
Abstract
Thyroid malignant neoplasm develops from follicular or parafollicular thyroid cells. A higher proportion of anaplastic thyroid cancer has an adverse prognosis. New drugs are being used in clinical treatment. However, for advanced thyroid malignant neoplasm such as anaplastic thyroid carcinoma, the major impediment to successful control of the disease is the absence of effective therapies. Oncolytic virotherapy has significantly progressed as therapeutics in recent years. The advance is that oncolytic viruses can be designed with biological specificity to infect, replicate and lyse tumor cells. Significant advances in virotherapy have being achieved to improve the accessibility, safety and efficacy of the treatment. Therefore, it is necessary to summarize and bring together the main areas covered by these investigations for the virotherapy of thyroid malignant neoplasm. We provide an overview of the progress in virotherapy research and clinical trials, which employ virotherapy for thyroid malignant neoplasm as well as the future prospect for virotherapy of thyroid malignant neoplasms.
Similar content being viewed by others


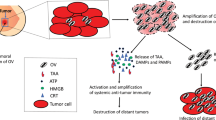
Background
Thyroid malignant neoplasm (TMN) occurs in the follicular or parafollicular thyroid cells. Thyroid, a butterfly-shaped gland located at the base of neck, produces hormones that regulate heart rate, blood pressure, body temperature and weight. Thyroid malignant neoplasm is the most common endocrine malignancy, accounting for 1.9% of all new malignant tumors diagnosed annually in the United States [1]. TMN can be classified according to their histopathological characteristics. Papillary (PTC), follicular (FTC), and anaplastic thyroid carcinomas (ATC) arise from endodermal-derived follicular cells, which represent the most abundant cellular population of the thyroid gland. PTC comprises 80% to 85% of all thyroid neoplasms, which is frequent in young females with excellent prognosis. FTC is accounting for approximately 10% to 15% of cases. PTC and FTC will lie in the low-risk group, have a low recurrence rate (<5%) and excellent survival rate (>98%) [2]. The least common histotype is ATC (1–2%), which has a rapid progression and a very poor prognosis with a mean survival time of 2–6 months. Surgery, radiotherapy and chemotherapy do not improve survival [3]. Medullary thyroid carcinoma (MTC), about 3% of all thyroid cancer cases, originates from parafollicular C cells, which produce the hormone calcitonin. Currently, the most effective management of aggressive thyroid malignant neoplasm is surgical removal of the thyroid, followed by radioactive iodine ablation and TSH-suppression therapy. However, more effective therapies are urgently needed to minimize dismal outcomes for the thyroid malignant neoplasm [4]. Oncolytic virotherapy is a promising anti-cancer strategy, because it can provide a local regional control, or eradication of tumors without cross-interference with standard therapies [5]. Oncolytic viruses can be divided into wild type viruses that are naturally oncolytic such as reovirus, measles virus, Newcastle disease virus (NDV), myxoma virus (MYXV), and vesicular stomatitis virus (VSV) and those that require genetic modification for selective oncolysis. Genetic modification for selective oncolysis involves either the insertion or deletion of genes [6]-[9].
In this review, we have recapitulated the important findings that were obtained in preclinical and clinical trials for the treatment of TMN, as well as the future goals, with a particular focus on the strategies in the design of virotherapy.
Virotherapy for TMN
The viruses used for TMN mainly include three DNA viruses (Adenovirus, Herpesvirus and Vaccinia virus) and five RNA viruses (Reovirus, Newcastle disease virus, Vesicular stomatitis virus, Measles virus and Retrovirus). These viruses have been studied as potential therapeutics for monotherapy, or combination therapy in conjunction with chemotherapy and/or radiation therapy in pre-clinical or clinical trials (Table 1). We discuss these viruses in the order of the clinical study progress for TMN treatment.
Adenovirus - preclinical and clinical studies
Adenoviruses (Adv) are medium-sized (90–100 nm), nonenveloped viruses with an icosahedral nucleocapsid containing a double stranded DNA (dsDNA) genome between 26 and 48 Kb. This allows the virus to encode for 22 to 40 genes [10]. The two currently established receptors are CD46 for the group B human adenovirus serotypes and the coxsackievirus adenovirus receptor (CAR) for all other serotypes. A co-receptor is called αv integrin that results in endocytosis of the virus particle via clathrin-coated pits [11]. In this process, a series of viral proteins co-operate to promote efficient replication of the virus and its release. These major viral proteins include E1A, E1B-55kD, E1B-19kD, E3-11.6 kD and other associated proteins [12]. Human adenovirus have 57 distinct adenoviral serotypes have been found to cause a wide range of illnesses, from mild respiratory infections in young children to life-threatening multi-organ disease in people with a weakened immune system. Nearly all adults have antibodies to endemic serotypes 1, 2, 5, or 6 [13].
Various Adenovirus strains have been explored extensively as a viral vector for gene therapy and also as an oncolytic virus. A common strategy in adenovirus work is to express an enzyme inhibitor, a suicide gene such as express herpes simplex virus (HSV) thymidine kinase (tk) and then administer ganciclovir (GCV), or express p53, interleukin-2 (IL-2) and interleukin-12 (IL-12), or human sodium iodide symporter (NIS) or pigment epitheliumderived, factor (PEDF) as a potent inhibitor of tumor angiogenesis [14] and then used in combination therapy. Combination approaches have been explored as means to promote adenovirus replication and killing at tumor sites.
Shen et al. [15] constructed an oncolytic adenovirus with a Survivin targeted small hairpin RNA and a reporter gene (ZD55-Sur-EGFP). The virus can replicate specifically in cancer cells, reduce effectively both the expression of mRNA and protein expression of Survivin (P <0.0001), induce cancer cell apoptosis and inhibit human colon adenocacinoma SW480 cell line growth both in vitro and in vivo [15]. Minemura et al. [16] characterized two adenovirus-derived vectors, which were combined with GCV to inhibit the growth of MTC cells. These two adenoviral-based vector systems were termed AdCMVtk and AdDCTtk. The latter contains human CALC-I minigene under the control of the CALC-I promoter, whereas the former carries the HSVtk gene driven by the human cytomegalovirus promoter. All cell lines transduced with AdCMVtk were rendered sensitive to GCV. Cell killing was also observed in HeLa, HepG2, rat MTC and TT (human MTC) cells by the combination of GCV and AdDCTtk vector [16]. Zhang et al. [17] evaluated cell-specific killing activity of replication defective adenovirus transducing TK gene under the control of the rat thyroglobulin (rTg) promoter (AdrTgtk) and then to transduce various cell lines. When FRTL-5 cells were infected with AdrTgtk followed by GCV treatment, more than 90% were killed [17]. Moreover, the enhanced effect of combined HSV-tk and ganciclovir with IL-2 [18] and combined HSV-tk/GCV, IL-12 [19] and p53 [20] have also been evaluated. The tumor suppressor p53 is a transcription factor that regulates cell cycling, DNA repair and apoptosis. However, p53 mutations are not frequent in thyroid cancer. The exogenous wild type p53 could increase chemo sensitivity in three anaplastic thyroid carcinoma cell lines (BHT-101, SW-1736, and KAT-4), which had nonfunctional p53 and ATC cell lines have been observed [21]. It is probable that the majority of thyroid cancers activate an alternative pathway that compromises the function of wild-type p53. Well-differentiated thyroid cancers generally do not express a mutation in p53 [22].
