of a selective PI3Kα inhibitor, pictilisib, as single agents or in combination. First, to evaluate the sensitivity to MEK and/or PI3KCA inhibitors between parental and derived cells, SW48, LIM1215, SW48-HER2 and LIM1215-HER2 cell lines were treated with both drugs as a single agent. As shown in Fig. 2-a, although refametinib and pictilisib treatments caused a dose-dependent cell growth inhibition in all colon cancer cell lines, the HER2-amplified cells were more sensitive to both drugs, as single agents, compared to parental cells at all concentrations tested. Next, we evaluated whether the treatment of refametinib in combination with pictilisib could induce a more effective anti-proliferative effect as compared to single treatments in HER2-amplifed colon cancer cell lines. The combined treatment with refametinib and pictilisib induced significant growth inhibition in both SW48-HER2 and LIM1215-HER2 cells (Fig. 2-b). To further evaluate if anti-HER2 blockade with trastuzumab or lapatinib could increase the anti-proliferative effects of refametinib plus pictilisib, SW48-HER2 and LIM1215-HER2 cells were treated with trastuzumab, lapatinib, refametinib and pictilisib, in different combinations. As depicted in Fig. 2-b, treatments with anti-HER2 agents, trastuzumab and lapatinib alone and in combination, had no effect when added to the refametinib plus pictilisib combined treatment. These results suggest that combination of refametinib plus pictilisib is more effective than lapatinib plus trastuzumab combined treatment and that the addition of anti-HER2 drugs to MEK and PI3KCA inhibitors does not significantly increase cell growth inhibition in these HER2-amplified colon cancer models. To better understand whether the anti-tumor activity obtained by the combined treatment with MEK and PI3KCA inhibitor was due to a more effective inhibition of key intracellular signals, EGFR downstream signaling pathways were evaluated. SW48-HER2 and LIM1215-HER2 cells were treated with refametinib, pictilisib and/or their combination. The combined treatment with refametinib plus pictilisib substantially inhibited the expression of EGFR, HER2, HER4 and their downstream effectors, such as AKT, MEK, and MAPK in their total forms and consequently the combination blocked their activation compared with single-agent treatments after 24 h of incubation. The concomitant block of PI3KCA/AKT and MAPK pathways could generate a negative feedback loop that led to the reduction in expression and activation of proteins. However, further investigation is needed to elucidate in detail these molecular mechanisms (Fig. 2c).

Effects of refametinib and pictilisib alone and in combination on colonies formation and on EGFR-dependent intracellular signaling in parental SW48 and LIM1215 human colon cancer cell lines and in their HER2-amplified derivatives (SW48-HER2 and LIM1215-HER2). a Both parental and HER2-amplified derivatives cells were treated with different concentrations of refametinib and pictilisib as single agents (range, 0.05 to 10 μM), for 96 h and evaluated byclonogenic assay. Data were represented as cell colony area percentage calculated by ImageJ software after staining with crystal violet, as described in Materials and Methods. b SW48-HER2 and LIM1215-HER2 cell lines were treated with lapatinib, trastuzumab, refametinib and pictilisib at 0.5 μM, as single agent and in their possible combinations (**p < 0.01 compared to other treatments). Colony area percentage was presented in the Table. c SW48-HER2 and LIM1215-HER2 cells were treated with refametinib and pictilisib (0.5 μM) alone and in combination for 24 h. Cell protein extracts were subjected to immunoblotting with the indicate antibodies, as described in Materials and Methods. α-Tubulin was used as the loading control. Results represent the mean of three separate experiments, each performed in duplicate
Effects of refametinib and pictilisib on HER2-amplified human cancer xenografts
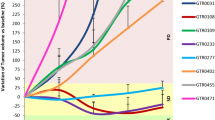
Subsequently, we investigated the in vivo antitumor activity of refametinib and pictilisib. Mice injected with LIM1215-HER2 and SW48-HER2 cells were randomly assigned to receive vehicle, refametinib (25 mg/kg), pictilisib (75 mg/Kg) or their combination for 4 weeks. As shown in Fig. 3, treatment with refametinib had little or no effect on tumor growth in both tumor xenografts. Similar results were obtained in the groups treated with pictilisib alone. On the contrary, the combined treatment suppressed almost completely LIM1215-HER2 and SW48-HER2 tumor growth at the end of the 4 weeks of therapy (Fig. 3 a-b). This complete suppression of tumor growth in the combined treatment group was long lasting up to week 20.

