
Abstract
Glioblastoma (GBM) is an aggressive nervous system tumor with a poor prognosis. Although, surgery, radiation therapy, and chemotherapy are the current standard protocol for GBM patients, there is still a poor prognosis in these patients. Temozolomide (TMZ) as a first-line therapeutic agent in GBM can easily cross from the blood-brain barrier to inhibit tumor cell proliferation. However, there is a high rate of TMZ resistance in GBM patients. Since, there are limited therapeutic choices for GBM patients who develop TMZ resistance; it is required to clarify the molecular mechanisms of chemo resistance to introduce the novel therapeutic targets. MicroRNAs (miRNAs) regulate chemo resistance through regulation of drug metabolism, absorption, DNA repair, apoptosis, and cell cycle. In the present review we discussed the role of miRNAs in TMZ response of GBM cells. It has been reported that miRNAs mainly induced TMZ sensitivity by regulation of signaling pathways and autophagy in GBM cells. Therefore, miRNAs can be used as the reliable diagnostic/prognostic markers in GBM patients. They can also be used as the therapeutic targets to improve the TMZ response in GBM cells.
Similar content being viewed by others


Background
Glioma is one of the aggressive nervous system tumors with a high rate of drug resistance [1, 2]. Glioblastoma (GBM) as the highest grade of astrocytoma is a malignant central nervous system disorder [3]. GBM is the primary cause of death among patients between the ages of 15 and 34 [4]. The current standard protocol for GBM therapy involves surgery, radiation therapy, and chemotherapy [5]. Apart from the therapeutic progresses in radiotherapy, chemotherapy, and surgical resection, there is still a poor prognosis in GBM patients [6, 7]. Resistance to therapeutic drugs is one of the most frequent causes of GBM recurrence [8, 9]. Temozolomide (TMZ) as a DNA-alkylating agent functions by promotion of DNA damage and double strand breaks (DSBs) that result in activation of caspase-mediated apoptosis in GBM cells [10,11,12]. Currently, regional fractionated radiation followed by TMZ are the first therapies for all GBM patients after surgery [13]. TMZ as a first-line therapeutic agent in GBM can easily cross from the blood-brain barrier to inhibit and induce the tumor cell proliferation and apoptosis, respectively [14]. However, due to the chemo and radiotherapeutic resistances, there is only a median survival of 14.6 months in GBM patients [15]. Regarding the limited repair mechanisms and anatomical complexity, treatment of the drug-resistant GBM is challenging [15,16,17]. Since, there are limited therapeutic choices for GBM patients who develop TMZ resistance; it is required to clarify the molecular mechanisms of chemo resistance to introduce the novel therapeutic targets. MicroRNAs (miRNAs) are involved in regulation of numerous biological processes such as autophagy, invasion, cell differentiation, proliferation, and apoptosis [18,19,20]. They function as either oncomiRs or tumor suppressive miRNAs during tumor progression [21, 22]. MiRNAs regulate chemo resistance through regulation of drug metabolism, absorption, DNA repair, apoptosis, and cell cycle [23, 24]. MiRNA deregulation has been found in GBM, which may be involved in tumor progression and therapeutic resistance [25,26,27]. Since, there are various reports about the miRNA profiles in TMZ-resistant GBM [28,29,30,31,32], we discussed the role of miRNAs in TMZ response through the regulation of signaling pathways, autophagy, and cell cycle to introduce them as the novel therapeutic options to improve prognosis among GBM patients (Table 1).
