
Abstract
EZH2 is the catalytic subunit of the polycomb repressive complex 2 (PRC2), which along with other PRC2 components mediates gene expression suppression via the methylation of Histone H3 at lysine 27. Recent studies have revealed a dichotomous role of EZH2 in physiology and in the pathogenesis of cancer. While it plays an essential role in the development of the lymphoid system, its deregulation, whether due to genetic or non-genetic causes, promotes B cell- and T cell-related lymphoma or leukemia. These findings triggered a boom in the development of therapeutic EZH2 inhibitors in recent years. Here, we discuss physiologic and pathogenic function of EZH2 in lymphoid context, various internal causes of EZH2 aberrance and how EZH2 modulates lymphomagenesis through epigenetic silencing, post-translational modifications (PTMs), orchestrating with surrounding tumor micro-environment and associating with RNA or viral partners. We also summarize different strategies to directly inhibit PRC2-EZH2 or to intervene EZH2 upstream signaling.
Similar content being viewed by others
Background
EZH2, the enzymatic component of polycomb repressive complex 2 (PRC2), is evolutionarily conserved from drosophila to mammals. It acts as a lysine methyltransferase to catalyze methylation of histone H3 at lysine 27, thus establishing and maintaining H3K27 tri-methylation repressive marks. Apart from EZH2, the realization of histone methylation also requires facilitation from other PRC2 core components like EED and SUZ12. Besides EZH2, EZH1 as EZH2 homolog also complexes with other PRC2 subunits to trigger H3K27 tri-methylation. In physiological conditions, EZH2 only expresses in actively dividing cells and plays a vital role in cellular proliferation and differentiation. Whereas, EZH1 expresses in both dividing and differentiated cells [1].
Structurally, EZH2 protein contains a C-terminal catalytic SET domain, an adjacent CXC domain, two SANT domains, and an in-between ncRBD domain. The SET and CXC domains are required for the realization of histone methyltransferase activity [2]; the SANT domains enable EZH2 to bind to DNA for chromatin remodeling and transcriptional regulation [3]; the ncRBD domain is the binding region with non-coding RNAs.
EZH2 aberration has been seen in a wide range of cancers, including several categories of B cell and T cell lymphoid malignancies, and it is associated with poor clinical prognosis and outcomes. In most lymphoid malignant contexts, EZH2 is overexpressed and tumorigenic, yet it is repressed and acts as a tumor suppressor in T cell acute lymphocytic leukemia (ALL). In this review, we summarize the indispensable role of EZH2 in lymphoid development, the variety of mechanisms of EZH2-related oncogenesis in lymphomas, lymphoid leukemias, and myeloma, how Epstein-Barr virus (EBV) impacts EZH2 signaling as a result of virus-host interplay and effective therapeutic strategies to harness EZH2 in different cancerous contexts.
Physiological function of EZH2 in lymphogenesis
EZH2 in T cells
Studies in recent years highlight a crucial role of EZH2 functioning as an epigenetic repressor via the catalyzing of H3K27 tri-methylation in T cell activation [4], differentiation [4], plasticity, and lineage commitment [5, 6] by safeguarding correct downstream gene expressions. The repressive targets include key transcriptional factors which function as pivotal regulators of differentiation or self-tolerance [7,8,9], cytokines like interleukins, and IFN-γ [7, 10,11,12] as well as cell cycle-regulatory CDK inhibitors [4, 13]. EZH2 depletion in T lymphocytes disrupts genomic transcriptome and gene bivalent states [14], rendering loss-of-defense to pathogens [15], autoimmune diseases [15,16,17], or cell death via apoptosis [12]. Modulatory functions of EZH2 have been investigated intensively in several different subtypes and developmental phases of T lymphocyte, including T helper, T regulatory, cytotoxic T, memory T, natural killer (NK) T, and naïve T cells. Detailed findings of how EZH2 deploys H3K27 tri-methylation to regulate T cell development are summarized in Table 1 [4,5,6,7,8,9,10,11,12,13,14,15,16,17,18].
Notably, the role of EZH2 in modulating T cell fate is not limited to epigenetic silencing but multifaceted. AKT-phosphorylated EZH2, recruited by TCF1, activates BCL6 expression in T follicular helper cell development [4]. In NK T cells, EZH2 as a methyltransferase directly methylates the lineage-defining transcription factor PLZF, leading to its ubiquitination and degradation, thus restricting the expansion of a subset of immune-perturbing NK T cell [19]. Plus, EZH2 is phosphorylated by the DNA-dependent protein kinase during T cell activation which then regulates DNA damage-mediated T cell apoptosis, therefore maintaining T cell genomic integrity [20]. Besides its nuclear functions of modulating gene expression and chromosomal integrity, cytoplasmic EZH2, along with other PRC2 components, directly regulates T cell receptor signaling in the course of T cell activation [21].
EZH2 in B cells
EZH2 impacts B cell lineage commitment, maintenance, and transition through governing a repertoire of silent genomic loci. In early B cell progenitors, the level of EZH2 is abundantly seen in pro-B cells, which dropped drastically at the pre-B cell stage; EZH2-deficient B cells are developmentally impaired mainly due to a lack of immunoglobulin μ chain production. This indicates that EZH2 is involved in the checkpoint mechanisms controlling the pro-B to pre-B transition [22]. Germinal center (GC), the central factory of the humoral immune response, refers to the specialized microstructure within secondary lymphoid tissues formed upon infection or immunization. It is where maturing B cells undergo somatic mutations of B cell receptor genes, thereby producing a series of sublines. The long-lived antibody-secreting plasma cells and durable memory B cells are selected from these clones based on their antigen-binding affinity which ensure potent immune response against pathogens [23]. EZH2 also has profound influences on preserving a well-ordered GC development process. When resting B cells enters into cell cycle for activation, H3K27 tri-methylation marks increase noticeably along with other histone H3 methylation forms to orchestrate gene expressions and establish B cell identity at different developmental stages [24, 25]. In this process, EZH2 silences anti-proliferative genes, including CDKN family cell cycle-related tumor suppressors (CDKN1A, CDKN1B, and CDKN2A) [26, 27], through the conferment of H3K27 tri-methylation marks, and helps establish bivalent chromatin at key regulatory loci to transiently repress GC B cell differentiation [28], making provisions for antibody diversification, affinity maturation [29], and secretion [30].
EZH2 deregulation
Genetic and epigenetic abnormalities of EZH2
Among all categories of lymphomas and lymphoid leukemias, gain-of-function somatic mutations of EZH2 were mostly detected in GC-derived diffuse large B cell lymphoma (DLBCL) and follicular lymphoma (FL) [31,32,33,34]. In these malignancies, EZH2 mutations are causally linked to disease pathogenesis and transformation [28, 31]. This corresponds to the physiological role of wild-type EZH2 in GC formation by which EZH2 imprints repressive marks on proliferation-checkpoint genes and terminal-differentiation genes to temporarily maintain GC centroblast phenotypes in order to allow for important immune events like immunoglobin affinity maturation [27, 28]. Heterozygous mutation on tyrosine 641 (Y641H, Y641S, Y641C, Y641F, and Y641N) is the most widely occurred EZH2 activating mutation in GC-originated non-Hodgkin lymphoma (NHL), accounting for approximately 22% of GC-DLBCL and FL [35]. This point mutation is located within the catalytic SET domain of EZH2. It is associated with overall increased H3K27 tri-methylation level, suppressed expression of PRC2 target genes [35,36,37,38], and a global redistribution of the tri-methylation mark [39]. Collaborations between wild-type and Y641 single allele-mutated EZH2 for intensified conversion to H3K27 tri-methylation form have been observed that wild-type EZH2 displays greatest catalytic efficiency for H3K27 mono-methylation but decreased activity for subsequent di- and tri-methylation, and in contrast, Y641-mutated EZH2 exhibits limited efficiency to catalyze H3K27 mono-methylation but augmented catalytic activity for subsequent reactions [40]. The Y641-mutated EZH2 is resistant to JAK2/BTRC-mediated degradation and is thus more stable than wild-type EZH2 [41]. Besides, EZH2 A677G activating mutation was also identified in GC-NHL, which is related to enhanced H3K27 tri-methylation [35, 42]. In contrast, loss-of-function and non-sense EZH2 mutations were found in ALL, along with deletions and mutations of other PRC2 subunits [43,44,45,46,47]. In addition, missense EZH2 point mutations were also identified in a few Burkitt lymphoma cases with functions not fully determined [48]. Frequencies of EZH2 mutations in different malignant contexts have been summarized before [37, 49].
Besides, genetic lesions of EZH2, including copy number amplification, chromosomal gain or loss, and DNA hypermethylation, were also identified. In DLBCL, a total of 7.7% of cases harbored chromosomal gain and 2.7% had a chromosomal loss of EZH2 [50]. Moreover, 24% of FL and 39% of transformed FL (histologically transformed from FL to high-grade and aggressive type of NHL) patients had gain or amplification of EZH2 at chromosome 7q [31]. Promoter hypermethylation of EZH2 gene body was found in pediatric ALL, with 60% of patients displaying higher methylation at EZH2 CpG island than healthy donors [44].