Effects of refametinib and pictilisib on LIM1215-HER2 and SW48-HER2 tumor xenografts. Mice were injected subcutaneously in the right flank with (a) LIM1215-HER2 and (b) SW48-HER2 cell lines, as described in Materials and Methods. After 2 weeks (average tumor size 200 mm3) mice were treated with refametinib (25 mg/kg every day, by o.g.) and pictilisib (75 mg/Kg every day, by o.g.), as single agents and in combination, for 4 weeks. Thereafter, tumor growth was followed without any further treatment until the 20th week. Data are means ± SD of ten mice in each group
Antitumor efficacy of oxaliplatin plus trastuzumab followed by maintenance treatment with different inhibitors alone or in combination with trastuzumab in HER2-amplified human cancer xenografts
In a subsequent in vivo experiment, two groups of 90 nude mice were injected subcutaneously with each HER2-amplified cancer cells and were treated for 4 weeks with the combination of oxaliplatin plus trastuzumab. At the end of this treatment, mice were randomized into nine groups and treated for 8 weeks with different inhibitors, including refametinib, pictilisib, lapatinib and trastuzumab alone or in combination (maintenance period) (Fig. 4) a-c. After the maintenance period, mice were followed for an additional 16 weeks. At the end of the maintenance treatment (week 14), among the single-agent treatments, the group treated with trastuzumab showed the greatest tumor growth inhibition in both xenograft models with a mean tumor volume of 971 and 1500 mm3, respectively (Fig. 4 a-c and Additional file 2: Table S2). In fact, the growth rate of tumors treated with pictilisib and refametinib as single agent was similar to those treated with vehicle regardless of cell lines injected reaching the maximum allowed tumor size of 2000 mm3 (Fig. 4 a-c and Additional file 2: Table S2). In the combined treatment groups, although the addition to refametinib and/or pictilisib to trastuzumab caused a strong antitumor activity in both xenograft models, the most effective antitumor activity was observed with the combined treatment of refametinib plus pictilisib. In particular, this combination caused an almost complete suppression of tumor growth in LIM1215-HER2 and SW48-HER2 tumor xenografts with a mean tumor volume of 75 and 103 mm3, respectively. Moreover, only in this treatment group mice with no evidence of tumors were observed (Fig. 4 a-c and Additional file 2: Table S2). Mice were followed for additional 16 weeks (follow-up period) from the end of the treatment to evaluate which maintenance strategy could determine a more sustained and prolonged tumor growth control. In the combined trastuzumab plus refametinib or pictilisib groups, tumors started to regrowth immediately after the cessation of treatment. Indeed, 3 to 5 weeks after cessation of treatment the tumor growth rate in these combined treatment groups was comparable to the tumor growth rate in the trastuzumab group for both HER2-amplified cancer models. On the contrary, the anti-tumor activity of the combined refametinib plus pictilisib treatment was still maintained within 13 to 16 weeks after the cessation of therapy. In particular, in this combined treatment group almost half of the mice were still alive at the end of the follow-up period (Fig. 4a-c and Additional file 2: Table S2). Moreover, in order to evaluate the mechanism(s) by which the combined treatment with refametinib plus pictilisib was able to induce a significant and long lasting antitumor activity, one mouse for each group treated with refametinib, pictilisib or with their combination was sacrificed at the end of the maintenance treatment. As shown in Fig. 4-d, the combined treatment with refametinib plus pictilisib was the only treatment able to substantially inhibit the expression and activation of HER receptors family and their downstream effectors in their total forms at the end of the maintenance treatment.