Clinical trials were carried out using recombinant adenoviral-encoded human p53 gene (rAd-p53), combined with intensity-modulated radiation therapy (IMRT) for treatment of 46 patients with historically-diagnosed stage IV PTC. The patients received intratumoral injection. A month later, 76% (19/25) of patients underwent radical surgery. One-year survival rate was 96% and no local recurrence and distant metastases were observed in the cohort of patients with surgery [23]. Currently, the clinical trials, rAd-p53 gene therapy for advanced malignant thyroid tumors (stage III/IV) is in phase IV [24]. When ONYX-015 was combined with the standard chemotherapeutic agents cisplatin and 5′-fluorouracil to combat TMN, a considerable efficacy was reported in a proportion of cases [25]. ONYX-015 proved safe at a dose up to 2 × 1012 viral particles [26]. So far, ONYX-015 is the only oncolytic virus product on the market [27]. E1A-defective adenovirus dl922-947 and DeltaE1B55K (dl1520) mutants were compared in human thyroid anaplastic carcinoma cells in vivo and in vitro. The efficacy of dl922-947 exceeded that of dl1520 in all tested anaplastic thyroid carcinoma cells. The combination of dl922-947 with bevacizumab reduced significantly tumor growth compared with single treatments alone [28]. Recently, an important genetically modified antiangiogenic adenovirus, termed VB-111, exhibited to have significant antitumor activity for thyroid cancer. VB-111 expresses Fas-c in angiogenic blood vessels. VB-111 treatment resulted in 26.6% (P =0.0596), 34.4% (P =0.0046), and 37.6% (P =0.0249) inhibition of tumor growth in follicular, papillary and anaplastic thyroid cancer cell line models, respectively. No toxicity was observed in these studies [29]. The clinical trial based on VB-111 for oncolysis is currently underway for patients bearing thyroid malignant neoplasm. The multi-dose VB-111 phase II clinical trial dealt with the treatment of patients with gliobastoma multiforme, differentiated thyroid cancer and ovarian cancer (NCT0120506).
Reovirus - preclinical and clinical studies
Mammalian orthoreoviruses (reoviruses) are ubiquitous viruses that infect cells in the respiratory and enteric tracts. Reoviruses have a 70-85 nm diameter and are non-enveloped nearly spherical icosahedral particles. The icosahedral capsid is composed of an outer and inner protein shell. The double stranded RNA (dsRNA) genomes contain 10–12 segments, which are grouped into three categories corresponding to their size: L (large), M (medium) and S (small). Segments range from approximately 1 Kb to 3.9 Kb and each segment encodes 1–3 proteins (the L segment encodes for λ proteins, the M segment encodes for μ proteins and the S segment encodes for σ proteins). Replication occurs in cytoplasm and viral particles begin to assemble in the cytoplasm 6–7 hours after infection [30]. The virus can enter the host cell via an unknown cell surface receptor. The receptor is thought to include sialic acid and junctional adhesion molecules (JAMs). Reovirus is not considered a human pathogen, due to the absence of obvious symptoms, or known clinical impact during natural or experimental infection [31]. The virus seropositivity approaches 100% in adults [32].
The reoviruses have oncolytic properties, which are a bonus for the development of reovirus-based therapies for cancer treatment. Ikeda et al. proved that reovirus was effective to inhibit the growth of human head and neck squamous cells in vitro and in vivo [33]. Moreover, the efficacy of reovirus can be enhanced by combination with chemotherapy. A triple reovirus, cisplatin and paclitaxel combination showed a significantly more effective targeting than reovirus combined with either cisplatin, or paclitaxel [34]. These studies were carried out in four head and neck cancer cell lines in vitro and in vivo. The in vivo studies consisted of subcutanoeus injection of four head and neck cancer cell lines in athymic nude mice [34]. A Phase II clinical trial investigated the antitumor effects of intratumoral administration of REOLYSIN® in combination with low-dose radiation in patients with advanced cancers. A total of sixteen patients with advanced cancer were enrolled in the trial. Of these, partial responses were observed in two patients (thyroid, ovarian), for a total disease control rate of 93% in the treated lesions [35]. RT3D was combined with carboplatin/paclitaxel in patients with advanced cancers. Thirty-one heavily pretreated patients received study therapy in the phase I/II studies [36]. In summary, of the current phase I/II studies, reovirus showed safety both as a single agent when administered intratumorally and intravenously, as well as in combination therapy, with multiple chemotherapeutics such as gemcitabine, carboplatin and/or paclitaxel, and docetaxel. Similar findings were observed with radiation. Reovirus was therapeutically formulated and used in a phase III registration study in metastatic squamous cell carcinoma of the head and neck [37],[38].
Herpes Simplex Virus – 1 - preclinical and clinical studies
The Herpes Simplex Virus – 1 (HSV-1) virion ranges from 180 to 300 nm in diameter and comprise an outer-lipid bilayer envelope embedded with glycoproteins. Beneath the envelope lies the tegument layer, which contains proteins and enzymes important in early virus infection. The capsid contains the large dsDNA genome, consisting of 152,261 base pairs in length and with a base composition of 67% G + C. HSV-1 has approximately 90 open reading frames, allowing for the transcription of at least 84 unique proteins [39]. HSV enters into the cell by fusion of the viral envelope proteins. Virus initial interactions occur when a viral envelope glycoprotein C binds to a cell surface particle called heparan sulfate. A second glycoprotein, glycoprotein D, binds specifically to at least one of three known entry receptors [40]. These include HSV entry mediator, nectin-1 and 3-O sulfated heparan sulfate. Once bound to the mediator, gD interacts with viral glycoproteins H and L complex. In cells that are permissive for the propagation of this virus, the replication cycle is usually completed within 20 hours, releasing viral progeny through cell lysis. HSV-1 infection is extremely common and approximately 90% of adults have been exposed to the virus. The virus can be spread through contact with saliva. Symptoms of herpes simplex virus infection include watery blisters in the skin, or mucous membranes of the mouth, lips or genitals. As neurotropic and neuroinvasive viruses, HSV-1 persists in the body by becoming latent in the cell bodies of neurons. Some infected people may experience sporadic episodes of viral reactivation [41].
The strategies used in adenovirus work were also utilized in designing oncolytic HSV, such as expression of the tk in the tumor, followed by GCV administration, or other chemotherapeutic drugs, which are converted by tk into a cell-lethal metabolite in human thyroid carcinoma cell lines and in nude mice [42]. Soler et al. [43] evaluated a strategy for treatment of MTC, combining “suicide” and IL-2 gene therapies. Tumors were produced in Wag/Rij rats by orthotopic injection of the rMTC 6–23 cell line, and/or derivatives expressing the HSV1-TK gene (rMTC-TK). GCV, which is selectively transformed into a toxic metabolite by HSV1-TK, totally eradicated rMTC-TK tumors in 60% of the animals [43]. In a similar study, the expression of both the HSV-TK and the hIL-2 therapeutic genes in infected cells was high and persistent with time and showed that treatment with GCV led to more than 90% tumor growth inhibition and 100% inhibition transduced cells proliferation [44]. Moreover, successful safety and efficacy preclinical studies were conducted in NV 1023 and G207 cell lines, both in vitro and in animal models. Yu et al. [45] tested the oncolytic HSV (NV1023) against seven different thyroid cancers, including one papillary (NPA-187), one follicular (WRO82-1), one medullary (DRO81-1) and four anaplastic (DRO90-1, ARO, KAT-4C and KAT-18) cell lines. With the exception of WRO82-1 cell line, all other cell lines at MOI 5 demonstrated >95% infection in vitro at day 2. DRO81-1 tumors demonstrated partial response and all NPA-187 tumors in athymic nude mice completely regressed following the administration of a single dose [45]. A preclincial study utilized HSV NV 1023 strain, which expresses HSV glycoprotein D receptors nectin-1 and HSV entry mediator [46]. The HSV NV 1023 strain exhibited cytotoxicity greater than 85% in anaplastic, medullary and papillary cancers [46]. Another study used HSV G207 and NV1023 strains in combination with paclitaxel and doxorubicin to induce oncolysis in three human anaplastic thyroid cancer cell lines [47]. A dose-dependent cytotoxicity was observed for all four agents. Interestingly, G207 exhibited synergistic cytotoxicity, when combined with paclitaxel. A single intra-tumor injection (it) of G207 combined with biweekly intraperitoneal (ip) injection of paclitaxel injections in athymic nude mice bearing human anaplastic KAT4 cell line in a flank showed significantly reduced mean tumor volume [47].