Non-genetic causes of abnormal EZH2 expression
Dysregulated signaling pathways also cause abnormal EZH2 expression. C-MYC (hereafter MYC) overexpression or rearrangements have been uncovered in different types of aggressive B cell or T cell lymphomas, and the MYC-miRNA-EZH2 oncogenic axis or loop has been demonstrated in several reports, including ours [51,52,53,54]. Mechanistically, MYC upregulates EZH2 through repression of the EZH2-targeting miRNAs (miR-26a, miR-26b, miR-101) [52,53,54], and EZH2 could induce MYC expression in reverse via inhibition of the MYC-targeting miR-494 [54]. In multiple myeloma (MM), downregulated miR-138 brings about the upregulation of EZH2 target gene [55]. Besides, the NF-ΚB family transcription factors REL [56] and IL-6 [57] are critical EZH2 activators that upregulate EZH2 expression level in T-ALL or MM cells. In addition, proteolytic system deregulation could stabilize EZH2 protein and lead to its overexpression. As stated above, the recurrent Y641 EZH2 mutation in DLBCL and FL prevents JAK2-mediated phosphorylation and BTRC-mediated proteasomal degradation, leading to stabilization of EZH2 expression [41]. In natural killer/T cell lymphoma (NKTL), overexpression of MELK kinase phosphorylates and stabilizes EZH2 via modulation of EZH2 ubiquitination status [58]. Moreover, in chronic lymphocytic leukemia, microenvironment-induced NF-ΚB regulates genes associated with cell cycle, reactive oxygen species (ROS), and protein stability, resulting in suppression of RB and induction of EZH2 [59].
EZH2 mediates lymphoid oncogenesis
As epigenetic modifier
The most commonly observed function of EZH2 as a modulator of tumorigenesis is through epigenetic modification at histone H3 lysine 27. As target gene clusters of EZH2 such as the SOX genes and KIF family [60] are essential to maintain the developmental integrity of T cells and B cells, transcriptional impairment of these genes by EZH2 deregulation in lymphoid cells may thus give rise to malignant progression. In B cell lymphomas, overexpressed EZH2 hypermethylates the promoter of FAS-AS1 lncRNA and represses the FAS-AS1 expression, leading to impaired FAS-mediated apoptosis [61]. EZH2 activation also represses the tumor-suppressive BLIMP1 in GC-B cell lymphoma [29]. In B cell ALL, up-regulated EZH2 was shown to transcriptionally hold back the expression of tumor suppressor PTEN and P21 [62]. In MM, a genome-wide profiling of H3K27 tri-methylation identified a positive correlation between PRC2-mediated gene silencing and disease progression or poor prognosis [63]. A number of EZH2 target genes relating to MM pathogenesis and progression have been characterized; for example, RBPMS [55], which confers chemo-resistance to MM cells. In T-ALL, deficiency of EZH2 and H3K27 tri-methylation results in activation of IGF1, which relates to leukemia stem cell property [64], and CRIP2, which represses apoptosis [65], as well as genes involved in the cytokine signaling pathways and early hematopoietic transcriptional regulators [66]. Importantly, EZH2 may transcriptionally coordinate with other oncogenic regulators. Aberrantly activated NOTCH1 in T-ALL binds to the promoter of HES1, a critical mediator of leukemogenesis [67], and forces considerable eviction of EZH2 and other PRC2 subunits as well as loss of H3K27 tri-methylation repressive marks [46], reinforcing the activation of HES1. Defectively mutated RB fails to recruit EZH2 to diverse repeat DNA sequences as RB-EZH2 complex, disperses H3K27 tri-methylation from these genomic locations and permits repeat expression, thus conferring lymphoma susceptibilities [68].
In recent years, the non-canonical role of EZH2 as a transcriptional co-activator has been demonstrated in several types of cancer [52, 69,70,71,72]. Our team has reported that in the NKTL oncogenic role of EZH2 is independent of its methyltransferase activity [52]; the Y244-phosphorylated EZH2 binds to RNA POLII and trans-activates a set of genes associated with proliferation, cell cycle, DNA replication, stemness, and invasiveness [69].
Associating with long non-coding RNAs
In recent decades, long non-coding RNAs (lncRNAs) have emerged as an indispensable regulator in almost every aspect of biology, and mounting evidence reveals that lncRNAs play an important role in tumorigenesis by modulating chromatin organization, transcription and post-transcriptional regulation [73]. The interplay between EZH2 and lncRNA is noticeable when it comes to EZH2-mediated oncogenesis. HOTAIR was originally demonstrated as a modular scaffold that provides binding surfaces to tether EZH2 with other histone-modifying complexes [74]. Later studies in several cancerous contexts revealed that HOTAIR modulates gene repression [75,76,77,78,79,80] or associates with other oncogenic players [81, 82] in both PRC2-dependent and independent manners to promote malignant phenotypes. In DLBCL, upregulated HOTAIR is validated as an independent indicator of poor prognosis [83] and thought as one of the possible mechanisms for inducing H3K27 tri-methylation via EZH2-related PRC2 activation [84]. MALAT1, another EZH2-binding lncRNA, regulates cancer cell metastasis [85,86,87,88] and chemo-resistance [89] in different solid tumors via EZH2-associated mechanisms. In mantle cell lymphoma, elevated MALAT1 expression correlates with poor prognosis and reduces overall survival, and knockdown of MALAT1 results in an enhanced level of P21 and P27 through the modulation of EZH2 [90]. In T and NK cell lymphoma, the EZH2-binding MALAT1 is highly expressed and linked to poor prognosis [91]. In addition, ANRIL, frequently overexpressed and tumorigenic in a panel of cancers [92,93,94,95], forms a ternary complex with EZH2 and P65, and epigenetically marks and represses P21/CDKN1A transcription in adult T cell leukemia [96].
Regulated by phosphorylation
Over the past few years, various forms of EZH2 post-translational modifications (PTMs) have been uncovered. These include phosphorylation, acetylation, ubiquitination, and O-GlcNAcylation, all of which confer marked biological impacts to EZH2 functions. Phosphorylation is the most well-studied EZH2 PTM, and EZH2 phosphorylation at multiple distinct residues yields critical influences or are even vital to EZH2-related lymphoid oncogenesis. The firstly reported EZH2 S21 phosphorylation, observed in CD138+ bone marrow cells of MM patients, endows cell-adhesion-mediated drug resistance to MM cells through H3K27 hypomethylation-induced activation of IGF1 [97]. As stated earlier, JAK2- and MELK-directed EZH2 phosphorylation each modulates stability and proteasome-mediated degradation of EZH2 in DLBCL/FL and NKTL, respectively [41, 58]. Moreover, EZH2 phosphorylation by JAK3 in NKTL, as we reported previously, functionally switch the role of EZH2 from PRC2-associated epigenetic repressor to PRC2-independent transcriptional activator [69]. CDK1-mediated EZH2 phosphorylation, initially demonstrated as a key mechanism that regulates cell cycle and differentiation in embryonic stem cells [98,99,100], is crucial for EZH2 binding to tumorigenic HOTAIR [98] and MALAT1 [90].
Modulating tumor microenvironment
Apart from mediating tumorigenicity in cancer cells, EZH2 also has a role in the regulation of tumor microenvironment. PRC2 is activated in hematopoietic bone marrow resident cells besides tumor cells in DLBCL patients and correlates with poor outcomes [101]. In cutaneous anaplastic T cell lymphoma, EZH2 not only boosts lymphoma cell survival but also represses CXCL10 to hinder the recruitment of effector CD4+ and CD8+ T cells into the microenvironment, suggesting EZH2 possesses dual roles here [102]. In DLBCL, the survival-favoring EZH2 Y641 mutation is also a genetic mechanism underlying MHC-II deficiency, which leads to escape from immune surveillance and inferior prognosis [103]. In CLL-associated fibroblasts, NF-ΚB-activated EZH2 antagonizes a subset of senescence-associated genes to promote tumorigenesis [59].
Connected with EBV infections
EBV is one of the human herpes virus that was firstly observed in Burkitt lymphoma cells [104]. It mostly transmits via saliva and oral contacts. Primary infection of EBV is widespread and tends to be latent and asymptomatic in most individuals. However, as lymphocytes are the major host cells for the EBV natural life cycle, EBV infection is closely associated with onsets of several types of lymphomas and lymphocytic leukemias both in the immunocompetent and immunosuppressive states [105, 106].
The life cycle of EBV is regulated by chromatin modifiers which epigenetically switch EBV protein expression on and off. EZH2 is one of these modifiers involved in the maintenance of EBV latency and reactivation acting by conferring H3K27 tri-methylation marks to EBV-associated genes [107]. Of note, EBV-encoded proteins also utilize or synergize with EZH2 to silence tumor suppressor genes to drive B cell lymphomagenesis. The viral oncoprotein LMP-1 induces the recruitment of inhibitory protein complexes containing EZH2 to the promoter of tumor suppressor gene DOK1, resulting in H3K27 tri-methylation-mediated repression [108]. LMP-1-dependent activation of JNK-1 facilitates the recruitment of P73 transcription factor to the promoter of ΔNP73α, a strong antagonist of P53, in accord with the displacement of EZH2 within the same locus [109]. Together with EZH2, EBV nuclear antigens EBNA3A and EBNA3C co-silence CDNK2A/B loci, which encode P14ARF, P15INK4B, and P16INK4A as well as the BIM enhancer hub, through inducing chromatin looping, disrupting physical interactions between the promoters or depositing H3K27 tri-methylation silencing mark [110, 111]. Importantly, EBV viral gene products may indirectly affect EZH2 expression by modulating EZH2 upstream gene NF-ΚB via multifaceted mechanisms. EBV viral proteins regulate NF-ΚB signaling cascade dynamics within each stage of entry-latency-host lysis infection cycle through mimicking host proteins, binding to host receptors or other forms of interplay [112].