Effect of maintenance treatment with different kinase inhibitors alone or in combination with trastuzumab, after induction therapy with oxaliplatin plus trastuzumab in HER2-amplified colon cancer xenograft models. HER2-amplified colon cancer cells (SW48-HER2 and LIM1215-HER2) were injected into the right flank of nude mice. After 2 weeks of subcutaneous injection, mice were treated with oxaliplatin (10 mg/kg once every 2 weeks, i.p.) in combination with trastuzumab (10 mg/kg twice a week, i.p.) for 4 weeks (induction treatment). Afterward, mice were randomized into nine groups and treated for 8 weeks (maintenance treatment). Ctrl: control; pictilisib (75 mg/Kg) and refametinib (25 mg/kg) were administrated every day for 5 days by o.g, as single agents and in different combinations; trastuzumab (10 mg/kg twice a week, by i.p.) and lapatinib (30 mg/kg every day, by o.g.) as single agents and in different combinations. a Treatment scheme. Red boxes: pictilisib treatment days; green boxes: refametinib treatment days; violet boxes: trastuzumab treatment days; light blue boxes: lapatinib treatment days. b–c Antitumor activity of maintenance treatment in SW48-HER2 and LIM1215-HER2 tumor-bearing mice. The indicated cancer cell lines were grown as subcutaneous tumor xenografts in nude mice and treated with different drugs as indicated above. The mean data are present. Tumor growth curves were calculated on the basis of three times a week tumor measurements during the treatment period and after 16 weeks of observation after termination of therapy. d Analysis of EGFR-dependent intracellular signaling by western blotting in LIM1215-HER2 colorectal cancer xenograft. At the end of maintenance treatment, 1 mouse per group treated with refamentinib, pictilisib or with their combination was sacrificed. Tumor samples were collected, and total cell protein extracts were subjected to immunoblotting with the indicated antibodies, as described in materials and methods. Anti-tubulin antibody was used for normalization of protein extract content
To further dissect the effects of refametinib plus pictilisib combined treatment on HER2-amplified xenograft models, LIM1215-HER2 and SW48-HER2 cells were injected subcutaneously into nude mice and were treated with veichle, lapatinib plus trastuzumab or pictilisib plus refametinib until tumors increased growth (Fig. 5 a-b). When tumors were growing despite treatment with lapatinib plus trastuzumab, treatment with refametinib plus pictilisib was started. As shown in Fig. 5, treatment with refametinib plus pictilisib provided further antitumor activity. However, this effect was transient. In fact, after 5 weeks of this therapy, tumors started to regrowth reaching the maximum allowed tumor size of 2000 mm3 at week 26. On the contrary, the group of mice treated with refametinib plus pictilisib from the beginning of the experiment experienced the best antitumor efficacy with no evidence of tumor progression up to week 26 (Fig. 5 a-b). Moreover, the combined treatment with refametinib plus pictilisib as initial therapy substantially inhibited phosphorylation of EGFR, HER2, HER3, HER4 and of downstream signaling pathways such as MEK, MAPK, and AKT, as measured by western blotting at the end of the treatment period (Fig. 5-c). Collectively, these data support the hypothesis that in HER2-amplified colon cancer xenografts treatment with refametinib plus pictilisib is more effective as initial therapy in order to better control the onset of cancer cell resistance mechanisms, that are more frequent and develop earlier with HER2 blockade.