The following clinical trials were not designed for the TMN treatment; however they may provide useful information about the employment of HSV as vector for virotherapy. SEPREHVIR (HSV-1716) was utilized in three clinical trials to treat 47 patients with glioma, 5 patients with melanoma and 20 patients with squamous cell carcinoma of the head and neck [48],[49]. No toxicity attributable to the use of HSV1716 was reported in any of these clinical studies. A new phase I/IIa clinical study for the treatment of mesothelioma commenced in 2012 [48]. This trial is based on HSV-1 and was previously tested in a Phase I trial for other solid tumors, such as melanoma, liver cancer, pancreatic cancer, lung cancer (NCT01935453) and head and neck cancer (NCT00931931). Efficacy and Safety Study of OncoVEXGM-CSF compared to GM-CSF in patients with melanoma is ongoing (NCT00769704) [49].
Vaccinia Virus - preclinical and clinical studies
Vaccinia virus (VV) is a member of the genus Orthopoxvirus of the family of Poxviridae. VV is a large dsDNA virus whose entire life cycle takes place within the cytoplasm of host cells. Vaccinia intercellular mature virus (IMV) particles are brick-shaped, approximately 300 × 240 × 120 nm in size, with a lipoprotein shell surrounding a complex core structure [50]. The core structure contains a linear, ds DNA genome of approximately 192 kb associated with a number of virus-encoded proteins [51]. VV has inverted terminal repeats (ITRs) that are required for VV DNA replication. The 192-kb genome encodes some 200 genes that are largely non-overlapping. VV produces three forms of infectious particles: IMV, cell-associated enveloped virus (CEV) and extracellular enveloped virus (EEV) with a different cell surface binding sites [50]. IMVs enter cells by fusion with the plasma membrane [52]. EEVs enter cells by endocytosis followed by low pH disruption of the EEV outer membrane and fusion of the released IMV with endosomal membranes [53]. It typically begins 1–2 h after infection and generates large numbers of genome copies per cell within hours of infection, of which half are ultimately packaged into infectious virions [54]. The host response to VV infection is multifactorial. Immediately after VV invasion, nonspecific mechanisms involving apoptosis induction, complement, interferons, cytokines and natural killer cells serve as the first-line host defense. [55]. Although neutralizing antibodies are also involved in host protection during VV infection, the cell-mediated immune responses are known to be particularly potent and may be most critical for viral clearance [56]. T helper 1 (Th1) immune response is mediated by antiviral cytokines such as IL-12, IL-18 and interferon (IFN)-γ, and plays a critical role in host defense [57],[58]. Th2 immune response is mediated by IL-4 and IL-10 and may actually suppress the host response to vaccinia [59]. In addition, due to VV infection, the host cell shut-off is the abrogation of class I and class II major histocompatibility complex (MHC) molecule production and presentation, thereby leading to poor recognition of the virus by T cells [60]. At least two virus-encoded factors were shown to inhibit the complement cascade: C3L and B5R. VV encodes at least three proteins to inhibit premature cell death caused by apoptosis [61].
The majority of studies for TMN in oncolytic mutant vaccinia virus were designed to assess the viral oncolysis efficacy in thyroid cell lines and animal models of thyroid malignant tumors [62]. For example, vaccinia virus strain recombinant GLV-1 h68 infection was detected in all cell lines in 24 hours and increased in intensity at 36 hours in six ATC cell lines after inoculation [63],[64]. In vivo studies showed an enhanced anti-tumor efficacy with one i.v. GLV-1 h68 injection followed by multiple i.p. injections of Avastin [65]. Expression of human NIS (hNIS) was also used in the VV work [66],[67]. Vaccinia virus (GLV-1 h153) carrying hNIS was able to infect, replicate and kill all ATC cell lines. At a MOI of 1.0, GLV-1 h153 reached near 100% cytotoxicity in 8305c and FRO at day 5. The efficacy of the treatment was also observed in infected ATC xenografts [66],[67]. In addition, GLV-1 h153 achieved more than 90% cytotoxicity in human gastric cancer cell lines and xenografts [68].
Currently, there are no virotherapy clinical trials based on VV for the treatment of TMN. However, the VV results obtained from several other trials are certainly interesting. The phase I clinical trial that utlized GL-ONC1 proved that the virus could safely be administered intravenously in patients with advanced solid tumors [69]. JX-594 (Pexa-Vec) is a VV engineered by addition of the GM-CSF gene and deletion of the thymidine kinase gene, which limits viral replication to cells with high levels of thymidine kinase, typically seen in cancer cells with a mutated RAS or p53 gene [70]. It has orphan drug designation from US FDA and EUMA for the treatment of hepatocellular carcinoma (HCC). Two phase I trials were concluded and a phase II trial for primary liver cancer, alone and in combination with sorafenib is starting. Both GL-ONC1 and JX-594 provided promising results in preclinical studies [71].
Newcastle disease virus - preclinical and clinical studies
Newcastle disease virus (NDV), also known as avian paramyxovirus-1 (APMV-1) and belongs to the genus Avulavirus of the family Paramyxoviridae. NDV is enveloped and spherical, with a diameter in the range of 150 nm and the genome consists of single stranded negative-sense RNA (ssRNA) that contains six open reading frames (ORF) and comprises 15,186 nucleotides. The six ORF encode the nucleoprotein (NP), the phosphoprotein (P), the matrix protein (M), the fusion protein (F), the haemagglutinin-neuraminidase (HN) and the large protein (L). At least one additional non-structural protein (V) and possibly a second one (W) are generated by RNA editing during P gene transcription [72]. Virus infection is initiated by attachment of the virion to the surface of the target cell. Binding of the viral HN glycoprotein to sialic acid-containing cell surface proteins, which serve as receptors, triggers the F protein promoted fusion of the viral envelope with the plasma membrane of the host cell through a pH-independent mechanism [73]. NDV has only one serotype and causes Newcastle disease, which is a contagious bird disease. The virus is transmissible to humans and can cause mild conjunctivitis and influenza-like symptoms [74]. NDV replicate up to 10,000 times faster in human cancer cells than in most normal human cells. NDV is a naturally oncolytic virus that has been shown to be safe and effective for cancer therapy. In 1999, promising results were reported using an attenuated strain of the NDV [75].
Recombinant NDV is an effective oncolytic agent against a panel of tumor cell lines including thyroid, head and neck, and mouse model [76]. For example, NDV (F3aa)-GFP, a genetically modified fusogenic NDV, was used for the treatment of head and neck squamous cell carcinoma. Four cell lines were highly susceptible to viral cytotoxicity, with more than 75% of cells lysed at day 6 at MOI 0.1, whereas two other cell lines were partially susceptible. Oncolytic NDV was used in a number of in vivo studies for the effective target of various solid tumors [77],[78].