Therapeutic interventions to EZH2 signaling
EZH2 inhibitors
The adenosine homolog DZnep, originally developed as an anti-HIV1 agent [113] and S-adenosylhomocysteine hydrolase inhibitor [114], was the first drug used to inhibit EZH2 expression [115]. DZnep effectively depletes cellular levels of EZH2, inhibits tri-methylation of Histone H3 at lysine 27, and induces cell apoptosis in a variety of cancerous contexts [115,116,117,118], including Burkitt lymphoma [119], ALL [120], and NKTL [52]. However, Dznep is a global histone methylation inhibitor rather than a H3K27-specific one [121].
Since the year 2012, several serials of specific EZH2 inhibitors, which compete with S-adenosylmethionine for the binding to EZH2 enzymatic SET domain, have been reported (Table 2). These inhibitors could suppress H3K27 tri-methylation, reactivate silenced PRC2 target genes and demonstrated effectiveness in inhibition of cell survival in GC-derived B cell lymphomas harboring EZH2-activating mutations amid several types of lymphoid malignancies [35, 122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140].
Among these inhibitors, EPZ-6438 (Tazemetostat) is a representative that has already entered clinical trial phase I/II for the treatment of multiple malignancies with EZH2 aberrance, including GC-derived and other types of B cell lymphoma (NCT03010982, NCT03028103, NCT01897571, and NCT02875548) [141].
As both EZH1-containing PRC2 and EZH2-containing PRC2 complexes contribute to the maintenance of H3K27 tri-methylation marks, dual inhibitors of EZH1/2, like UNC1999, were developed to target the homologous enzymatic SET domain [142, 143]. In addition, a group of Tanshindiol compounds, major active components of the root of Salvia miltiorrhiza, also displayed inhibitory effects on EZH2 methyltransferase activity [144].
Differential response to EZH2 small-molecule inhibitors like EI1 and GSK126 were observed in GC B cell lymphoma cell lines, owing to the activation of IGF-1R, MEK, and PI3K pathways [145], MLL1-P300/CBP-directed H3K27 acetylation gain [146] as well as secondary EZH2 mutations in wild-type and mutant EZH2 alleles [147, 148].
Inhibiting EED
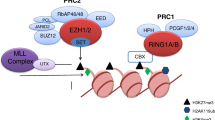
Another strategy to harness H3K27 hypermethylation for therapeutics is to target the regulatory PRC2 subunit EED or EED-EZH2 interaction. SAH-EZH2, a peptide inhibitor which dissociates bound EZH2-EED, degrades EZH2, diminishes H3K27 tri-methylation, and inhibits cell proliferation in MLL-AF9 cells [149]. Treatment with this peptide in NKTL cells also inhibits H3K27 tri-methylation, but leaves EZH2 expression intact [69]. Astemizole, originally a second-generation anti-histamine, was withdrawn due to its potential to cause arrhythmia. Later studies uncovered that Astemizole could disrupt EZH2-EED interaction and arrest cell proliferation in some B cell lymphoma cell lines [150]. Moreover, several selective EED inhibitors binding to the H3K27 tri-methylation pocket on EED have been synthesized [151]. Among them, EED226 could induce a conformational change upon its binding to EED and demonstrated sustained sustain tumor regression effects in EZH2-mutant preclinical DLBCL model [151,152,153]. Therapeutic strategies for targeting EZH1/2 or EED are depicted in Fig. 1.

Schematic diagram showing agents specifically inhibit EZH1/2 or EED
Inhibiting EZH2 upstream signaling
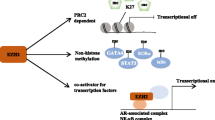
Apart from directly targeting the PRC2 complex, recent studies on EZH2 signaling pathways, including ours, also implied potentials of targeting EZH2 upstream factors for therapeutic purposes (Fig. 2). Proteasome inhibitors Bortezomib and Carfilzomib block proteasome-mediated degradation of CDK inhibitors P21 and P27, therefore depleting EZH2 expression level via CDK-RB-E2F axis in MM [55, 154]. In NKTL, selective JAK3 inhibitor PF-956980 removes EZH2-Y244 phosphorylation and thus obliterates oncogenic and downstream activating function of EZH2 [69]. MELK inhibition in this malignancy effectively diminishes EZH2 level by eliminating EZH2 S220 phosphorylation and promoting associated K222 ubiquitination [58].

Schematic diagram showing strategies of inhibiting EZH2 upstream signaling
Combinatorial targeting with other chemotherapeutic agents
In recent years, therapeutics combining individual EZH2 inhibitor with other drugs have been explored. Synergistic or re-sensitizing effects between EZH2 inhibitors and traditional cytotoxic anti-cancer drugs have been seen [155, 156]. In view of the abundantly present epigenetic abnormalities of H3K27 tri-methylation and histone acetylation, a couple of studies have combined EZH2 inhibitors with different HDAC inhibitors for the treatment in MM [157, 158] and B cell lymphoma cells [54, 123, 159, 160], and favorable therapeutic coordination were observed. Dual-targeting of MYC and EZH2 has been employed to disrupt MYC-EZH2 oncogenic axis in aggressive B cell lymphoma cells [51]. In addition, EZH2 inhibition has also been combined with inhibition of REL, a transcriptional activator of EZH2 expression [56]. Details of combinational regimens are summarized in Table 3.
Conclusions and future directions
The research evidence accumulated in this review demonstrates an indispensable role of physiological EZH2 in mediating normal B cell and T cell lymphogenesis and reveals how deregulated EZH2 modulates pathogenesis of lymphoid malignancies. Major causes dictating EZH2 aberrance are genetic abnormalities including somatic mutations, chromosomal gain/loss, and promoter hypermethylation as well as translational and post-translational causes via multiple signaling pathways. Pathogenic EZH2 modulates lymphoid oncogenesis by epigenetic repression of tumor suppressors, orchestrating with lncRNAs, site-specific PTMs, affecting microenvironment and EBV-host interplay. In recent years, an emerging interest in investigating how EZH2 assists tumor cells to escape immune surveillance has developed, and more efforts are required in future studies to clarify the exact role of EZH2 in facilitating a tumorigenic microenvironment in different types of lymphoid malignancies.
In the recent decade, a couple of strategies have been adopted to harness EZH2 deregulation for therapeutic intervention. Although the oncogenic mechanisms of EZH2 have already been uncovered by a number of in-depth studies, PRC2-based EZH2 therapeutics still have a long way to go. Dozens of chemotherapeutic agents have been developed to target the EZH2 enzymatic SET domain for therapeutics; yet, for most of these drugs, satisfactory effectiveness was only seen in B cell lymphoma cell lines or xenografts with EZH2 gain-of-function mutations. Although several compounds of EZH2-SET inhibitors have entered into clinical trials, some have already failed in phase I at least partly due to the negative mediation of anti-tumor immunity [161]. Development of EZH1/2 inhibitors and EED inhibitors represents a big leap, as these agents effectively overcome chemo-resistance of EZH2-SET inhibitors GSK126 and EPZ-6438 in DLBCL [145].
Due to the fact that none of the commercialized EZH2-specific inhibitors was able to bring down EZH2-mediated lymphomagenesis in NKTL, JAK3, or MELK inhibition has been exploited for dual-targeting of the kinase and EZH2 as an alternative strategy [52, 58, 69]. Future studies are still required to precisely deplete tumorigenic EZH2. Given that EBV infections manifest in all cases of NKTL as well as in some cases of Burkitt lymphoma and DLBCL [105], anti-EBV treatment may well complement EZH2-based therapeutics. Studies determining whether combining antivirals and EZH2 inhibitors could yield synergism are therefore needed.
Availability of data and materials
Data sharing is not applicable to this review article as no datasets were analyzed.
Abbreviations
- ALL:
-
Acute lymphocytic leukemia
- DLBCL:
-
Diffuse large B cell lymphoma
- EBV:
-
Epstein-Barr virus
- FL:
-
Follicular lymphoma
- GC:
-
Germinal center
- LncRNAs:
-
Long non-coding RNAs
- MM:
-
Multiple myelomas
- NHL:
-
Non-Hodgkin lymphoma
- NK:
-
Natural killer
- NKTL:
-
Natural killer/T cell lymphoma
- PRC2:
-
Polycomb repressive complex 2
- PTMs:
-
Post-translational modifications
- ROS:
-
Reactive oxygen species
References
Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–9.
Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutation Res. 2008;647:21–9.