Effect of refametinib and pictilisib combined treatment after tumor progression due to lapatinib plus trastuzumab therapy in HER2-amplified colon cancer xenograft models. a-b SW48-HER2 and LIM1215-HER2 were injected into right flank of nude mice. After 2 weeks from subcutaneous injection, mice were divided in four groups and treated for 26 weeks. Group 1: ctrl. Group 2: lapatinib (30 mg/kg every day, o.g.) and trastuzumab (10 mg/kg twice a week, i.p.) were administrated in combination. Group 3: refametinib (25 mg/kg every day, o.g.) and pictilisib (75 mg/Kg every day, o.g.) were administrated in combination. Group 4: mice were treated with combination of lapatinib (30 mg/kg every day, o.g.) and trastuzumab (10 mg/kg twice a week, i.p.) until progression (defined as > 30% increment of tumor volume from baseline). After tumor progression of group 4, mice were treated in second-line with combination of refametinib and pictilisib. c Tumor samples were collected and total protein extracts were subjected to immunoblotting with all antibodies, as described in materials and methods section. Anti-tubulin antibody was used for normalization of protein extract content
Effect of refametinib and pictilisib combined treatment with or without trastuzumab in HER2-amplified metastatic colorectal cancer patient derived tumor xenografts
One way to proceed with efficient, high-fidelity drug development at the stage of in vivo validation while minimizing the effects of uncharacterized tumor heterogeneity is to perform preclinical population-based studies by using human cancer specimens directly transplanted into mice (“xenopatients”) [17]. For this reason, to further evaluate the antitumor efficacy of refametinib plus pictilisib as a therapeutic strategy for HER2-amplified colon cancer we used patients derived tumor xenografts (PDTXs), by selecting three representative cases (CRC 1432, CRC 1430 and CRC 0186) of HER2-amplified tumors from mCRC patients [17]. For each experiment, mice were divided into four groups and treated for 5 weeks with vehicle, trastuzumab, refametinib plus pictilisib or the triple combination of refametinib, pictilisib and trastuzumab (Fig. 6 a-b). The combination treatment with refametinib plus pictilisib and triple combination of refametinib, pictilisib and trastuzumab were highly effective in suppressing tumor growth, as compared to either the vehicle-treated controls or the trastuzumab single agent treatment group. Moreover, the combined treatment with refametinib plus pictilisib exhibited an even greater antitumor activity as compared to the triple combination.

Effect of refametinib and pictilisib combined treatment with or without trastuzumab in HER2-amplified metastatic colorectal cancer patient derived tumor xenografts. Tumor samples with HER2 amplification derived from three different patients were directly implanted and expanded in mice. a Representative tumor growth curves of tumors in xenopatients derived from quadruple-negative HER2-amplified cases CRC 1432, CRC 130, CRC 0186 treated with the indicated modalities for 5 weeks: trastuzumab 10 mg/kg i.p., twice weekly; refametinib 25 mg/kg by o.g., daily; pictilisib 75 mg/kg by o.g., daily. b Histogram represents the tumor volume of each treatment compared to the maximum growth of the control at the fourth week
Discussion
In the past decade, the development of targeted therapies has provided new options for the personalized management of patients with advanced solid tumors [1]. In particular, CRC represents a heterogeneous group of diseases with different sets of genetic events, accompanying immune response, and influences of exogenous factors, providing a challenge for personalized therapeutic approaches [1]. The mAbs directed against the EGFR, such as cetuximab and panitumumab, are currently approved for the treatment of patients with mCRC [1,2,3,4,5]. Despite significant progress in strategies for cancer treatment, these therapies have improved patient responses and their use is limited by the presence of pre-existing intrinsic resistance mechanisms or by the ability of cancer cells to acquire resistance [6,7,8,9,10].
One of major mechanism of acquired resistance to anti-EGFR mAbs is the activation of growth-factor signaling pathways by upregulation of alternative and compensatory signaling cascades through receptors other than EGFR [14,15,16]. In particular, HER2 amplification has been suggested as a mechanism of resistance [14, 17]. One explanation could be that pre-existing infrequent HER2-amplified clones might be expanded under the selective pressure of anti-EGFR therapy, leading to disease progression [20]. In this regard, HER2 amplification is more likely to confer acquired anti-EGFR therapy resistance. HER2 amplification was found in 5% of mCRC patients harboring no mutation in KRAS, NRAS, BRAF, PI3KCA genes [17,18,19,20] and together with “xenopatients” preclinical data provided the rational for clinical studies with HER2-targeting therapies after failure of anti-EGFR treatments [17, 23, 24]. The HERACLES-A clinical trial reported clinical meaningful responses to dual inhibitor HER2-directed therapy in a subset of HER2-amplified mCRC patients, whose disease was refractory to chemotherapy and anti-EGFR antibodies [23, 36].