So far, no clinical investigation has been initiated with NDV for TMN. However, four clinical trials have been conducted in the United States studied the use of NDV oncolysates in patients with metastatic melanoma. Some positive results were found in these studies [79].
Vesicular stomatitis virus - preclinical and clinical studies
The vesicular stomatitis virus (VSV) is a single negative ssRNA virus. VSV is a member of the Rhabdoviridae family, which includes the rabies virus. The virus particle carries a single approximately 11-kb (−) RNA encoding five genes: the nucleocapsid protein N, the phosphor-protein P, the matrix protein M, glyco protein G and the large polymerase protein L. The G spikes on the outside of the virus particles will help to attach to other cells that can be infected. The VSV G protein mediates viral attachment either to Low-Density Lipoprotein (LDL) receptor (LDLR), or an LDLR family member present on the host cell. Following binding, the VSV-LDLR complex is rapidly endocytosed. It then mediates fusion of the viral envelope with the endosomal membrane [80]. The M protein is part of the inner membrane of the virus and is responsible for shutting down the host and the viral translation. VV replication occurs in the cytoplasm [81]. There are two distinct immunological classes of VSV recognized: New Jersey (NJ) and Indiana (IND). In humans, the incubation period may vary from 24 hours to 6 days but it is usually 3–4 days. VSV grows rapidly, is zoonotic and non-pathogenic to humans [82]. VSV only causes a flu-like illness in infected humans [82].
The efficiency of VSV to express either human or murine IFN-β, or a methionine deletion at residue 51 of the matrix protein was been observed in vitro and in vivo [83]. Currently, there is no clinical investigation that utilizes VSV for the treatment of TMN. A VSV-based phase I trial is currently underway for the treatment of patients with liver cancer (NCT01628640). In this clinical, VSV particles expressing human interferon beta will be administered into patients via intratumoral injection.
Measles Virus - preclinical and clinical studies
Measles virus (MV) is a single-stranded, negative-sense, enveloped RNA virus of the genus Morbillivirus of the family Paramyxoviridae. The measles virus has two envelope glycoproteins on the viral surface: hemagglutinin (H) and membrane fusion protein (F). These proteins are responsible for host cell binding and invasion. Three receptors for the H protein have been so far identified: complement regulatory molecule CD46, the signaling lymphocyte activation molecule (SLAM) and the cell adhesion molecule Nectin-4 [84]. Humans are the natural hosts of the virus. No animal reservoirs are known to exist. This virus is the cause of measles, an infection of the respiratory system. Approximately 500,000 persons with measles are reported each year in the United States [85]. The vaccine-attenuated strains of MV show a promising potential for cancer therapy [86]. There is a study that utilizes MV-Edm, which was modified to include the NIS gene to be combined with radiology treatment. This study demonstrated the oncolytic efficacy of MV-NIS in BHT-101 and KTC-3, ATC-derived cell lines and mouse model [87].
There is no MV clinical investigation for TMN yet. However, there are two phase I trials that are recruiting patients with other types of malignancies. One trial will studying the side effects and best dose of the MV derivative producing CEA (MV-CEA) to treat patients with recurrent glioblastoma multiforme (NCT00390299). Another MV-NIS phase I trial will study the side effects and the best dose of viral therapy to treat patients with recurrent or metastatic squamous cell carcinoma of the head and neck (NCT01846091).
Retrovirus - preclinical and clinical studies
The murine leukemia virus (MLV) is one of the simplest retroviruses. The virus has a spherical shape that has a diameter of 80 to 100 nm. The envelope of the virus is covered with glycoprotein spikes. The viral genome is a single stranded, linear, positive-sense RNA molecule of approximately 8,000 nucleotides and 4 ORF. The genome contains gag, pol, and env regions, which encode for structural proteins, enzymes including the RNA-dependent DNA polymerase (reverse transcriptase), and coat proteins, respectively [88].
A MLV-derived retroviral vector termed pMFGIL-2TKSN expresses two therapeutic genes, such as HSVtk and IL-2. This retroviral-based vector system exhibited enhanced tumor growth inhibition in animal models [89]. In addition, Barzon et al. [44] used a similar retroviral vector system and evaluated its efficiency in vitro and in vivo, following GCV treatment [90]. This study showed an 80% reduction of the size of differentiated thyroid carcinomas [44]. Two patients with end-stage anaplastic thyroid carcinoma were treated with direct intratumor injection of retroviral vector expressing the human IL-2 gene and the suicide gene tk of HSV 1, followed by GCV administration. The treatment was safe and demonstrated local tumor necrosis [91]. To date, the RV clinical investigation has not been reported for TMN. A phase I study will test the safety of different doses of the patients own immune cells, which will be genetically modified to help recognize and destroy the cancer cells for treatment of castrate metastatic prostate cancer (NCT01140373).
Consideration for the oncolytic virus design
Based on previous research, oncolytic virus replication in normal cells remains a concern and, therefore, the efforts should be focused on improving the oncolytic viral tumor selectivity and specificity [92], as the specificity of oncolytic virus is a most important factor that would eventually determine the clinical safety and efficacy for the therapeutics. Specific targeting of tumour cell can be achieved by taking advantage of the fact that tumour cells have altered microenvironment, display certain tumour specific receptors and modified cellular pathways [93]. In fact, the specificity of oncolytic virus depends on the oncolytic virus tropism, which can be modified. For example, expression profiles of multiple microRNAs can reshape the tropism of MV, which could be safer and efficient for a high-dose systemic administration [94]. Indeed, viruses with altered tropism may be used for the targeting of tumor-specific surface biomarkers. Several TMN immuno-histochemical and molecular biomarkers have been proposed. Some of them might be useful for this purpose, or for diagnosis. Biomarkers for TMN comprise: RET, vascular endothelial growth factor receptor, hepatocyte growth factor receptor, epidermal growth factor receptor, ATP-binding cassette sub-family G member 2, cytokeratin 19 (CK19), galectin-3, oncofetal fibronectin (OnfFN), hector battifora mesothelial cell 1, CD44, CD56, CD63, CD113, CD133, lysosomal enzymes cathepsin B, cathepsin L and lysosomal dipeptidyl peptidase II [95]-[97]. The distribution of these biomarkers in TMN is variable. Demellawy et al. [98] reported that the CD56 positive in all the lesions and tumors except for PTC in all cases (100%). CD56 was negative in all PTC cases (100%). CK19 showed positive expression in PTC accounting for 85% of cases and in 26% of non-PTC lesions/tumors. P63 expression was in 70% of cases of PTC and was consistently absent in all the non-PTC cases [98]. Medullary thyroid carcinomas account for less than 5% of thyroid nodules and exhibit a specific circulating biomarker, i.e. calcitonin (CT). Another major issue is the relevance of oncogene mutation and signaling transduction in TMN. The genetic alteration described in PTC is the RET/PTC rearrangement, encoding for a transmembrane tyrosine kinase receptor. The incidence of RAS mutations in these different histotypes ranges from 0%–50% in PTC, 14%–62% in FTC and 0%–60% in ATC. The over-expression of the MET/HGF receptor is 100-fold higher in PTC than in FTC, ATC and MTC [99]. In conclusion, a panel of biomarkers was studied for TMN. However, there is no validated biomarker that is specific for TMN.