Boyer LA, Latek RR, Peterson CL. The SANT domain: a unique histone-tail-binding module? Nat Rev Mol Cell Biol. 2004;5:158–63.
Li F, Zeng Z, Xing S, Gullicksrud JA, Shan Q, Choi J, Badovinac VP, Crotty S, Peng W, Xue HH. Ezh2 programs TFH differentiation by integrating phosphorylation-dependent activation of Bcl6 and polycomb-dependent repression of p19Arf. Nat Commun. 2018;9:5452.
Gray SM, Amezquita RA, Guan T, Kleinstein SH, Kaech SM. Polycomb repressive complex 2-mediated chromatin repression guides effector CD8(+) T cell terminal differentiation and loss of multipotency. Immunity. 2017;46:596–608.
He S, Liu Y, Meng L, Sun H, Wang Y, Ji Y, Purushe J, Chen P, Li C, Madzo J, Issa JP, Soboloff J, Reshef R, Moore B, Gattinoni L, Zhang Y. Ezh2 phosphorylation state determines its capacity to maintain CD8(+) T memory precursors for antitumor immunity. Nat Commun. 2017;8:2125.
Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, Hosokawa H, Koseki H, Tokoyoda K, Suzuki Y, Motohashi S, Nakayama T. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity. 2013;39:819–32.
Xiong Y, Khanna S, Grzenda AL, Sarmento OF, Svingen PA, Lomberk GA, Urrutia RA, Faubion WA Jr. Polycomb antagonizes p300/CREB-binding protein-associated factor to silence FOXP3 in a Kruppel-like factor-dependent manner. J Biol Chem. 2012;287:34372–85.
Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, Rudensky AY. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol. 2014;15:580–7.
Koyanagi M, Baguet A, Martens J, Margueron R, Jenuwein T, Bix M. EZH2 and histone 3 trimethyl lysine 27 associated with Il4 and Il13 gene silencing in Th1 cells. J Biol Chem. 2005;280:31470–7.
Wang A, Pan D, Lee YH, Martinez GJ, Feng XH, Dong C. Cutting edge: Smad2 and Smad4 regulate TGF-beta-mediated Il9 gene expression via EZH2 displacement. J Immunol. 1950;191(2013):4908–12.
Zhang Y, Kinkel S, Maksimovic J, Bandala-Sanchez E, Tanzer MC, Naselli G, Zhang JG, Zhan Y, Lew AM, Silke J, Oshlack A, Blewitt ME, Harrison LC. The polycomb repressive complex 2 governs life and death of peripheral T cells. Blood. 2014;124:737–49.
Chen G, Subedi K, Chakraborty S, Sharov A, Lu J, Kim J, Mi X, Wersto R, Sung MH, Weng NP. Ezh2 regulates activation-induced CD8(+) T cell cycle progression via repressing Cdkn2a and Cdkn1c expression. Frontiers Immunol. 2018;9:549.
Dobenecker MW, Kim JK, Marcello J, Fang TC, Prinjha R, Bosselut R, Tarakhovsky A. Coupling of T cell receptor specificity to natural killer T cell development by bivalent histone H3 methylation. J Exp Med. 2015;212:297–306.
Yang XP, Jiang K, Hirahara K, Vahedi G, Afzali B, Sciume G, Bonelli M, Sun HW, Jankovic D, Kanno Y, Sartorelli V, O’Shea JJ, Laurence A. EZH2 is crucial for both differentiation of regulatory T cells and T effector cell expansion. Sci Rep. 2015;5:10643.
DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, Zhang R, Turka L, Marson A, Bluestone JA. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity. 2015;42:227–38.
Sarmento OF, Svingen PA, Xiong Y, Sun Z, Bamidele AO, Mathison AJ, Smyrk TC, Nair AA, Gonzalez MM, Sagstetter MR, Baheti S, McGovern DP, Friton JJ, Papadakis KA, Gautam G, Xavier RJ, Urrutia RA, Faubion WA. The role of the histone methyltransferase enhancer of zeste homolog 2 (EZH2) in the Pathobiological mechanisms underlying inflammatory bowel disease (IBD). J Biol Chem. 2017;292:706–22.
Hou S, Clement RL, Diallo A, Blazar BR, Rudensky AY, Sharpe AH, Sage PT. FoxP3 and Ezh2 regulate Tfr cell suppressive function and transcriptional program. J Exp Med. 2019;216:605–20.
Vasanthakumar A, Xu D, Lun AT, Kueh AJ, van Gisbergen KP, Iannarella N, Li X, Yu L, Wang D, Williams BR, Lee SC, Majewski IJ, Godfrey DI, Smyth GK, Alexander WS, Herold MJ, Kallies A, Nutt SL, Allan RS. A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep. 2017;18:619–31.
Wang Y, Sun H, Wang J, Wang H, Meng L, Xu C, Jin M, Wang B, Zhang Y, Zhang Y, Zhu T. DNA-PK-mediated phosphorylation of EZH2 regulates the DNA damage-induced apoptosis to maintain T-cell genomic integrity. Cell Death Dis. 2016;7:e2316.
Dobenecker MW, Park JS, Marcello J, McCabe MT, Gregory R, Knight SD, Rioja I, Bassil AK, Prinjha RK, Tarakhovsky A. Signaling function of PRC2 is essential for TCR-driven T cell responses. J Biol Chem. 2018;215:1101–13.
Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT, Tarakhovsky A. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003;4:124–31.
Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, Graca L. Regulation of the germinal center response. Frontiers Immunol. 2018;9:2469.
Baxter J, Sauer S, Peters A, John R, Williams R, Caparros ML, Arney K, Otte A, Jenuwein T, Merkenschlager M, Fisher AG. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J. 2004;23:4462–72.
Herviou L, Jourdan M, Martinez AM, Cavalli G, Moreaux J. EZH2 is overexpressed in transitional preplasmablasts and is involved in human plasma cell differentiation. Leukemia. 2019;12:1.
Velichutina I, Shaknovich R, Geng H, Johnson NA, Gascoyne RD, Melnick AM, Elemento O. EZH2-mediated epigenetic silencing in germinal center B cells contributes to proliferation and lymphomagenesis. Blood. 2010;116:5247–55.
Beguelin W, Rivas MA, Calvo Fernandez MT, Teater M, Purwada A, Redmond D, Shen H, Challman MF, Elemento O, Singh A, Melnick AM. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat Commun. 2017;8:877.
Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, Martinez-Garcia E, Zhang H, Zheng Y, Verma SK, McCabe MT, Ott HM, Van Aller GS, Kruger RG, Liu Y, McHugh CF, Scott DW, Chung YR, Kelleher N, Shaknovich R, Creasy CL, Gascoyne RD, Wong KK, Cerchietti L, Levine RL, Abdel-Wahab O, Licht JD, Elemento O, Melnick AM. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23:677–92.
Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, George L, Alberghini F, Ferrarini L, Talukder AK, Ponzoni M, Testa G, Nojima T, Doglioni C, Kitamura D, Toellner KM, Su IH, Casola S. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest. 2013;123:5009–22.
Guo M, Price MJ, Patterson DG, Barwick BG, Haines RR, Kania AK, Bradley JE, Randall TD, Boss JM, Scharer CD. EZH2 represses the B cell transcriptional program and regulates antibody-secreting cell metabolism and antibody production. J Immunol. 1950;200(2018):1039–52.
Bouska A, Zhang W, Gong Q, Iqbal J, Scuto A, Vose J, Ludvigsen M, Fu K, Weisenburger DD, Greiner TC, Gascoyne RD, Rosenwald A, Ott G, Campo E, Rimsza LM, Delabie J, Jaffe ES, Braziel RM, Connors JM, Wu CI, Staudt LM, D'Amore F, McKeithan TW, Chan WC. Combined copy number and mutation analysis identifies oncogenic pathways associated with transformation of follicular lymphoma. Leukemia. 2017;31:83–91.
Majer CR, Jin L, Scott MP, Knutson SK, Kuntz KW, Keilhack H, Smith JJ, Moyer MP, Richon VM, Copeland RA, Wigle TJ. A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 2012;586:3448–51.
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R, Tam A, Varhol R, Smailus D, Moksa M, Zhao Y, Delaney A, Qian H, Birol I, Schein J, Moore R, Holt R, Horsman DE, Connors JM, Jones S, Aparicio S, Hirst M, Gascoyne RD, Marra MA. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–5.
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, Firme MR, Rogic S, Griffith M, Chan S, Yakovenko O, Meyer IM, Zhao EY, Smailus D, Moksa M, Chittaranjan S, Rimsza L, Brooks-Wilson A, Spinelli JJ, Ben-Neriah S, Meissner B, Woolcock B, Boyle M, McDonald H, Tam A, Zhao Y, Delaney A, Zeng T, Tse K, Butterfield Y, Birol I, Holt R, Schein J, Horsman DE, Moore R, Jones SJ, Connors JM, Hirst M, Gascoyne RD, Marra MA. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303.
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, Brandt M, Miller WH, Dhanak D, Verma SK, Tummino PJ, Creasy CL. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12.