Wnt/β-catenin and PI3K/AKT signaling pathways
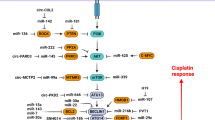
Wnt/β-catenin is an important oncogenic signaling pathway in tumor cells that can be triggered by the WNT ligands following the binding with FZD/LRP receptor [33]. WNT ligands promotes the accumulation of cytoplasmic β-catenin that finally enters to the nucleus to regulate WNT target genes [33]. It has been shown that miRNAs have a key role in TMZ response of GBM cells by regulation of Wnt/β-catenin pathway (Fig. 1). There was miR-505 down regulation in GBM tissues. It functioned as a tumor suppressor by targeting WNT7B. Moreover, TMZ had the ability to elevate the levels of miR-505 to enhance its inhibitory roles in GBM cells [34]. GSK3β as a component of degradation complex in WNT signaling regulates cell growth and survival [35,36,37]. MiR-101 increased TMZ sensitivity in GBM cells by GSK3β targeting. There was also a correlation between miR-101 down regulation and poor survival in GBM patients [38]. Wnt/β-catenin is a key regulator of EMT process in cancer [39,40,41]. There was miR-137 down regulation in recurrent GBM tissues. MiR-137 up regulated E-cadherin while down regulated vimentin and N-cadherin in GBM cells. MiR-137 increased TMZ sensitivity by targeting LRP6. Hypoxia-induced down regulation of miR-137 significantly regulated TMZ resistance and GBM growth via LRP6/β-catenin pathway [42]. LINC00511 activated Wnt/β-catenin via miR-126-5p sponging. There was a correlation between LINC00511 up regulation and poor prognosis in GBM patients. LINC00511/miR-126-5p/DVL3 axis induced the TMZ resistance in GBM cells [43]. The downstream pathways of Wnt5a are planar cell polarity (PCP) and Wnt/Ca2 + pathways, which have a key role in cell physiology and tumor progression [44, 45]. PCP pathway activates JNK through the regulation of morphogenetic motions and cell polarity. Wnt/Ca2 + pathway activate PKC and cam kinase II, which can modulate cell adhesion and motility [44,45,46]. PKC/ERK/NF-κB and JNK pathways were activated by dysregulation of miR-129-5p/Wnt5a signaling, resulting in a more malignant phenotype with TMZ resistance. MiR-129-5p suppressed angiogenesis, tumor invasion, and TMZ resistance by Wnt5a targeting in GBM cells [47]. SOX2 up regulation is observed in various cancers that increases tumor aggressiveness and poor prognosis [48]. It regulates glioma cell invasion and proliferation as an oncogene [49, 50]. In addition, SOX2 has been linked with the development of resistance to several chemotherapeutic medications by regulating numerous signaling pathways [51,52,53]. SOX2 is involved in regulation of cisplatin response in tumor cells by regulation of Wnt-β-catenin pathway [54]. The decreased expression of miR-126-3p was observed in GBM cells and samples that exhibited TMZ resistance. MiR-126-3p enhanced the GBM sensitivity to TMZ by SOX2 targeting and inhibition of the Wnt/β-catenin pathway [55]. SOX2 mediated miR-486-5p expression enhanced the self-renewal capacity of GBM by PTEN and FoxO1 down regulations [56]. Glioma-associated microglial cells (GAMs) interact closely with GBM cells through intracellular communication and share similar functions with tumor-associated macrophages in the peripheral system [57, 58]. GAMs release various signaling molecules and cytokines that inhibit apoptosis, while promote metastasis and angiogenesis [59, 60]. TMZ-resistant GBM samples had a notable SNHG15 up regulation, which was correlated with the aggressive characteristics of GBM. Elevated expression of lncSNHG15 was associated with increased levels of stemness markers such as β-catenin and Sox2, as well as oncogene markers such as EGFR and CDK6. Glioma-associated microglia (M2-GAMs) could be more easily M2-polarized by TMZ-resistant GBM cells than by their sensitive counterparts. SNHG15 down-regulation led to a decrease in carcinogenesis, self-renewal, and heightened TMZ sensitivity. TMZ resistance could be overcome through SNHG15/CDK6/miR-627 by reducing the M2 polarization of glioma-associated microglia in GBM [61].