It has been previously reported that BRAF mutations and rearrangements of RET (RET/PTC) prevent the differentiation of thyroblasts into prothyrocytes, resulting in the generation of papillary carcinomas, whereas PAX8-PPARγ1 rearrangement prevents differentiation of prothyrocytes into thyrocytes, resulting in the generation of follicular tumors [100]. We understand that human thyroid ancestor cells or embryonic stem cells (ES) cell-derived, endoderm-positive cells have been shown to express several early thyroid markers. For example, anaplastic carcinoma (Tg -, OnfFN +) derives from thyroid stem cells, papillary carcinoma (Tg+, onfFN +) from thyroblast cells, and follicular tumor (Tg+, onfFN -) from prothyrocytes, respectively [101]. According to this hypothesis, the thyroid tumors were caused by different mutations during thyroid fetal development. Thus, any event that prevents fetal thyroid cells from differentiation might stop carcinogenesis. Some thyroid specific biomarkers were expressed during normal human fetal development. Between E60 and E70, a series of events lead the thyroid primordial toward a functional thyroid gland able to produce and release hormones during the normal thyroid development in embryology [102]. The thyroid markers appear in that period. These include thyroglobulin (Tg), NIS, thyroid peroxidase (TPO), thyroid-stimulating hormone (TSH), and TSH receptor (TSHR) [102] (Figure 1) [103]. This information suggests that the thyroid markers, such as TSH, may be more useful for TMN virotherapy. TSH is a glycoprotein and consists of two subunits, the alpha (α) and the beta (β) subunit. The α subunit, a 92-amino acid sequence, is thought to be the effector region responsible for stimulation of adenylate cyclase (involved the generation of cAMP). The β subunit (TSHB), a 118-amino acid sequence, is unique to TSH, and therefore determines its receptor specificity [104],[105]. The TSHR is mainly found on thyroid follicular cells. TSH binds TSHR with high affinity. TSHR is widely expressed in a variety of extrathyroidal tissues [102]. The level and the tissues distribution of TSHR and TPO are very similar to the distribution of NIS. Therefore, TSHR (or TPO) might still be considered as a fetal thyroid marker, which can be used for oncolytic virotherapy. Another strategy consists of expressing the Fab fragment of antibodies anti-TSHR, or anti-TPO, or anti-NIS. This might increase the oncolytic virus binding and/or entry specifically for cells expressing TSHR, or TPO, or NIS.

TSHR localization and structure. (A) Thyroid follicle, where the TSH-stimulated synthesis of thyroid hormone occurs following iodide uptake and organification into thyroglobulin. (B) TSHR protein structure, showing the β subunit composed of leucine-rich repeats and the N-terminus, and the β subunit composed of 7 trans-membrane domains, the intra-cellular and extra-cellular loops and the C-terminus. Figure adapted with permission from Atlas Genet Cytogenet Oncol Haematol [103].
In addition, previous studies that utilized VSV and NDV as oncolytic agents provided encouraging results. Moreover, VSV and NDV are zoonotic pathogens. Humans have little to no immunity against these viruses, which facilitates their escape from host immune system. This allows for the oncolytic viruses to reach and kill malignant cells. Virtually, the final step to success is the selection of the site and route for virus delivery. Current studies have emphasized intratumoral delivery. The direct tumor injection cannot kill the tumor cells in blood stream, which may cause the tumor recurrence. Therefore, the systemic delivery through the bloodstream to reach cancer cells will be necessary. Studies are ongoing to improve efficient delivery of oncolytic viruses into the host. To this end, preclinical studies are focusing on the use of cellular carriers of oncolytic viruses [106]. The cellular carriers may shield oncolytic virus from neutralizing antibodies during delivery, providing a simple and effective tool to enhance the therapeutic efficacy of oncolytic viruses [106]. However, as a human therapeutic agent, the maximum purity that related with agent’s safety and efficacy is very important. An improper agent preparation might cause some unacceptable adverse events. Therefore, it is important to improve the safety of therapeutic biological agents used in therapy. In this respect, the techniques used in the pharmaceutical industry to optimize the formulation and lyophilization of live viral vaccines might be applied to oncolytic virus preparations.
Authors’ contributions
MG drafted the manuscript, and EEH approved it. GR, RC, and EEH provided conceptual input. All authors participated in the discussion and approved the final submitted version of the manuscript.
References
Davidson BJ, Burman KD: Cancer of the thyroid and parathyroid. Head and Neck Cancer: A Multidisciplinary Approach. Edited by: Harrison LB, Sessions RB, Hong WK. 2009, Lippincott, Williams & Wilkins, Philadelphia, PA, 690-742. 3
Cunningham MP, Duda RB, Recant W, Chmiel JS, Sylvester JA, Fremgen A: Survival discriminants for differentiated thyroid cancer. Am J Surg. 1990, 160: 344-347.
Nagaiah G, Hossain A, Mooney CJ, Parmentier J, Remick SC: Anaplastic thyroid cancer: A review of epidemiology, pathogenesis, and treatment. J Oncol. 2011, 542358: 1-13.
Davies L, Welch HG: Increasing incidence of thyroid cancer in the United States, 1973–2002. Jama. 2006, 295 (18): 2164-2167.
Kelly E, Russell SJ: History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007, 15 (4): 651-659.
Leja J, Yu D, Nilsson B, Gedda L, Zieba A, Hakkarainen T, Åkerström G, Öberg K, Giandomenico V, Essand M: Oncolytic adenovirus modifed with somatostatin motifs for selective infection of neuroendocrine tumor cells. Gene Ther. 2011, 18 (11): 1052-1062.
Perett L: Measles viruses genetically modified to treat ovarian cancer. (30 June 2011) National Cancer Institute, Benchmarks. Retrived 5 September. 2012
Breitbach CJ, Thorne SH, Bell JC, Kirn DH: Targeted and armed oncolytic poxviruses for cancer: The lead example of JX-594?. Curr Pharm Biotechnol. 2012, 13 (9): 1768-1772.
Vaha-Koskela MJ, Heikkilä JE, Hinkkanen AE: Oncolytic viruses in cancer therapy. Cancer Lett. 2007, 254 (2): 178-216.
Meier O, Greber UF: Adenovirus endocytosis. J Gene Med. 2004, 6 (suppl 1): S152-S163.
Wu E, Nemerow GR: Virus yoga: the role of flexibility in virus host cell recognition. Trends Microbiol. 2004, 12 (4): 162-168.
White E, Gooding LR: Regulation of apoptosis by human. Adenoviruses Curr Commun Cell Mol Bio. 1994, 8: 111-114.
Wold WM, Horwitz MS: Adenovirus . Fields Virology . Edited by: Knipe DM, Howley PM. 2007, Lippincott Williams & Wilkins, Philadelphia, 2395-5,
Yang LP, Cheng P, Peng XC, Shi HS, He WH, Cui FY, Luo ST, Wei YQ, Yang L: Anti-tumor effect of adenovirus- mediated gene transfer of pigment epithelium-derived factor on mouse B16-F10 melanoma. J Exp Clin Can Res. 2009, 28: 75.
Shen W, Wang CY, Wang XH, Fu ZX: Oncolytic adenovirus mediated Survivin knockdown by RNA interference suppresses human colorectal carcinoma growth in vitro and in vivo. J Exp Clin Can Res. 2009, 28: 81.
Minemura K, Takeda T, Minemura K, Nagasawa T, Zhang R, Leopardi R, DeGroot LJ: Cell-specific induction of sensitivity to ganciclovir in medullary thyroid carcinoma cells by adenovirus-mediated gene transfer of herpes simplex virus thymidine kinase. Endocrinology. 2000, 141 (5): 1814-1822.