Wigle TJ, Knutson SK, Jin L, Kuntz KW, Pollock RM, Richon VM, Copeland RA, Scott MP. The Y641C mutation of EZH2 alters substrate specificity for histone H3 lysine 27 methylation states. FEBS Lett. 2011;585:3011–4.
Lunning MA, Green MR. Mutation of chromatin modifiers; an emerging hallmark of germinal center B-cell lymphomas. Blood Cancer J. 2015;5:e361.
Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, Marquez VE, Marra MA, Gascoyne RD, Humphries RK, Arrowsmith CH, Morin GB, Aparicio SA. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–9.
Souroullas GP, Jeck WR, Parker JS, Simon JM, Liu JY, Paulk J, Xiong J, Clark KS, Fedoriw Y, Qi J, Burd CE, Bradner JE, Sharpless NE. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med. 2016;22:632–40.
Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010;107:20980–5.
Sahasrabuddhe AA, Chen X, Chung F, Velusamy T, Lim MS, Elenitoba-Johnson KS. Oncogenic Y641 mutations in EZH2 prevent Jak2/beta-TrCP-mediated degradation. Oncogene. 2015;34:445–54.
McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, Chen SB, Della Pietra A 3rd, Dul E, Hughes AM, Gilbert SA, Thrall SH, Tummino PJ, Kruger RG, Brandt M, Schwartz B, Creasy CL. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A. 2012;109:2989–94.
Burgos S, Montalban-Bravo G, Fuente L, Jabbour EJ, Kanagal-Shamanna R, Soltysiak KA, Garcia-Manero G, Mela-Osorio MJ. Novel EZH2 mutation in a patient with secondary B-cell acute lymphocytic leukemia after deletion 5q myelodysplastic syndrome treated with lenalidomide: a case report. Medicine. 2019;98:e14011.
Schafer V, Ernst J, Rinke J, Winkelmann N, Beck JF, Hochhaus A, Gruhn B, Ernst T. EZH2 mutations and promoter hypermethylation in childhood acute lymphoblastic leukemia. J Cancer Res Clin Oncol. 2016;142:1641–50.
Neumann M, Vosberg S, Schlee C, Heesch S, Schwartz S, Gokbuget N, Hoelzer D, Graf A, Krebs S, Bartram I, Blum H, Bruggemann M, Hecht J, Bohlander SK, Greif PA, Baldus CD. Mutational spectrum of adult T-ALL. Oncotarget. 2015;6:2754–66.
Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, Asp P, Hadler M, Rigo I, De Keersmaecker K, Patel J, Huynh T, Utro F, Poglio S, Samon JB, Paietta E, Racevskis J, Rowe JM, Rabadan R, Levine RL, Brown S, Pflumio F, Dominguez M, Ferrando A, Aifantis I. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012;18:298–301.
Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, Collins-Underwood JR, Ma J, Roberts KG, Pounds SB, Ulyanov A, Becksfort J, Gupta P, Huether R, Kriwacki RW, Parker M, McGoldrick DJ, Zhao D, Alford D, Espy S, Bobba KC, Song G, Pei D, Cheng C, Roberts S, Barbato MI, Campana D, Coustan-Smith E, Shurtleff SA, Raimondi SC, Kleppe M, Cools J, Shimano KA, Hermiston ML, Doulatov S, Eppert K, Laurenti E, Notta F, Dick JE, Basso G, Hunger SP, Loh ML, Devidas M, Wood B, Winter S, Dunsmore KP, Fulton RS, Fulton LL, Hong X, Harris CC, Dooling DJ, Ochoa K, Johnson KJ, Obenauer JC, Evans WE, Pui CH, Naeve CW, Ley TJ, Mardis ER, Wilson RK, Downing JR, Mullighan CG. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–63.
Richter J, Schlesner M, Hoffmann S, Kreuz M, Leich E, Burkhardt B, Rosolowski M, Ammerpohl O, Wagener R, Bernhart SH, Lenze D, Szczepanowski M, Paulsen M, Lipinski S, Russell RB, Adam-Klages S, Apic G, Claviez A, Hasenclever D, Hovestadt V, Hornig N, Korbel JO, Kube D, Langenberger D, Lawerenz C, Lisfeld J, Meyer K, Picelli S, Pischimarov J, Radlwimmer B, Rausch T, Rohde M, Schilhabel M, Scholtysik R, Spang R, Trautmann H, Zenz T, Borkhardt A, Drexler HG, Moller P, MacLeod RA, Pott C, Schreiber S, Trumper L, Loeffler M, Stadler PF, Lichter P, Eils R, Kuppers R, Hummel M, Klapper W, Rosenstiel P, Rosenwald A, Brors B, Siebert R. Recurrent mutation of the ID3 gene in Burkitt lymphoma identified by integrated genome, exome and transcriptome sequencing. Nat Genet. 2012;44:1316–20.
Lue JK, Amengual JE. Emerging EZH2 inhibitors and their application in lymphoma. Curr Hematol Malig Rep. 2018;13:369–82.
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, Hodson DJ, Xiao W, Yu X, Yang Y, Zhao H, Xu W, Liu X, Zhou B, Du W, Chan WC, Jaffe ES, Gascoyne RD, Connors JM, Campo E, Lopez-Guillermo A, Rosenwald A, Ott G, Delabie J, Rimsza LM, Wei KTK, Zelenetz AD, Leonard JP, Bartlett NL, Tran B, Shetty J, Zhao Y, Soppet DR, Pittaluga S, Wilson WH, Staudt LM. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378:1396–407.
Zhao X, Lwin T, Zhang X, Huang A, Wang J, Marquez VE, Chen-Kiang S, Dalton WS, Sotomayor E, Tao J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia. 2013;27:2341–50.
Yan J, Ng SB, Tay JL, Lin B, Koh TL, Tan J, Selvarajan V, Liu SC, Bi C, Wang S, Choo SN, Shimizu N, Huang G, Yu Q, Chng WJ. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood. 2013;121:4512–20.
Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, Moller P, Stilgenbauer S, Pollack JR, Wirth T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112:4202–12.
Zhang X, Zhao X, Fiskus W, Lin J, Lwin T, Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, Chen-Kiang S, Moscinski LC, Seto E, Dalton WS, Wright KL, Sotomayor E, Bhalla K, Tao J. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-cell lymphomas. Cancer Cell. 2012;22:506–23.
Rastgoo N, Pourabdollah M, Abdi J, Reece D, Chang H. Dysregulation of EZH2/miR-138 axis contributes to drug resistance in multiple myeloma by downregulating RBPMS. Leukemia. 2018;32:2471–82.
Neo WH, Lim JF, Grumont R, Gerondakis S, Su IH. C-Rel regulates Ezh2 expression in activated lymphocytes and malignant lymphoid cells. J Biol Chem. 2014;289:31693–707.
Croonquist PA, Van Ness B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene. 2005;24:6269–80.
Li B, Yan J, Phyu T, Fan S, Chung TH, Mustafa NB, Lin B, Wang L, Eichhorn PJA, Goh BC, Ng SB, Kappei D, Chng WJ. MELK mediates the stability of EZH2 through site-specific phosphorylation in extranodal natural killer/T-cell lymphoma. Blood. 2019.
Iannetti A, Ledoux AC, Tudhope SJ, Sellier H, Zhao B, Mowla S, Moore A, Hummerich H, Gewurz BE, Cockell SJ, Jat PS, Willmore E, Perkins ND. Regulation of p53 and Rb links the alternative NF-kappaB pathway to EZH2 expression and cell senescence. PLoS Genet. 2014;10:e1004642.
Wang C, Oshima M, Sato D, Matsui H, Kubota S, Aoyama K, Nakajima-Takagi Y, Koide S, Matsubayashi J, Mochizuki-Kashio M, Nakano-Yokomizo T, Bai J, Nagao T, Kanai A, Iwama A, Sashida G. Ezh2 loss propagates hypermethylation at T cell differentiation-regulating genes to promote leukemic transformation. J Clin Invest. 2018;128:3872–86.
Sehgal L, Mathur R, Braun FK, Wise JF, Berkova Z, Neelapu S, Kwak LW, Samaniego F. FAS-antisense 1 lncRNA and production of soluble versus membrane FAS in B-cell lymphoma. Leukemia. 2014;28:2376–87.
Chen J, Li J, Han Q, Sun Z, Wang J, Wang S, Zhao RC. Enhancer of zeste homolog 2 is overexpressed and contributes to epigenetic inactivation of p21 and phosphatase and tensin homolog in B-cell acute lymphoblastic leukemia. Exp Biol Med. 2012;237:1110–6.
Agarwal P, Alzrigat M, Parraga AA, Enroth S, Singh U, Ungerstedt J, Osterborg A, Brown PJ, Ma A, Jin J, Nilsson K, Oberg F, Kalushkova A, Jernberg-Wiklund H. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget. 2016;7:6809–23.
Giambra V, Gusscott S, Gracias D, Song R, Lam SH, Panelli P, Tyshchenko K, Jenkins CE, Hoofd C, Lorzadeh A, Carles A, Hirst M, Eaves CJ, Weng AP. Epigenetic restoration of fetal-like IGF1 signaling inhibits leukemia stem cell activity. Cell Stem Cell. 2018;23:714–726.e717.