Nevertheless, the mechanisms of resistance to HER2-directed and EGFR-directed therapies are still unclear. An improved understanding of the molecular characteristics of HER2-amplified CRC models and their potential mechanisms of resistance to HER2-directed therapy may influence the direction of future research on targeted therapies and inform future therapeutic decision-making.With this aim, starting from quadruple wild-type human CRC cells for KRAS, NRAS, BRAF and PI3KCA genes (LIM1215 and SW48), we have generated HER2-amplified (LIM1215-HER2 and SW48-HER2) cells. In the current study, we report the up-regulation of HER family receptors and over-expression of several markers involved in RAS/RAF/MAPK and PI3KCA-AKT pathways in HER2-amplified cells compared to parental colon cancer cells. Interestingly, these oncogenic pathways were activated by the interaction between HER2/HER3 and HER2/EGFR, consequent to the heterodimerization of these receptors. In agreement with these findings, several studies have demonstrated that the acquired resistance to anti-EGFR mAbs was associated with the increased activation not only of HER2, but also of HER3 and/or other alternative receptor tyrosine kinases with consequent activation of EGFR-independent intracellular downstream pathways [37, 38]. Based on these data, in order to inhibit the compensatory feedback effect due to activation of downstream signaling pathways, we have tested as therapeutic strategy the combination of two selective MEK and PI3KCA inhibitors, refametinib and pictilisib, respectively, in HER2-amplified colon cancer cells (LIM1215-HER2 and SW48-HER2). The combined treatment with refametinib plus pictilisib determined a strong antitumor activity both in vitro and in vivo, providing the rationale for the further clinical development of this combination. In particular, in nude mice bearing LIM1215-HER2 or SW48-HER2 CRC tumor xenografts, the combined treatment with refametinib plus pictilisib caused complete tumor regression that lasted up to 20 weeks of follow up after the end of treatment. These results suggest that the concomitant blockade of two key intracellular signaling hubs that could be involved in the development of cancer cell resistance to anti-HER2 inhibitors might be a possible strategy to delay or prevent its onset. Moreover, to extend the validation and the potential clinical relevance of these findings, we have performed an additional in vivo experiment. We treated mice first with a combination of oxaliplatin plus trastuzumab for 4 weeks as a strategy to treat HER2-amplified mCRC patients [23]. Before tumor started to regrowth and eventually possible resistance mechanisms to targeted agents would occur, mice were randomized to different maintenance treatments with several kinase inhibitors, such as refametinib, pictilisib, lapatinib or trastuzumab alone or in combination. The combined treatment with refametinib plus pictilisib again was the best therapeutic strategy also in this experimental maintenance therapy setting. Moreover, these findings have been further validated in three HER2-amplified mCRC patient derived xenografts, which could better resemble the complexity of a human mCRC, since these models have been demonstrated as a better surrogate of human cancer and may represent a valid experimental tool to overcome the limitations of in vitro models by faithfully recapitulating the histological and functional heterogeneity observed in primary tumor samples. The combined treatment with refametinib plus pictilisib exhibited a significant antitumor activity, that was accompanied by a sustained tumor growth inhibition in all three HER2-amplified mCRC patient derived xenografts.
Collectively, these data suggest that the dual and combined inhibition of MEK and PI3KCA significantly inhibit tumor growth in several different models of HER2-amplified CRC both in vitro and in vivo. In this respect, early clinical trials have evaluated the combined inhibition of MEK and PI3KCA signaling pathways in different tumor types [39,40,41,42]. In particular, although the effect of PI3KCA pathway activation as a mechanism of resistance to HER2-directed therapy in CRC has not been directly investigated, activating mutations of PI3KCA and decreased expression of PTEN have been identified as potential mechanisms of resistance to trastuzumab and lapatinib in breast cancer [43, 44]. A study using human breast cancer cell lines and mouse tumor xenografts has shown that activating mutations of PIK3CA and/or decreased expression of PTEN could be responsible of resistance to lapatinib and that this resistance is reversible by double blockade of PI3KCA and mTOR [45].
The ongoing Personalized Oncogenomics Group (POG) trial (NCT02155621) has undertaken complete molecular characterization of a total of 60 patients with mCRC, two of which showed high-level of HER2 amplification. With the aim of better understanding mechanisms of resistance to HER2-directed and EGFR-directed therapies, Owen et al. reported the complete molecular characterization of these two cases of HER2-amplified mCRC. Their findings included increased expression of MUC1 and MET, decreased expression of PTEN, and an activating mutation in PIK3CA [46]. These data support the hypothesis that a potentially promising alterative to overcome resistance mechanisms would be to apply a therapy in the upfront setting in order to suppress and ideally eradicate pre-existing resistant clones while they still are present in a low frequency subpopulation.