Zhang R, Straus FH, DeGroot LJa: Adenoviral-mediated gene therapy for thyroid carcinoma using thymidine kinase controlled by thyroglobulin promoter demonstrates high specificity and low toxicity. Thyroid. 2001, 11 (2): 115-123.
Zhang R, DeGroot LJ: An adenoviral vector expressing functional heterogeneous proteins herpes simplex viral thymidine kinase and human interleukin-2 has enhanced in vivo antitumor activity against medullary thyroid carcinoma. Endocr Relat Cancer. 2001, 8 (4): 315-325.
Yamazaki M, Straus FH, Messina M, Robinson BG, Takeda T, Hashizume K, DeGroot LJ: Adenovirus-mediated tumor-specific combined gene therapy using Herpes simplex virus thymidine/ganciclovir system and murine interleukin-12 induces effective antitumor activity against medullary thyroid carcinoma. Cancer Gene Ther. 2004, 11 (1): 8-15.
Blagosklonny MV, Giannakakou P, Wojtowicz M, Romanova LY, Ain KB, Bates SE, Fojo T: Effects of p53-Expressing adenovirus on the chemosensitivity and differentiation of anaplastic thyroid cancer cells. J Clin Endocrinol and Metabol. 1998, 83 (7): 2516-2522.
Portella G, Scala S, Vitagliano D, Vecchio G, Fusco A: ONYX-015, an E1B gene- defective adenovirus, induces cell death in human anaplastic thyroid carcinoma cell lines. J Clin Endocrinol Metab. 2002, 87 (6): 2525-2531.
Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, Koeffler HP: High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J Clin Invest. 1993, 91: 179-184.
Zhu J: Preoperative recombinant adenoviral human p53 gene combined with intensity-modulated radiation therapy in treatment of stage IV papillary thyroid carcinoma: A randomized clinical study. ASCO Annual Meeting, suppl; abstr. 2012, ?: 5550.
Clinical trial (PDQ), rAd-p53 Gene Therapy for advanced malignant thyroid tumors phase IV, rAd-p53-003. NLM Identifer NCT00902122, National Cancer Institute, Clinical Trials.gov processed this data on October 17. 2013
Khuri F, Nemunaitis J, Ganly I, Arseneau J, Tannock I, Romel L, Gore M, Ironside J, MacDougall R, Heise C, Randlev B, Gillenwater AM, Bruso P, Kaye SB, Hong WK, Kirn DH: A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000, 6 (8): 879-885.
McCormick F: Cancer-specific viruses and the development of ONYX-15. Can Bio Ther. 2003, 2 (4 sippl 1): S157-S160.
Crompton AM, Kirn DH: From ONYX-015 to armed vaccinia viruses: the education and evolution of oncolytic virus development. Curr Cancer Drug Targets. 2007, 7 (2): 133-139.
Libertini S, Iacuzzo I, Perruolo G, Scala S, Ieranò C, Franco R, Hallden G, Portella G: Bevacizumab increases viral distribution in human anaplastic thyroid carcinoma xenografts and enhances the effects of E1A-defective adenovirus dl922-947. Clin Cancer Res. 2008, 14 (20): 6505-6514.
Reddi HV, Madde P, Cohen YC, Bangio L, Breitbart E, Harats D, Bible KC, Eberhardt NL: Antitumor Activity of VB-111, a Novel Antiangiogenic virotherapeutic, in thyroid cancer xenograft mouse models. Genes Can. 2011, 2 (10): 993-995.
Nibert ML, Schiff LA, Fields BN: Reoviruses and their replication. Fields Virology. 1996, Lippincott-Raven, Philadelphia, 1557-1596. 3
Rosen LH, Evans E, Spickard A: Reovirus infections in human volunteers. Am J Hyg. 1963, 77: 29-37.
Selb B, Weber B: A study of human reovirus IgG and IgA antibodies by ELISA and Western blot. J Virol Methods. 1994, 47: 15-25.
Ikeda Y, Nishimura G, Yanoma S, Kubota A, Furukawa M, Tsukuda M: Reovirus oncolysis in human head and neck squamous carcinoma cells. Auris Nasus Larynx. 2004, 31 (4): 407-412.
Roulstone V, Twigger K, Zaidi S, Pencavel T, Kyula JN, White C, McLaughlin M, Seth R, Karapanagiotou EM, Mansfield D, Coffey M, Nuovo G, Vile RG, Pandha HS, Melcher AA, Harrington KJ: Synergistic cytotoxicity of oncolytic reovirus in combination with cisplatin–paclitaxel doublet chemotherapy. Gene Therapy. 2013, 20: 521-528.
Intratumoral Administration of REOLYSIN® in Combination with Low-Dose Radiation for Patients with Advanced Malignancies. Oncolytics Biotech Inc News 2009. Trail number REO 008. ,
Karapanagiotou EM, Roulstone V, Twigger K, Ball M, Tanay M, Nutting C, Newbold K, Gore ME, Larkin J, Syrigos KN, Coffey M, Thompson B, Mettinger K, Vile RG, Pandha HS, Hall GD, Melcher AA, Chester J, Harrington KJ: Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012, 18 (7): 2080-2089.
Black AJ, Morris DG: Clinical trials involving the oncolytic virus, reovirus: ready for prime time?. Expert Rev Clin Pharmacol. 2012, 5 (5): 517-520.
Maitra R, Ghalib MH, Goel S: Reovirus: a targeted therapeutic-progress and potential. Mol Cancer Res. 2012, 10 (12): 1514-1525.
Akhtar J, Shukla D: Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2000, 276 (24): 7228-7236.
Subramanian RP, Geraghty RJ: Herpes simplex virus type 1 mediates fusion through ahemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc Natl Acad Sci U S A. 2007, 104 (8): 2903-2908.
Usatine RP, Tinitigan R: Nongenital herpes simplex virus. Am Fam Physician. 2010, 82 (9): 1075-1082.
Nishihara E, Nagayama Y, Mawatari F, Tanaka K, Namba H, Niwa M, Yamashita S: Retrovirus-mediated herpes simplex virus thymidine kinase gene transduction renders human thyroid carcinoma cell lines sensitive to ganciclovir and radiation in vitro and in vivo. Endocrinol. 1997, 138 (11): 4577-4583.
Soler MN, Milhaud G, Lekmine F, Treilhou-Lahille F, Klatzmann D, Lausson S: Treatment of medullary thyroid carcinoma by combined expression of suicide and interleukin-2 genes. Cancer Immunol Immunother. 1999, 48 (2–3): 91-99.
Barzon L, Bonaguro R, Castagliuolo I, Chilosi M, Franchin E, Vecchio CD, Giaretta I, Boscaro M, Palu G: Gene therapy of thyroid cancer via retrovirally-driven combined expression of human interleukin-2 and Herpes Simplex Virus thymidine kinase. Euro J Endocrinol. 2003, 148: 73-80.
Yu Z, Eisenberg DP, Singh B, Shah JP, Fong Y, Wong RJ: Treatment of aggressive thyroid cancer with an oncolytic herpes virus. Int J Cancer. 2004, 112 (3): 525-532.
Huang YY, Yu Z, Lin SF, Li S, Fong Y, Wong RJ: Nectin-1 is a marker of thyroid cancer sensitivity to herpes oncolytic therapy. J Clin Endocrinol Metab. 2007, 92 (5): 1965-1970.
Lin SF, Gao SP, Price DL, Li S, Chou TC, Singh P, Huang YY, Fong Y, Wong RJ: Synergy of a herpes oncolytic virus and paclitaxel for anaplastic thyroid cancer. Clin Cancer Res. 2008, 14 (5): 1519-1528.