Aries IM, Bodaar K, Karim SA, Chonghaile TN, Hinze L, Burns MA, Pfirrmann M, Degar J, Landrigan JT, Balbach S, Peirs S, Menten B, Isenhart R, Stevenson KE, Neuberg DS, Devidas M, Loh ML, Hunger SP, Teachey DT, Rabin KR, Winter SS, Dunsmore KP, Wood BL, Silverman LB, Sallan SE, Van Vlierberghe P, Orkin SH, Knoechel B, Letai AG, Gutierrez A. PRC2 loss induces chemoresistance by repressing apoptosis in T cell acute lymphoblastic leukemia. J Exp Med. 2018;215:3094–114.
Danis E, Yamauchi T, Echanique K, Zhang X, Haladyna JN, Riedel SS, Zhu N, Xie H, Orkin SH, Armstrong SA, Bernt KM, Neff T. Ezh2 controls an early hematopoietic program and growth and survival signaling in early T cell precursor acute lymphoblastic leukemia. Cell Rep. 2016;14:1953–65.
Schnell SA, Ambesi-Impiombato A, Sanchez-Martin M, Belver L, Xu L, Qin Y, Kageyama R, Ferrando AA. Therapeutic targeting of HES1 transcriptional programs in T-ALL. Blood. 2015;125:2806–14.
Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, Ferwati S, MacDonald WA, Howlett CJ, Welch ID, Rubin SM, Mann MRW, Dick FA. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol Cell. 2016;64:1074–87.
Yan J, Li B, Lin B, Lee PT, Chung TH, Tan J, Bi C, Lee XT, Selvarajan V, Ng SB, Yang H, Yu Q, Chng WJ. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood. 2016;128:948–58.
Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, Liou YC, Yu Q. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43:798–810.
Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, Thornton JE, Gregory RI, Morrissey C, Vessella RL, Montironi R, Magi-Galluzzi C, Kantoff PW, Balk SP, Liu XS, Brown M. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–9.
Kim J, Lee Y, Lu X, Song B, Fong KW, Cao Q, Licht JD, Zhao JC, Yu J. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep. 2018;25:2808–2820.e2804.
Yang G, Lu X, Yuan L. LncRNA: a link between RNA and cancer. Biochim Biophys Acta. 2014;1839:1097–109.
Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science. 2010;329:689–93.
Wang L, Zeng X, Chen S, Ding L, Zhong J, Zhao JC, Wang L, Sarver A, Koller A, Zhi J, Ma Y, Yu J, Chen J, Huang H. BRCA1 is a negative modulator of the PRC2 complex. EMBO J. 2013;32:1584–97.
Kim K, Jutooru I, Chadalapaka G, Johnson G, Frank J, Burghardt R, Kim S, Safe S. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene. 2013;32:1616–25.
Liu YW, Sun M, Xia R, Zhang EB, Liu XH, Zhang ZH, Xu TP, De W, Liu BR, Wang ZX. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015;6:e1802.
Yang SZ, Xu F, Zhou T, Zhao X, McDonald JM, Chen Y. The long non-coding RNA HOTAIR enhances pancreatic cancer resistance to TNF-related apoptosis-inducing ligand. J Biol Chem. 2017;292:10390–7.
Li CH, Xiao Z, Tong JH, To KF, Fang X, Cheng AS, Chen Y. EZH2 coupled with HOTAIR to silence MicroRNA-34a by the induction of heterochromatin formation in human pancreatic ductal adenocarcinoma. Int J Cancer. 2017;140:120–9.
Chen WM, Chen WD, Jiang XM, Jia XF, Wang HM, Zhang QJ, Shu YQ, Zhao HB. HOX transcript antisense intergenic RNA represses E-cadherin expression by binding to EZH2 in gastric cancer. World J Gastroenterol. 2017;23:6100–10.
Sun S, Wu Y, Guo W, Yu F, Kong L, Ren Y, Wang Y, Yao X, Jing C, Zhang C, Liu M, Zhang Y, Zhao M, Li Z, Wu C, Qiao Y, Yang J, Wang X, Zhang L, Li M, Zhou X. STAT3/HOTAIR signaling Axis regulates HNSCC growth in an EZH2-dependent manner. Clin Cancer Res. 2018;24:2665–77.
Ding J, Yeh CR, Sun Y, Lin C, Chou J, Ou Z, Chang C, Qi J, Yeh S. Estrogen receptor beta promotes renal cell carcinoma progression via regulating LncRNA HOTAIR-miR-138/200c/204/217 associated CeRNA network. Oncogene. 2018;37:5037–53.
Yan Y, Han J, Li Z, Yang H, Sui Y, Wang M. Elevated RNA expression of long noncoding HOTAIR promotes cell proliferation and predicts a poor prognosis in patients with diffuse large B cell lymphoma. Mol Med Rep. 2016;13:5125–31.
Oh EJ, Kim SH, Yang WI, Ko YH, Yoon SO. Long non-coding RNA HOTAIR expression in diffuse large B-cell lymphoma: in relation to Polycomb repressive complex pathway proteins and H3K27 trimethylation. J Pathol Transl Med. 2016;50:369–76.
Chen M, Xia Z, Chen C, Hu W, Yuan Y. LncRNA MALAT1 promotes epithelial-to-mesenchymal transition of esophageal cancer through Ezh2-Notch1 signaling pathway. Anti-Cancer Drugs. 2018;29:767–73.
Huo Y, Li Q, Wang X, Jiao X, Zheng J, Li Z, Pan X. MALAT1 predicts poor survival in osteosarcoma patients and promotes cell metastasis through associating with EZH2. Oncotarget. 2017;8:46993–7006.
Han T, Jiao F, Hu H, Yuan C, Wang L, Jin ZL, Song WF, Wang LW. EZH2 promotes cell migration and invasion but not alters cell proliferation by suppressing E-cadherin, partly through association with MALAT-1 in pancreatic cancer. Oncotarget. 2016;7:11194–207.
Wang D, Ding L, Wang L, Zhao Y, Sun Z, Karnes RJ, Zhang J, Huang H. LncRNA MALAT1 enhances oncogenic activities of EZH2 in castration-resistant prostate cancer. Oncotarget. 2015;6:41045–55.
Li P, Zhang X, Wang H, Wang L, Liu T, Du L, Yang Y, Wang C. MALAT1 is associated with poor response to oxaliplatin-based chemotherapy in colorectal cancer patients and promotes chemoresistance through EZH2. Mol Cancer Ther. 2017;16:739–51.
Wang X, Sehgal L, Jain N, Khashab T, Mathur R, Samaniego F. LncRNA MALAT1 promotes development of mantle cell lymphoma by associating with EZH2. J Transl Med. 2016;14:346.
Kim SH, Kim SH, Yang WI, Kim SJ, Yoon SO. Association of the long non-coding RNA MALAT1 with the polycomb repressive complex pathway in T and NK cell lymphoma. Oncotarget. 2017;8:31305–17.
Kangarlouei R, Irani S, Noormohammadi Z, Memari F, Mirfakhraie R. ANRIL and ANRASSF1 long noncoding RNAs are upregulated in gastric cancer. J Cell Biochem. 2019;120:12544–8.
Chen F, Li M, Zhu X. Propofol suppresses proliferation and migration of papillary thyroid cancer cells by down-regulation of lncRNA ANRIL. Exp Mol Pathol. 2019;107:68–76.
Yang Y, Peng XW. The silencing of long non-coding RNA ANRIL suppresses invasion, and promotes apoptosis of retinoblastoma cells through the ATM-E2F1 signaling pathway. Biosci Rep. 2018;38.
Sun LY, Li XJ, Sun YM, Huang W, Fang K, Han C, Chen ZH, Luo XQ, Chen YQ, Wang WT. LncRNA ANRIL regulates AML development through modulating the glucose metabolism pathway of AdipoR1/AMPK/SIRT1. Mol Cancer. 2018;17:127.
Song Z, Wu W, Chen M, Cheng W, Yu J, Fang J, Xu L, Yasunaga JI, Matsuoka M, Zhao T. Long noncoding RNA ANRIL supports proliferation of adult T-cell leukemia cells through cooperation with EZH2. J Virology. 2018;92:e00909–18.
Kikuchi J, Koyama D, Wada T, Izumi T, Hofgaard PO, Bogen B, Furukawa Y. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J Clin Invest. 2015;125:4375–90.
Kaneko S, Li G, Son J, Xu CF, Margueron R, Neubert TA, Reinberg D. Phosphorylation of the PRC2 component Ezh2 is cell cycle-regulated and up-regulates its binding to ncRNA. Genes Dev. 2010;24:2615–20.
Wei Y, Chen YH, Li LY, Lang J, Yeh SP, Shi B, Yang CC, Yang JY, Lin CY, Lai CC, Hung MC. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol. 2011;13:87–94.
Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem. 2011;286:28511–9.
Oh EJ, Kim EK, Yang WI, Yoon SO. Activation of the polycomb repressive complex pathway in the bone marrow resident cells of diffuse large B-cell lymphoma patients. Leukemia Lymphoma. 2016;57:1921–32.