Conclusions
In summary, the present study provides experimental evidence that the combined treatment with refametinib plus pictilisib could be a potential novel therapeutic strategy to treat HER2-amplified mCRC patients with the aim of improving the outcome of this aggressive disease subgroup which is mostly refractory to standard therapies.
Abbreviations
- Akt:
-
Protein Kinase-B
- CRC:
-
colorectal cancer
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
epithelial- mesenchymal transition
- HER family:
-
Epidermal growth factor receptor family
- HER2:
-
Human epidermal growth factor receptor 2
- HER3:
-
Human epidermal growth factor receptor 3
- HER4:
-
Human epidermal growth factor receptor 4
- HERACLES:
-
HER2 Amplification for ColorectaL Cancer Enhanced Stratification
- mAbs:
-
monoclonal antibodies
- MAPK:
-
mitogen-activated protein kinase
- mCRC:
-
metastatic colorectal cancer
- PDTX:
-
patient derived tumor xenograft
- PI3KCA:
-
Phosphatidylinositol 4,5-bisphosphate 3-kinase
- WT:
-
wild-type
References
Pietrantonio F, Vernieri C, Siravegna G, Mennitto A, Berenato R, Perrone F, et al. Heterogeneity of Acquired Resistance to Anti-EGFR Monoclonal Antibodies in Patients with Metastatic Colorectal Cancer. Clin Cancer Res. 2017; 15;23(10):2414–2422.
Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34.
Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol. 2011;29:2011–9.
Fakih MG. Metastatic colorectal cancer: current state and future directions. J Clin Oncol. 2015;33:1809–24.
Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27:1386–422.
Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–40.
Misale S, Arena S, Lamba S, Siravegna G, Lallo A, Hobor S, et al. Blockadeof EGFR and MEK intercepts heterogeneous mechanisms of acquired resistance to anti-EGFR therapies in colorectal cancer. Sci Transl Med. 2014;6:224ra26.
Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–3.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, et al. Effects of KRAS, BRAF, NRAS, and PI3KCA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–62.
Troiani T, Martinelli E, Napolitano S, Morgillo F, Belli G, Cioffi L, et al. Molecular aspects of resistance to biological and non-biological drugs and strategies to overcome resistance in colorectal cancer. Curr Med Chem. 2014;21:1639–53.
Altorta E, Misale S, Sartore-Bianchi A, Nagtegaal ID, Paraf F, Lauricella C, et al. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int J Cancer. 2013;133:1259–65.
Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer. 2007;97:1139–45.
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–6.
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activationn of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. SciTransl Med. 2011;3:99–59.
Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravenga G, et al. Amplification of MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3:658–73.
Troiani T, Martinelli E, Napolitano S, Vitagliano D, Ciuffreda LP, Costantino S, et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013;19:6751–65.
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011;1:508–23.
Parikh A, Atreya C, Korn WM, Venook AP. Prolonged response to HER2-directed therapy in a patient with HER2-amplified, rapidly progressive metastatic colorectal cancer. J Natl Compr Cancer Netw. 2017;15:3–8.
Martinelli E, Troiani T, Sforza V, Martini G, Cardone C, Vitiello PP, et al. Sequential HER2 blockade as effective therapy in chemorefractory, HER2 gene-amplified, RAS wild-type, metastatic colorectal cancer: learning from a clinical case. ESMO Open 2018; 10;3(1):e000299.
Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526:263–7.
Leto SM, Sassi F, Catalano I, Torri V, Migliardi G, Zanella ER, et al. Sustained inhibition of HER3 and EGFR is necessary to induce regression of HER2-amplified gastrointestinal carcinomas. Clin Cancer Res. 2015;21:5519–31.
Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5(8):832–41.
Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/ 13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17(6):738–46.
Siena A, Sartore-Bianchi S, Marsoni HI, Hurwitz S, McCalln J, Penault-Llorca F, et al. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann Oncol. 2018;29(5):1108–19.