Growth factors activate the PI3K/AKT pathway through binding with receptor tyrosine kinases (RTKs) that results in cell growth, proliferation, drug resistance, and tumor progression [62, 63]. It has been reported that miRNAs have an important role in TMZ response of GBM cells via regulation of PI3K/AKT pathway (Fig. 1). RTKs promote resistance to both chemotherapy and irradiation in GBM cells [64,65,66]. EGFR belongs to the HER family of RTKs [67, 68]. Growth factors bind with EGFR to activate downstream pathways that regulate differentiation, cell growth, and survival [69, 70]. MiR-181b increased TMZ sensitivity in GBM cell by regulation of the EGFR pathway [71]. There was EGFR down regulation in TMZ and irradiation-resistant GBM cells. MiR-221 increased TMZ and radio therapeutic resistances in GBM cells by EGFR targeting [72]. CircCABIN1 induced TMZ resistance and stemness in GBM cells by miR-637 sponging and subsequent OLFML3 up regulation that activated ErbB pathway [73]. LRIG1 as an inhibitor of EGFR promoted the TMZ sensitivity in GBM cells [74]. MiR-20a mediated TMZ resistance by suppressing LRIG1 in GBM cells [75]. c-Met as a RTK is involved in embryogenesis [76]. Deregulation of c-Met promoted cell proliferation and angiogenesis, while inhibited apoptosis in brain tumors [77]. Overexpression of c-Met affects chemo sensitivity, causing GBM cells to become resistant to drugs [78, 79]. Moreover, miR-128-3p prevents the EMT process through vimentin down regulation while CDH1 up regulation. MiR-128-3p promoted the inhibitory role of TMZ in cell migration and proliferation via EMT suppression. miR-128-3p induced the TMZ sensitivity of GBM cells through c-Met targeting and EMT suppression [80]. AKT2 as a serine/threonine kinase has key roles in tumor metastasis, metabolism, radio-resistance, and drug resistance [81]. Down regulation of miR-625 was observed in human glioma in contrast to normal human brain tissues. MiR-625 increased apoptosis while reduced the TMZ resistance of glioma cells by targeting AKT2 [82]. Activation of Akt can efficiently suppress GSK-3β, leading to a decrease in β-catenin degradation [83]. Down regulation of β-catenin also suppresses cell growth while induces cell death. CD133(+) glioma cells have demonstrated a significant self-renewal ability, resulting in subcutaneous tumors and the generation of stem cell spheres in nude mice [84]. The concurrent administration of LY294002 (PI3K inhibitor), TMZ, and miR-125b inhibitor demonstrated the most significant impact on p-β-catenin up regulation while reduction of p-GSK-3β. This suggests that the combined treatment was highly influential in inactivation of Wnt/β-catenin pathway. TMZ resistance of Glioblastoma stemcells (GSCs) cells could be successfully reversed by treating them with both miR-125b inhibitors and PI3K/Akt inhibition [85]. MALAT1 inhibition increased TMZ sensitivity in GBM cells by miR-101/GSK3β and MGMT [86]. IGF signaling among the several other tumor microenvironmental factors has been found to increase the risk of brain tumor progression [87]. The binding of the IGF-1 with IGF-1R results in its activation by autophosphorylation that promotes cell migration and proliferation in gliomas [88]. IGF-1 also reduced etoposide-induced apoptosis of glioma cells via Bcl-2 up regulation while inhibition of caspase-3 activity [89]. Mammalian target of rapamycin (mTOR) belongs to the serine/threonine kinases that regulate cell growth and proliferation [90]. IGF-1 trigger the mTOR and its downstream targets such as 4E-BP1 and p70S6K1 via the PI3K/PDK1/AKT axis [91]. mTORC1 regulates protein translation in the brain to control learning, memory, synaptic plasticity, and the pathogenesis of GBM [92]. The production of ROS, MMP loss, apoptosis, and non-protective autophagy were significantly increased by miR-128 via mTOR, PIK3R1, IGF1, and RICTOR targeting. Temozolomide can promote apoptosis in glioblastoma cells by miR-128 up regulation. TMZ increased apoptosis through JNK2/c-Jun mediated miR128 up regulation in GBM cells. MiR-128 targeted p70S6K1 and down regulated its substrates such as HIF-1 and VEGF [93]. HOXA-AS2 promoted TMZ resistance through regulation of miR-302a-3p/IGF1 axis in GBM cells [94]. Disintegrin and metalloproteinase-17 (ADAM17) has ability to cleave the membrane-bound TNF-α which activates EGFR pathway [95, 96]. However, the function of ADAM17 goes beyond the release of soluble TNF-α and is able to process various substrates, such as EGFR, cytokines, and adhesion molecules [97, 98]. Furthermore, ADAM17 is responsible for pathological and physiological processes, such as cell growth, inflammation, differentiation, regeneration, and tumor progression [97]. It was identified that miR-145-5p increased TMZ sensitivity in GBM cells by ADAM17 targeting [99].