Yi S, Sun J, Qiu L, Fu W, Wang A, Liu X, Yang Y, Kadin ME, Tu P, Wang Y. Dual role of EZH2 in cutaneous anaplastic large cell lymphoma: promoting tumor cell survival and regulating tumor microenvironment. J Invest Dermatol. 2018;138:1126–36.
Ennishi D, Takata K, Beguelin W, Duns G, Mottok A, Farinha P, Bashashati A, Saberi S, Boyle M, Meissner B, Ben-Neriah S, Woolcock BW, Telenius A, Lai D, Teater M, Kridel R, Savage KJ, Sehn LH, Morin RD, Marra MA, Shah SP, Connors JM, Gascoyne RD, Scott DW, Melnick AM, Steidl C. Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Dis. 2019;9:546–63.
Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet. 1964;1:702–3.
Shannon-Lowe C, Rickinson AB, Bell AI. Epstein-Barr virus-associated lymphomas, philosophical transactions of the Royal Society of London. Series B. Biol Sci. 2017;372.
Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–68.
Murata T, Kondo Y, Sugimoto A, Kawashima D, Saito S, Isomura H, Kanda T, Tsurumi T. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. J Virol. 2012;86:4752–61.
Siouda M, Frecha C, Accardi R, Yue J, Cuenin C, Gruffat H, Manet E, Herceg Z, Sylla BS, Tommasino M. Epstein-Barr virus down-regulates tumor suppressor DOK1 expression. PLoS Pathog. 2014;10:e1004125.
Accardi R, Fathallah I, Gruffat H, Mariggio G, Le Calvez-Kelm F, Voegele C, Bartosch B, Hernandez-Vargas H, McKay J, Sylla BS, Manet E, Tommasino M. Epstein - Barr virus transforming protein LMP-1 alters B cells gene expression by promoting accumulation of the oncoprotein DeltaNp73alpha. PLoS Pathog. 2013;9:e1003186.
Jiang S, Zhou H, Liang J, Gerdt C, Wang C, Ke L, Schmidt SCS, Narita Y, Ma Y, Wang S, Colson T, Gewurz B, Li G, Kieff E, Zhao B. The Epstein-Barr virus regulome in lymphoblastoid cells. Cell Host Microbe. 2017;22:561–573.e564.
Wood CD, Veenstra H, Khasnis S, Gunnell A, Webb HM, Shannon-Lowe C, Andrews S, Osborne CS, West MJ. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLife. 2016;5:e18270.
de Oliveira DE, Ballon G, Cesarman E. NF-kappaB signaling modulation by EBV and KSHV. Trends Microbiol. 2010;18:248–57.
Mayers DL, Mikovits JA, Joshi B, Hewlett IK, Estrada JS, Wolfe AD, Garcia GE, Doctor BP, Burke DS, Gordon RK, et al. Anti-human immunodeficiency virus 1 (HIV-1) activities of 3-deazaadenosine analogs: increased potency against 3′-azido-3′-deoxythymidine-resistant HIV-1 strains. Proc Natl Acad Sci U S A. 1995;92:215–9.
Gordon RK, Ginalski K, Rudnicki WR, Rychlewski L, Pankaskie MC, Bujnicki JM, Chiang PK. Anti-HIV-1 activity of 3-deaza-adenosine analogs. Inhibition of S-adenosylhomocysteine hydrolase and nucleotide congeners. Eur J Biochem. 2003;270:3507–17.
Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, Karuturi RK, Tan PB, Liu ET, Yu Q. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–63.
Rao M, Chinnasamy N, Hong JA, Zhang Y, Zhang M, Xi S, Liu F, Marquez VE, Morgan RA, Schrump DS. Inhibition of histone lysine methylation enhances cancer-testis antigen expression in lung cancer cells: implications for adoptive immunotherapy of cancer. Cancer Res. 2011;71:4192–204.
Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, Balusu R, Koul S, Atadja P, Marquez VE, Bhalla KN. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009;114:2733–43.
Crea F, Hurt EM, Mathews LA, Cabarcas SM, Sun L, Marquez VE, Danesi R, Farrar WL. Pharmacologic disruption of polycomb repressive complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol Cancer. 2011;10:40.
Bouzelfen A, Kora H, Alcantara M, Bertrand P, Latouche JB, Jardin F. Heterogeneous epigenetic regulation of HACE1 in Burkitt- lymphoma-derived cells. Leukemia Res. 2017;60:53–7.
Shen J, Su J, Wu D, Zhang F, Fu H, Zhou H, Xu M. Growth inhibition accompanied by MOB1 upregulation in human acute lymphoid leukemia cells by 3-deazaneplanocin a. Biochem Genet. 2015;53:268–79.
Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8:1579–88.
Xu L, Tang H, Wang K, Zheng Y, Feng J, Dong H, Jin Y, Cao C, Chen X, Gao G. Pharmacological inhibition of EZH2 combined with DNAdamaging agents interferes with the DNA damage response in MM cells. Mol Med Rep. 2019;19:4249–55.
Lue JK, Prabhu SA, Liu Y, Gonzalez Y, Verma A, Mundi PS, Abshiru N, Camarillo JM, Mehta S, Chen EI, Qiao C, Nandakumar R, Cremers S, Kelleher NL, Elemento O, Amengual JE. Precision targeting with EZH2 and HDAC inhibitors in epigenetically dysregulated lymphomas. Clin Cancer Res. 2019.
Adamik J, Jin S, Sun Q, Zhang P, Weiss KR, Anderson JL, Silbermann R, Roodman GD, Galson DL. EZH2 or HDAC1 inhibition reverses multiple myeloma-induced epigenetic suppression of osteoblast differentiation. Mol Cancer Res. 2017;15:405–17.
Zeng D, Liu M, Pan J. Blocking EZH2 methylation transferase activity by GSK126 decreases stem cell-like myeloma cells. Oncotarget. 2017;8:3396–411.
Ott HM, Graves AP, Pappalardi MB, Huddleston M, Halsey WS, Hughes AM, Groy A, Dul E, Jiang Y, Bai Y, Annan R, Verma SK, Knight SD, Kruger RG, Dhanak D, Schwartz B, Tummino PJ, Creasy CL, McCabe MT. A687V EZH2 is a driver of histone H3 lysine 27 (H3K27) hypertrimethylation. Mol Cancer Ther. 2014;13:3062–73.
Ezponda T, Dupere-Richer D, Will CM, Small EC, Varghese N, Patel T, Nabet B, Popovic R, Oyer J, Bulic M, Zheng Y, Huang X, Shah MY, Maji S, Riva A, Occhionorelli M, Tonon G, Kelleher N, Keats J, Licht JD. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell Rep. 2017;21:628–40.
Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, Gu J, Ardayfio O, Zhang JH, Yan X, Fang J, Mi Y, Zhang M, Zhou T, Feng G, Chen Z, Li G, Yang T, Zhao K, Liu X, Yu Z, Lu CX, Atadja P, Li E. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360–5.
Herviou L, Kassambara A, Boireau S, Robert N, Requirand G, Muller-Tidow C, Vincent L, Seckinger A, Goldschmidt H, Cartron G, Hose D, Cavalli G, Moreaux J. PRC2 targeting is a therapeutic strategy for EZ score defined high-risk multiple myeloma patients and overcome resistance to IMiDs. Clin Epigenetics. 2018;10:121.
Dimopoulos K, Sogaard Helbo A, Fibiger Munch-Petersen H, Sjo L, Christensen J, Sommer Kristensen L, Asmar F, Hermansen NEU, O'Connel C, Gimsing P, Liang G, Gronbaek K. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol Oncol. 2018;12:180–95.
Brach D, Johnston-Blackwell D, Drew A, Lingaraj T, Motwani V, Warholic NM, Feldman I, Plescia C, Smith JJ, Copeland RA, Keilhack H, Chan-Penebre E, Knutson SK, Ribich SA, Raimondi A, Thomenius MJ. EZH2 inhibition by Tazemetostat results in altered dependency on B-cell activation signaling in DLBCL. Mol Cancer Ther. 2017;16:2586–97.
Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, Kadowaki T, Uesugi M, Kuznetsov G, Kumar N, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Waters NJ, Smith JJ, Porter-Scott M, Chesworth R, Moyer MP, Copeland RA, Richon VM, Uenaka T, Pollock RM, Kuntz KW, Yokoi A, Keilhack H. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13:842–54.
Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, Richon VM, Copeland RA, Keilhack H, Pollock RM, Kuntz KW. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6.
Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, Kawano S, Minoshima Y, Choi HW, Porter Scott M, Waters NJ, Smith JJ, Chesworth R, Moyer MP, Copeland RA. EPZ011989, a potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS Med Chem Lett. 2015;6:491–5.
Lu B, Shen X, Zhang L, Liu D, Zhang C, Cao J, Shen R, Zhang J, Wang D, Wan H, Xu Z, Ho MH, Zhang M, Zhang L, He F, Tao W. Discovery of EBI-2511: a highly potent and orally active EZH2 inhibitor for the treatment of non-Hodgkin's lymphoma. ACS Med Chem Lett. 2018;9:98–102.