Haslem DS, Ji HP, Ford JM, Nadauld LD. Precision oncology strategy in trastuzumab-resistant human epidermal growth factor receptor 2-positive colon cancer: case report of durable response to ado-trastuxumabemtansine. JCO Precis Oncol. 2017. https://doi.org/10.1200/PO.16.00055.
Hainsworth JD, Meric-Bernstam F, Swanton C, Hurwitz H, Spigel DR, Sweeney C, et al. Targeted therapy for advanced solid tumors on the basis of molecular profiles: results from Mypathway, an open-label, phase IIa multiple basket study. J Clin Oncol. 2018;36(6):536–42.
Franken NA, Rodermond HM, Stap J, Haveman J, Van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1(5):2315–9.
Troiani T, Napolitano S, Martini G, Martinelli E, Cardone C, Normanno N, et al. Maintenance treatment with Cetuximab and BAY86-9766 increases antitumor efficacy of irinotecan plus Cetuximab in human colorectal Cancer xenograft models. Clin Cancer Res. 2015;21(18):4153–64.
Galimi F, Torti D, Sassi F, Isella C, Corà D, Gastaldi S, et al. Genetic and expression analysis of MET, MACC1, and HGF in metastatic colorectal cancer: response to MET inhibition in patient xenografts and pathologic correlations. Clin Cancer Res. 2011;17(10):3146–56.
Baralis E, Bertotti A, Fiori A, Grand A. LAS: a software platform to support oncological data management. J Med Syst. 2012;36(Suppl1):S81–90.
Janmaat ML, Rodriguez JA, Gallegos-Ruiz M, Kruyt FA, Giaccone G. Enhanced cytotoxicity induced by gefitinib and specific inhibitors of the Ras or phosphatidyl inositol-3 kinase pathways in non-small cell lung cancer cells. Int J Cancer. 2006;118(1):209–14.
Gandhi J, Zhang J, Xie Y, Soh J, Shigematsu H, Zhang W, et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS One. 2009;4(2):e4576.
Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5(5):341–54.
Napolitano S, Martini G, Martinelli E, Belli V, Parascandolo A, Laukkanen MO, et al. Therapeutic efficacy of SYM004, a mixture of two anti-EGFR antibodies in human colorectal cancer with acquired resistance to cetuximab and MET activation. Oncotarget. 2017;8(40):67592–604.
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, et al. Activation of ERBB2 signaling causes resistance to the EGFR-directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86.
Siena S, Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, et al. D01* final results of the Heracles trial in Her2 amplified colorectal cancer. Ann Oncol. 2016;27(Suppl. 4):iv39.
Khelwatty SA, Essapen S, Seddon AM, Fan Z, Modjtahedi H. Acquired resistance to anti-EGFR mAb ICR62 in cancer cells is accompanied by an increased EGFR expression; HER-2/HER-3 signalling and sensitivity to pan HER blockers. Br J Cancer. 2015;113(7):1010–9.
Zhang L, Castanara C, Luan B, Fan L, Fairhurst JL, Rafique A, et al. ERBB3-HER2 signaling promotes resistance to EGFR blockade in head and neck and colorectal cancer models. Mol Cancer Ther. 2014;13(5):1345–55.
Sato H, Yamamot H, Sakaguchi M, Shien K, Tomida S, Shien T, et al. Combined inhibition of MEK and PI3K pathways overcomes acquired resistance to EGFR-TKIs in non-small lung cancer. Cancer Sci. 2018;109(10):3183–96.
Elmohk O, Ruffieux-Daidie D, Roelli MA, Stooss A, Phillips WA, Gertsch J, et al. Combined MEK and Pi3’-kinase inhibition reveals synergy in targeting thyroid cancer in vitro and in vivo. Oncotarget. 2017;8:24604–20.
Siravegna S, Lazzari L, Crisafulli G, Sartore-Bianchi A, Mussolin B, Cassingena A, et al. Radiologic and genomic evolution of individual metastases during HER2 blockade in colorectal cancer. Cancer Cell. 2018;34:148–62.
Britten CD. PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol. 2013;71(6):1395–409.
Hanker AB, Pfefferle AD, Balko JM, Kuba MG, Young CD, Sanchez V, et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc Natl Acad Sci. 2013;110(35):14372–7.