TGF-β, NF-kB, and hedgehog signaling pathways
MiRNAs have a key role in TMZ response of GBM cells by regulation of TGF-β, NF-kB, and Hedgehog signaling pathways (Fig. 2). TGF-β is a multifaceted regulatory cytokine with cell proliferation, differentiation, and tissue homeostasis functions [100, 101]. It enhances the GBM growth, metastasis, and angiogenesis. TGF-β enables GBM cells to evade growth suppression and immune checkpoint blockade and develop resistance to chemotherapy [102,103,104]. There was lncRNA-MUF up regulation in GBM tissues that was correlated with histological grading. It serves as an oncogenic lncRNA and sponges miR-34a which suppresses Snail1 to promote glioma cell growth and invasion. Inhibition of lncRNA-MUF increased TMZ-mediated apoptosis in GBM cells by reducing TGF-β-induced phosphorylation of SMAD2/3 [105]. The most direct cause of drug tolerance is the expression of Methylguanine DNA methyltransferase (MGMT), as it reverses DNA alkylation in TMZ-induced O6-methyguanine lesions by eliminating methyl groups [106, 107]. Patients with higher levels of MGMT had less effective outcomes from TMZ chemotherapy in comparison to those with lower MGMT levels [108]. It was shown that TGF-β1 was correlated with TMZ resistance in GBM cells with MGMT hypomethylation. TGF-β1 up regulated the lncRNAs that attached competitively to KSRP, thereby blocking KSRP from taking part in switching of miR-198 and finally up regulated MGMT. H19 or HOXD-AS2 sponged KSRP and prohibited it from engaging in the FSTL1/miR-198 cascade, therefore resulting in miR-198 down regulation and MGMT up regulation. Consequently, HOXD-AS2 or H19 are responsible for TMZ resistance through KSRP/miR-198/MGMT axis. TGF-β1 up regulated the MGMT by miR-198 inhibition that conferred TMZ resistance in GBM cells [104]. MGMT can be regulated by promoter methylation, transcription factors, histone acetylation, and microRNAs [109]. There was miR-198 down regulation in GBM tissues that was correlated with poor prognosis. MiR-198 enhanced the TMZ sensitivity in GBM cells via MGMT targeting [110]. NF-kB has a key role in chemo resistance of malignant tumor cells [111, 112]. The inactivated NF-kB comprises p50 (NFKB1)/p65 (RelA) subunits. Regularly, they are held in the cytoplasm by IkB, which is the NF-kB inhibitor. External stimuli or stress activate the IKK to phosphorylate IkB for protein degradation through ubiquitin. Subsequently, the activated NF-kB migrates to the nucleus and interacts with different genes which affect apoptosis, invasion, and proliferation. Tumor necrosis factor alpha-induced protein 3 (TNFAIP3) negatively operates in a feedback loop to prevent NF-kB from being activated. This protein also catalyzes the fragmentation of ubiquitin chains linked to K63 and the attachment of K48-linked polyubiquitin chains, which aids in the degradation of receptor-interacting serine-threonine kinase 1 [113, 114]. NF-kB inhibitors interacting with RAS-like (NKIRAS) 1 and 2 intervene with IkB proteasomal degradation and are implied in the activity of NF-kB as well [115,116,117,118]. MiR-125b induced TMZ resistance by TNFAIP3 and NKIRAS2 targeting in GBM cells [119]. The activation of SMO is triggered by the interaction between SHH and PTCH1 receptor, which results in the de-repression of SMO [120]. Glioma-associated oncogene 1 (Gli1), which is a transcriptional factor and downstream of SMO, is the main player in the SHH signaling pathway. Several cancers have been associated with the overexpression of SHH signaling [121]. MiR-9 induced TMZ resistance by PTCH1 targeting that up regulated the drug efflux pumps in GBM cells [122].