Song X, Zhang L, Gao T, Ye T, Zhu Y, Lei Q, Feng Q, He B, Deng H, Yu L. Selective inhibition of EZH2 by ZLD10A blocks H3K27 methylation and kills mutant lymphoma cells proliferation. Biomed Pharmacother. 2016;81:288–94.
Wu Y, Hu J, Ding H, Chen L, Zhang Y, Liu R, Xu P, Du D, Lu W, Liu J, Liu Y, Liu YC, Lu J, Zhang J, Yao Z, Luo C. Identification of novel EZH2 inhibitors through pharmacophore-based virtual screening and biological assays. Bioorg Med Chem Lett. 2016;26:3813–7.
Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M, Iyer P, Balasubramanian S, Zhao F, Good AC, Campbell R, Lee C, Cantone N, Cummings RT, Normant E, Bellon SF, Albrecht BK, Harmange JC, Trojer P, Audia JE, Zhang Y, Justin N, Chen S, Wilson JR, Gamblin SJ. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1 -(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for phase I clinical trials for B-cell lymphomas. J Med chemistry. 2016;59:9928–41.
Nasveschuk CG, Gagnon A, Garapaty-Rao S, Balasubramanian S, Campbell R, Lee C, Zhao F, Bergeron L, Cummings R, Trojer P, Audia JE, Albrecht BK, Harmange JC. Discovery and optimization of Tetramethylpiperidinyl Benzamides as inhibitors of EZH2. ACS Med Chem Lett. 2014;5:378–83.
Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, Romeril SP, Burgess JL, Grant SW, Brackley JA, Graves AP, Scherzer DA, Shu A, Thompson C, Ott HM, Aller GS, Machutta CA, Diaz E, Jiang Y, Johnson NW, Knight SD, Kruger RG, McCabe MT, Dhanak D, Tummino PJ, Creasy CL, Miller WH. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. 2012;3:1091–6.
Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, Coindre JM, Blakemore SJ, Clawson A, Suttle B, McDonald AA, Woodruff M, Ribich S, Hedrick E, Keilhack H, Thomson B, Owa T, Copeland RA, Ho PTC, Ribrag V. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19:649–59.
Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, Liu F, Gao C, Huang XP, Kuznetsova E, Rougie M, Jiang A, Pattenden SG, Norris JL, James LI, Roth BL, Brown PJ, Frye SV, Arrowsmith CH, Hahn KM, Wang GG, Vedadi M, Jin J. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8:1324–34.
Garapaty-Rao S, Nasveschuk C, Gagnon A, Chan EY, Sandy P, Busby J, Balasubramanian S, Campbell R, Zhao F, Bergeron L, Audia JE, Albrecht BK, Harmange JC, Cummings R, Trojer P. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem Biol. 2013;20:1329–39.
Woo J, Kim HY, Byun BJ, Chae CH, Lee JY, Ryu SY, Park WK, Cho H, Choi G. Biological evaluation of tanshindiols as EZH2 histone methyltransferase inhibitors. Bioorg Med Chem Lett. 2014;24:2486–92.
Bisserier M, Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood. 2018;131:2125–37.
Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, Wei R, Zheng XL, Liu Z, Zhang X, Yang H, Hao B, Shen YY, Su Y, Cong X, Huang M, Tan M, Ding J, Geng M. Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors. Cell. 2018;175:186–199.e119.
Gibaja V, Shen F, Harari J, Korn J, Ruddy D, Saenz-Vash V, Zhai H, Rejtar T, Paris CG, Yu Z, Lira M, King D, Qi W, Keen N, Hassan AQ, Chan HM. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene. 2016;35:558–66.
Baker T, Nerle S, Pritchard J, Zhao B, Rivera VM, Garner A, Gonzalvez F. Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget. 2015;6:32646–55.
Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 2013;9:643–50.
Kong X, Chen L, Jiao L, Jiang X, Lian F, Lu J, Zhu K, Du D, Liu J, Ding H, Zhang N, Shen J, Zheng M, Chen K, Liu X, Jiang H, Luo C. Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2. J Med Chem. 2014;57:9512–21.
Li L, Zhang H, Zhang M, Zhao M, Feng L, Luo X, Gao Z, Huang Y, Ardayfio O, Zhang JH, Lin Y, Fan H, Mi Y, Li G, Liu L, Feng L, Luo F, Teng L, Qi W, Ottl J, Lingel A, Bussiere DE, Yu Z, Atadja P, Lu C, Li E, Gu J, Zhao K. Discovery and molecular basis of a diverse set of Polycomb repressive complex 2 inhibitors recognition by EED. PLoS One. 2017;12:e0169855.
Huang Y, Zhang J, Yu Z, Zhang H, Wang Y, Lingel A, Qi W, Gu J, Zhao K, Shultz MD, Wang L, Fu X, Sun Y, Zhang Q, Jiang X, Zhang J, Zhang C, Li L, Zeng J, Feng L, Zhang C, Liu Y, Zhang M, Zhang L, Zhao M, Gao Z, Liu X, Fang D, Guo H, Mi Y, Gabriel T, Dillon MP, Atadja P, Oyang C. Discovery of first-in-class, potent, and orally bioavailable embryonic ectoderm development (EED) inhibitor with robust anticancer efficacy. J Med Chem. 2017;60:2215–26.
Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, Zhang M, Zhang J, Yu Z, Li L, Teng L, Chuai S, Zhang C, Zhao M, Chan H, Chen Z, Fang D, Fei Q, Feng L, Feng L, Gao Y, Ge H, Ge X, Li G, Lingel A, Lin Y, Liu Y, Luo F, Shi M, Wang L, Wang Z, Yu Y, Zeng J, Zeng C, Zhang L, Zhang Q, Zhou S, Oyang C, Atadja P, Li E. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol. 2017;13:381–8.
Rizq O, Mimura N, Oshima M, Saraya A, Koide S, Kato Y, Aoyama K, Nakajima-Takagi Y, Wang C, Chiba T, Ma A, Jin J, Iseki T, Nakaseko C, Iwama A. Dual inhibition of EZH2 and EZH1 sensitizes PRC2-dependent tumors to proteasome inhibition. Clin Cancer Res. 2017;23:4817–30.
Smonskey M, Lasorsa E, Rosario S, Kirk JS, Hernandez-Ilizaliturri FJ, Ellis L. EZH2 inhibition re-sensitizes multidrug resistant B-cell lymphomas to etoposide mediated apoptosis. Oncoscience. 2016;3:21–30.
D’Angelo V, Iannotta A, Ramaglia M, Lombardi A, Zarone MR, Desiderio V, Affinita MC, Pecoraro G, Di Martino M, Indolfi P, Casale F, Caraglia M. EZH2 is increased in paediatric T-cell acute lymphoblastic leukemia and is a suitable molecular target in combination treatment approaches. J Exp Clin Cancer Res. 2015;34:83.
Harding T, Swanson J, Van Ness B. EZH2 inhibitors sensitize myeloma cell lines to panobinostat resulting in unique combinatorial transcriptomic changes. Oncotarget. 2018;9:21930–42.
Kalushkova A, Fryknas M, Lemaire M, Fristedt C, Agarwal P, Eriksson M, Deleu S, Atadja P, Osterborg A, Nilsson K, Vanderkerken K, Oberg F, Jernberg-Wiklund H. Polycomb target genes are silenced in multiple myeloma. PLoS One. 2010;5:e11483.
Fiskus W, Rao R, Balusu R, Ganguly S, Tao J, Sotomayor E, Mudunuru U, Smith JE, Hembruff SL, Atadja P, Marquez VE, Bhalla K. Superior efficacy of a combined epigenetic therapy against human mantle cell lymphoma cells. Clin Cancer Res. 2012;18:6227–38.
Johnson DP, Spitz GS, Tharkar S, Quayle SN, Shearstone JR, Jones S, McDowell ME, Wellman H, Tyler JK, Cairns BR, Chandrasekharan MB, Bhaskara S. HDAC1,2 inhibition impairs EZH2- and BBAP-mediated DNA repair to overcome chemoresistance in EZH2 gain-of-function mutant diffuse large B-cell lymphoma. Oncotarget. 2015;6:4863–87.
Huang S, Wang Z, Zhou J, Huang J, Zhou L, Luo J, Wan YY, Long H, Zhu B. EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res. 2019;79:2009–20.
Acknowledgements
We thank Jennie Wong (National University of Singapore) for revising the English Language. We sincerely apologize to those authors whose work was not cited due to space constraints.
Funding
This work was supported by the National Research Foundation Singapore and the Singapore Ministry of Education under the Research Centres of Excellence initiative as well as the RNA Biology Centre at the Cancer Science Institute of Singapore, National University of Singapore (MOE2014-T3-1-006). W.-J.C. was also supported by National Medical Research Council (NMRC) Singapore Translational Research (STaR) Investigatorship.
Author information
Authors and Affiliations
Contributions
BL and W-JC reviewed the literature and wrote the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
All readers read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, B., Chng, WJ. EZH2 abnormalities in lymphoid malignancies: underlying mechanisms and therapeutic implications. J Hematol Oncol 12, 118 (2019). https://doi.org/10.1186/s13045-019-0814-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-019-0814-6