Loibl S, Von Minckwitz G, Schneeweiss A, Paepke S, Lehmann A, Rezai M, et al. PIK3CA mutations are associated with lower rates of pathologic complete response to anti-human epidermal growth factor receptor 2 (her2) therapy in primary HER2-overexpressing breast cancer. J Clin Oncol. 2014;32:3212–20.
Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, et al. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008;68(22):9221–30.
Owen DR, Wong HL, Bonakdar M, Jones M, Hughes CS, et al. Molecular characterization of ERBB2-amplified colorectal cancer identifies potential mechanisms of resistance to targeted therapies: a report of two instructive cases. Cold Spring Harb Mol Case Stud. 2018;4(2).
Acknowledgments
We thank all individuals who take part in this research.
Funding
This research has been supported by a grant from Associazione Italiana per la Ricerca sul Cancro (AIRC) to FC (AIRC IG 18972).
Availability of data and materials
All data generated or analyzed during this study are included either in this article or in the supplementary information files.
Author information
Authors and Affiliations
Contributions
VB, TT, and FC conceived and designed the experiments; VB and NM performed the experiments; GM, FC, AB and LV also contributed to performing PDTX experiments and data interpretation. VB, TT, EM and FM analyzed the data; DC, VDF, EFG and UB contributed to statistical analysis; VB, SN, TT and FC wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The University of Campania “Luigi Vanvitelli” (Naples, Italy) and Candiolo Cancer Institute (Torino, Italy) approved this study.
Consent for publication
All authors agree on publication of the results of the present manuscript.
Competing interests
TT: advisory board for Amgen, Bayer, Merck, Servier, Roche, Sanofi. EM: advisory board for Amgen, Bayer, Merck, Roche, Sanofi, Servier, Biocartis and expert opinion for ESMO (European Society of Medical Oncology).FM: advisory boards: MSD, Lilly; institutional research grants: AstraZeneca. FC: Advisory Boards, Bayer, Merck KgA, Roche, Servier, Amgen, Symphogen, Pfizer; Research funds, Roche, Bayer, Merck KgA, Ipsen, Amgen.LV: Symphogen, Merus, Pfizer, Servier, Eli Lilly, AstraZeneca, Merck. AB: Relay Therapeutics. VB, NM, SN, GM, FC, DC, VDF, EFG and UB declare no competing conflict of interest regarding the following manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. Expression and phosphorylation of HER2 in parental SW48 and LIM1215 human colon cancer cell lines and in their HER2-amplified derivatives (SW48-HER2 and LIM1215-HER2) cells (Additional file 3: Supplementary Methods). Figure S2. Phenotypic characterization of parental SW48 and LIM1215 human colon cancer cell lines and of their HER2-amplified derivatives (SW48-HER2 and LIM1215-HER2) cells. Figure S3. Expression and phosphorylation of HER family receptors and their downstream signaling pathways in parental SW48 and LIM1215 human colon cancer cell lines and in their HER2-amplified derivatives (SW48-HER2 and LIM1215-HER2) cells. Figure S4. Effects of chemotherapeutic agents and of anti-EGFR monoclonal antibodies on cell proliferation in parental SW48 and LIM1215 human colon cancer cell lines and in their HER2-amplified derivatives (SW48-HER2 and LIM1215-HER2) cells. (DOCX 5258 kb)
Additional file 2:
Table S1. Evaluation of protein expression level in parental (SW48 and LIM1215) and in HER2-amplified human colon cancer cell lines. Legend: Negative symbol (-) no protein expression detected; Positive symbols: (+) expression and (++) over-expression levels of each protein detected. Table S2. Antitumor efficacy of oxaliplatin plus trastuzumab followed by maintenance treatment in human HER2-amplified colon cancer xenograft. (DOCX 613 kb)
Additional file 3:
Supplementary Methods. (DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Belli, V., Matrone, N., Napolitano, S. et al. Combined blockade of MEK and PI3KCA as an effective antitumor strategy in HER2 gene amplified human colorectal cancer models. J Exp Clin Cancer Res 38, 236 (2019). https://doi.org/10.1186/s13046-019-1230-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-019-1230-z