Oncolytic Virus Specialist Virttu Biologics Initiates Phase I/II SEPREHVIR™ Study in Mesothelioma. VIRTTU BIOLOGICS LIMITED - Company information. 2012, September 10th. ,
Efficacy and Safety Study of OncoVEXGM-CSF Compared to GM-CSF in Melanoma. Clinical Trials gov. 2014. ClinicalTrials.gov Identifier: NCT00769704.,
Moss B: Poxviridae: The viruses and their replication . Fields Virology . Edited by: Knipe DM, Howley PM. 2007, Lippincott Williams and Wilkins, Philadelphia, 2905-5,
Smith GL, Vanderplasschen A, Law M: The formation and function of extracellular enveloped vaccinia virus. J Gen Virol. 2002, 83 (12): 2915-2931.
Doms RW, Blumenthal R, Moss B: Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J Virol. 1990, 64 (10): 4884-4892.
Vanderplasschen A, Hollinshead M, Smith GL: Intracellaular and extracellular vaccinia virions enter cells by different mechanisms. J Gen Virol. 1998, 79 (4): 877-887.
Joklik WK, Becker Y: The replication and coating of vaccinia DNA. J Mol Biol. 1964, 10: 452-474.
Buller RM, Palumbo GJ: Poxvirus pathogenesis. Microbiol Rev. 1991, 55: 80-122.
Smith GL, Symons JA, Khanna A, Vanderplasschen A, Alcami A: Vaccinia virus immune evasion. Immunol Rev. 1997, 159: 137-154.
van Den Broek M, Bachmann MF, Kohler G, Barner M, Escher R, Zinkernagel R, Kopf M: IL-4 and IL-10 antagonize IL-12-mediated protection against acute vaccinia virus infection with a limited role of IFN-gamma and nitric oxide synthetase 2. J Immunol. 2000, 164: 371-378.
Zeh HJ, Bartlett DL: Development of a replication-selective, oncolytic poxvirus for the treatment of human cancers. Cancer Gene Ther. 2002, 9 (12): 1001-1012.
Sharma DP, Ramsay AJ, Maguire DJ, Rolph MS, Ramshaw IA: Interleukin-4 mediates down regulation of antiviral cytokine expression and cytotoxic T-lymphocyte responses and exacerbates vaccinia virus infection in vivo. J Virol. 1996, 70 (10): 7103-7107.
Shen Y, Nemunaitis J: Fighting cancer with vaccinia virus: teaching new tricks to an old dog. Mol Ther. 2005, 11: 180-195.
Kettle S, Alcami A, Khanna A, Ehret R, Jassoy C, Smith GL: Inhibits interleukin-1beta-converting enzyme and protects virus-infected cells from TNF- and Fas-mediated apoptosis, but does not prevent IL-1beta-induced fever. J Gen Virol. 1997, 78 (Pt 3): 677-685.
Yu YA, Galanis C, Woo Y, Chen N, Zhang Q, Fong Y, Szalay AA: Regression of human pancreatic tumor xenografts in mice after a single systemic injection of recombinant vaccinia virus GLV-1 h68. Mol Cancer Ther. 2009, 8 (1): 141-151.
Lin SF, Yu Z, Riedl C, Woo Y, Zhang Q, Yu YA, Timiryasova T, Chen N, Shah JP, Szalay AA, Fong Y, Wong RJ: Treatment of anaplastic thyroid carcinoma in vitro with a mutant vaccinia virus. Surgery. 2007, 142 (6): 976-983.
Lin SF, Price DL, Chen CH, Brader P, Li S, Gonzalez L, Zhang Q, Yu YA, Chen N, Szalay AA, Fong Y, Wong RJ: Oncolytic vaccinia virotherapy of anaplastic thyroid cancer in vivo. J Clin Endocrinol Metab. 2008, 93 (11): 4403-4407.
Frentzen A, Yu YA, Chen N, Zhang Q, Weibel S, Raab V, Szalay AA: Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proc Natl Acad Sci U S A. 2009, 106 (31): 12915-12920.
Yu Z, Li S, Brader P, Chen N, Yu YA, Zhang Q, Szalay AA, Fong Y, Wong RI: Oncolytic vaccinia therapy of squamous cell carcinoma. Molecular Can. 2009, 8 (45): 1-9.
Gholami S, Haddad D, Chen CH, Chen NG, Zhang Q, Zanzonico PB, Szalay AA, Fong Y: Novel therapy for anaplastic thyroid carcinoma cells using an oncolytic vaccinia virus carrying the human sodium iodide symporter. Surgery. 2011, 150 (6): 1040-1047.
Jun KH, Gholami S, Song TJ, Au J, Haddad D, Carson J, Chen CH, Mojica K, Zanzonico P, Chen NG, Zhang Q, Szalay A, Fong Y: A novel oncolytic viral therapy and imaging technique for gastric cancer using a genetically engineered vaccinia virus carrying the human sodium iodide symporter. J Experi Clini Can Res. 2014, 33: 2.
Safety study of GL-ONC1, an oncolytic virus, in patients with advanced solid tumors. Clinical Trials gov. 2014. Clinical Trials gov Identifier: NCT00794131. ,
Bos JL: Ras oncogenes in human cancer: a review. Cancer Res. 1989, 49 (17): 4682-4689.
Liu TC, Hwang T, Park BH, Bell J, Kirn DH: The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008, 16 (9): 1637-1642.
Steward M, Vipond IB, Millar NS, Emmerson PT: RNA editing in Newcastle disease virus. J Gen Virol. 1993, 74: 2539-2547.
Dortmans JC, Koch G, Rottier PJ, Peeters BP: Virulence of Newcastle disease virus: what is known so far?. Vet Res. 2011, 42 (1): 122.
Nelson CB, Pomeroy BS, Schrall K, Park WE, Lindeman RJ: An outbreak of conjunctivitis due to Newcastle disease virus (NDV) occurring in poultry workers. Am J pub health nation’s health. 1952, 42 (6): 672-678.
Csatary LK, Moss RW, Beuth J, Töröcsik B, Szeberenyi J, Bakacs T: Beneficial treatment of patients with advanced cancer using a Newcastle disease virus vaccine (MTH-68/H). Anticancer Res. 1999, 19 (1B): 635-638.
Zamarin D, Martínez-Sobrido L, Kelly K, Mansour M, Sheng G, Vigil A, García-Sastre A, Palese P, Fong Y: Enhancement of oncolytic properties of recombinant Newcastle disease virus through antagonism of cellular innate immune responses. Mol Ther. 2009, 17 (4): 697-706.
Li P, Chen CH, Li S, Givi B, Yu Z, Zamarin D, Palese P, Fong Y, Wong RJ: Therapeutic effects of a fusogenic Newcastle disease virus in treating head and neck cancer. Head Neck. 2011, 33 (10): 1394-1399.
Wei D, Sun N, Nan G, Wang Y, Liu HQ, Peeters B, Chen ZN, Bian H: Construction of recombinant Newcastle disease virus Italian strain for oncolytic virotherapy of tumors. Hum Gene Ther. 2012, 23 (7): 700-710.
Laurie SA, Bell JC, Atkins HL, Roach J, Bamat MK, O’Neil JD, Roberts MS, Groene WS, Lorence RM: A phase 1 clinical study of intravenous administration of PV701, an oncolytic virus, using two-step desensitization. Clin Cancer Res. 2006, 12 (8): 2555-2562.