MAPK signaling pathway
MAPK/ERK is a key pathway during tumor progression that can be activated by the external mitogens and growth factors [123]. It has been reported that miRNAs have key roles in TMZ response of GBM cells by regulation of MAPK pathway (Fig. 3). MAPK1 is involved in chemo resistance and malignant phenotype in various cancers [124, 125] .E2F7 induces glioma cell proliferation [126, 127]. It was shown that SNHG12 was epigenetically activated by DNA methylation that regulated the MAPK/ERK pathway and cell proliferation by miR-129-5p sponging in GBM cells. SNHG12 increased TMZ resistance by miR-129-5p sponging and subsequent MAPK1 and E2F7 up regulations [128]. NRAS is an important member of the RAS family, which functions as an on/off switch through GDP/GTP regulation. It is a membrane-bound protein that has a vital function in the signal transduction mechanisms of hormones, cytokines, and growth factors. NRAS regulates tumor cell proliferation and is often overstimulated in various cancers [125, 129]. CircASAP1 induced TMZ resistance by miR-502-5p sponging and NRAS up regulation that activated MEK1/ERK1-2 signaling. EIF4A3-mediated circASAP1 increased TMZ resistance and tumor progression in GBM cells [130]. RAP1 is a Ras GTPase that regulates cellular adhesion, growth, and migration. Two highly related isoforms of RAP1 are RAP1B and RAP1A [131]. Rap1B is linked to the cytoskeleton during cell activation [132]. Rap1B down regulated is correlated with reduced glioma cell migration that is induced by lysophosphatidic acid [133]. Cell division cycle 42 (Cdc42), RhoA, and Rac1 are also Rho family members that have key roles cell migration, adhesion, and actin cytoskeletal reorganization [134, 135]. MiR-128 and miR-149 improved TMZ sensitivity in GBM cells through Rap1B-mediated cytoskeletal remodeling. There was miR-149 down regulation in GBM tissues that was correlated with grades of astrocytomas. MiR-128 and miR-149 reduced cell invasion and proliferation by Rap1B regulation in GBM cells [136]. Adrenomedullin (ADM) as a vasodilator hormone significantly affects many vital pathways including PI3K/Akt and ERK. ADM triggers relaxation of blood vessels through PI3K/Akt signaling that is mediated by the endothelium [137], and its infusion alleviates reperfusion injury or myocardial ischemia [138]. ADM plays a regulatory role in modulating various downstream pathways that promote the proliferation and viability of endothelial cells through MAPK/ERK activation [139]. Moreover, ADM has the ability to increase the expression of Bcl-2 by autocrine or paracrine mechanisms of action to protect cancer cells from hypoxia-mediated apoptosis [140]. An up regulation of ADM was observed in TMZ resistant glioma tissues and cells. ADM inhibition promoted TMZ effects on Bax/Bcl-2, ERK1/2, and Akt phosphorylation. Moreover, miR-1297 induced TMZ sensitivity in glioma cells by ADM targeting [141]. MAPK14 could decrease the accumulation of reactive oxygen species, which can subsequently prevent hepatocarcinogenesis and liver fibrogenesis [142]. The p38-MAPK pathway has essential role in cell response to stress and cancer [143]. MiR-155 knockdown reduced cell invasion by p38 targeting. MiR-155 induced MMP9 and MMP2 secretions in the SF767 cell supernatant. Moreover, miR-155 knockdown improved the anti-tumor effect of TMZ on gliomas via MAPK14 and MAPK13-induced ROS generation [144].
Autophagy and cell cycle
Autophagy is a conserved cellular process that is responsible for degradation of intracellular proteins and organelles [145, 146]. It can be activated in both normal and stress condition to provide required metabolic substrates for the cell survival. Autophagy is also activated to preserve cellular homeostasis in infection, aging, neurodegenerative diseases, myopathies, and cancer [147, 148]. The mechanism of autophagy is the creation of autophagosome which merges with lysosomes to produce autolysosomes for intracellular degradation [149, 150]. Autophagy is activated following the TMZ treatment that results in chemo resistance in tumor cells [151,152,153]. In certain instances, autophagy induces TMZ-mediated apoptosis in GBM cells. Additionally, rapamycin, which induces autophagy, could increase apoptosis caused by chemotherapy [154,155,156,157,158]. STAT3 as a transcription factor regulates the autophagy from autophagosome formation to maturation [159]. MiR-519a down regulation was observed in TMZ resistant GBM tissues and cells. It improved the TMZ response of GBM cells through enhanced GBM cell autophagy by facilitating the separation of the Bcl-2/Beclin-1 complex. Moreover, miR-519a induced autophagy by suppressing the STAT3/Bcl-2 axis [160]. MiR-17 regulated autophagosome formation via ATG7 targeting that improved TMZ sensitivity in GBM cells [161]. MiR-30a significantly reduced TMZ-mediated autophagy while induced apoptosis by BECN1 targeting in GBM cells [162].