Miisch A, Xu H, Shields D, Rodriguez-Boulan E: Transport of vesicular stomatitis virus G protein to the cell surface is signal mediated in polarized and nonpolarized cells. J Cell Biol. 1996, 133: 543-588.
Lyles DS, Rupprecht CE: Rhabdoviridae . Fields Virology. Edited by: Knipe DM, Howley PM. 2007, Lippincott Williams & Wilkins, Philadelphia, 1363-5,
Csatary LK, Eckhardt S, Bukosza I, Czegledi F, Fenyvesi C, Gergely P, Bodey B, Csatary CM: Attenuated veterinary virus vaccine for the treatment of cancer. Cancer Detect Prev. 1993, 17 (6): 619-627.
Liu YP, Steele MB, Suksanpaisan L, Federspiel MJ, Russell SJ, Peng KW, Bakkum-Gamez JN: Oncolytic measles and vesicular stomatitis virotherapy for endometrial cancer. Gynecol Oncol. 2014, 132 (1): 194-202.
Lu G, Gao GF, Yan J: The receptors and entry of measles virus: a review. Sheng Wu Gong Cheng Xue Bao. 2013, 29 (1): 1-9.
Chen TH, Kutty P, Lowe LE, et al: Measles outbreak associated with an international youth sporting event in the United States, 2007. Pediatr Infect Dis J. 2010, 29: 794-800.
Pasquinucci G: Possible effect of measles on leukaemia. Lancet. 1971, 1: 136.
Reddi HV, Madde P, McDonough SJ, Trujillo MA, Morris JC, Myers RM, Peng KW, Russell SJ, McIver B, Eberhardt NL: Preclinical efficacy of the oncolytic measles virus expressing the sodium iodide symporter in iodine non-avid anaplastic thyroid cancer: a novel therapeutic agent allowing noninvasive imaging and radioiodine therapy. Cancer Gene Ther. 2012, 19 (9): 659-665.
D’souza V, Dey A, Habib D, Summers MF: NMR structure of the 101-nucleotide core encapsidation signal of the moloney murine leukemia virus. J Mol Biol. 2004, 337 (2): 427-442.
Nagayama Y, Nishihara E, Iitaka M, Namba H, Yamashita S, Niwa M: Enhanced efficacy of transcriptionally targeted suicide gene/prodrug therapy for thyroid carcinoma with the Cre-loxP system. Cancer Res. 1999, 59 (13): 3049-3052.
Barzon L, Bonaguro R, Castagliuolo I, Chilosi M, Elisa F, Vecchio CD, Giaretta I, Boscaro M, Palu G: Gene therapy of thyroid cancer via retrovirally-driven combined expression of human interleukin-2 and Herpes Simplex Virus thymidine kinase. Eur J Endocrinol. 2003, 148: 73-80.
Barzon L, Pacenti M, Taccaliti A, Franchin E, Bruglia M, Boscaro M, Palu G: A pilot study of combined suicide/cytokine gene therapy in two patients with end-stage anaplastic thyroid carcinoma. J Clin Endocrinol Metab. 2005, 90 (5): 2831-2834.
Singh PK, Doley J, Ravi Kumar G, Sahoo AP, Tiwari AK: Oncolytic viruses & their specific targeting to tumour cells. Indian J Med Res. 2012, 136 (4): 571-584.
Parato KA, Breitbach CJ, Boeuf FL, Wang J, Storbeck C, Ilkow C, Diallo SJ, Falls T, Burns J, Garcia V, Kanji F, Evgin L, Hu K, Paradis F, Knowles S, Hwang TH, Vanderhyden BC, Auer R, Kirn DH, Bell JC: The Oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in Cancers. Mol Ther. 2012, 20 (4): 749-758.
Baertsch MA, Leber MF, Bossow S, Singh M, Engeland CE, Albert J, Grossardt C, Jäger D, von Kalle C, Ungerechts G: MicroRNA-mediated multi-tissue detargeting of oncolytic measles virus. Cancer Gene Therapy advance online publication. 2014
Nechifor-Boila A, Borda A, Sassolas G, Hafdi-Nejjari Z, Borson-Chazot F, Lifante JC, Sturm N, Lavérriere MH, Berger N, Decaussin-Petrucci M: Immunohistochemical markers in the diagnosis of papillary thyroid carcinomas:The promising role of combined immunostaining using HBME-1 and CD56. Pathol Res Pract. 2013, 209 (9): 585-592.
O hara CJ, Heimann R, Murnane MJ, Cathepsin B: Activity and protein levels in thyroid carcinoma, Graves’ disease, and multinodular goiters. Thyroid. 1999, 9: 569-577.
Fr hlich E, Maier E, Wahl R: Interspecies differences in membrane-associated protease activities of thyrocytes and their relevance for thyroid cancer studies. J Exp Clin Cancer Res. 2012, 31: 45.
Demellawy DE, Nasr A, Alowami S: Application of CD56, P63 and CK19 immunohistochemistry in the diagnosis of papillary carcinoma of the thyroid. Diagnostic Pathol. 2008, 3 (5): 1-12.
Di Renzo MF, Olivero M, Serini G, Orlandi F, Pilotti S, Belfiore A, Costantino A, Vigneri R, Angeli A, Pierotti MA, Comoglio M: Overexpression of the c-MET/HGF receptor in human thyroid carcinomas derived from the follicular epithelium. J Endocrinol Invest. 1995, 18 (2): 134-139.
Trovato M, Villari D, Bartolone L, Spinella S, Simone A, Violi MA, Trimarchi F, Batolo D, Benvenga A: Expression of the hepatocyte growth factor and c-met in normal thyroid, non-neoplastic, and neoplastic nodules. Thyroid. 1998, 8: 125-131.
Takano T: Review Fetal cell carcinogenesis of the thyroid: theory and practice. Cancer Biol. 2007, 17 (3): 233-240.
Davies TF, Takao A, Reigh-Yi L, Yaron T, Rauf L: Thyrotropin receptor–associated diseases: from adenomata to Graves disease. J Clin Invest. 2005, 115: 1972-1983.
Iosco C, Rhoden KJ: Thyroid stimulating hormone receptor. Atlas Genet Cytogenet Oncol Haematol. 2009
Wondisford FE, Radovick S, Moates JM, Usala SJ, Weintraub BD: Isolation and characterization of the human thyrotropin beta-subunit gene. Differences in gene structure and promoter function from murine species. J Biol Chem. 1988, 5; 263 (25): 12538-12542.
Fiddes JC, Goodman HM: The gene encoding the common alpha subunit of the four human glycoprotein hormones. J Mol Appl Genet. 1981, 1 (1): 3-18.
Power AT, Wang J, Falls TJ, Paterson JM, Parato KA, Lichty BD, Stojdl DF, Forsyth PAJ, Atkins H, Bell JC: Carrier Cell-based Delivery of an Oncolytic Virus. Circumvents Antiviral Immunity Molecular Ther. 2007, 15 (1): 123-130.
Acknowledgments
We are grateful for the permission to adapt the figure from Atlas Genet Cytogenet Oncol Haematol, November 2009; Iosco, C; Rhoden, KJ. TSHR (thyroid stimulating hormone receptor).
http://atlasgeneticsoncology.org/Genes/TSHRID290ch14q31.html by permission of the Atlas.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Guan, M., Romano, G., Coroniti, R. et al. Progress in oncolytic virotherapy for the treatment of thyroid malignant neoplasm. J Exp Clin Cancer Res 33, 91 (2014). https://doi.org/10.1186/s13046-014-0091-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-014-0091-8