Hypoxia has been suggested to cause resistance to chemotherapy or radiotherapy in a number of malignant tumors [163, 164]. Critical cellular responses to hypoxia include the stability and activation of HIF1α and HIF2α that have a vital role in tumor progression [165, 166]. It is speculated that hypoxia supports the preservation of GSCs’ undifferentiated status and resistance to treatment as they often reside in hypoxic microenvironments [167]. GSCs interact with immune cells, astrocytes, vascular cells, and neurons in the hypoxic microenvironment to support the tumor maintenance. RHOB belongs to the Rho small GTPase family that has a vital role in regulation of apoptosis and cell cycle progression [168, 169]. Hypoxia may trigger RHOB via GSK-3 in GBM cells [170]. MiR-30b-3p targeted RHOB, which decreased cell cycle arrest by CDK6 and CDK2 up regulations while reduced apoptosis by BCL-2 up regulation and Bax down regulation. HIF-1α and STAT3 transcriptionally enhanced the expression of miR-30b-3p in GSCs under hypoxic conditions. MiR-30b-3p increased TMZ resistance by RHOB targeting. miR-30b-3p up regulation was correlated with poor response to TMZ in GBM tissues [171]. WEE1 kinase as a G2/M checkpoint arrest has key role for pre-mitotic DNA repair [172]. FOXD3-AS1 conferred TMZ resistance through miR-128-3p/WEE1 axis in GBM cells [173]. MiR-125b induced TMZ resistance by STAT3 targeting in GSC cells [174].
Conclusions
TMZ is the first-line therapeutic agent in GBM; however there is a high rate of TMZ resistance among GBM patients. Since, there are limited therapeutic choices for GBM patients who develop TMZ resistance; it is required to clarify the molecular mechanisms of chemo resistance to introduce the novel therapeutic targets. In the present review we discussed the role of miRNAs in TMZ response of GBM cells. It has been reported that miRNAs mainly increased TMZ sensitivity by regulation of signaling pathways and autophagy in GBM cells. Therefore, miRNAs can be used as the reliable tumor markers and therapeutic targets in GBM patients. Regarding the role of miRNAs as the TMZ sensitizers, a miRNA mimic strategy can be suggested to increase the TMZ response among GBM patients. However, further clinical trials and animal studies are needed to use the miRNAs as the therapeutic targets to improve the TMZ response in GBM patients.

Role of miRNAs in TMZ response of GBM cells by regulation of Wnt/β-catenin and PI3K/AKT pathways. (Created with BioRender.com)

Role of miRNAs in TMZ response of GBM cells by regulation of TGF-β, NF-kB, and hedgehog pathways. (Created with BioRender.com)

Role of miRNAs in TMZ response of GBM cells by regulation of MAPK pathway. (Created with BioRender.com)
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ADM:
-
Adrenomedullin
- Cdc4:
-
Cell division cycle 42
- ADAM1:
-
Disintegrin and metalloproteinase-17
- DSBs:
-
Double strand breaks
- GBM:
-
Glioblastoma
- GSCs:
-
Glioblastoma stemcells
- GAMs:
-
Glioma-associated microglial cells
- Gli1:
-
Glioma-associated oncogene 1
- mTOR:
-
Mammalian target of rapamycin
- MGMT:
-
Methylguanine DNA methyltransferase
- miRNAs:
-
MicroRNAs
- NKIRAS:
-
NF-kB inhibitors interacting with RAS-like
- PCP:
-
Planar cell polarity
- RTKs:
-
Receptor tyrosine kinases
- TMZ:
-
Temozolomide
- TNFAIP:
-
Tumor necrosis factor alpha-induced protein 3
References
Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–50.
Schaff LR, Mellinghoff IK. Glioblastoma and other primary brain malignancies in adults: a review. JAMA. 2023;329(7):574–87.
Fettweis G, Di Valentin E, L’homme L, Lassence C, Dequiedt F, Fillet M, et al. RIP3 antagonizes a TSC2-mediated pro-survival pathway in glioblastoma cell death. Biochim et Biophys Acta (BBA)-Molecular Cell Res. 2017;1864(1):113–24.
Hanif F, Muzaffar K, Perveen K, Malhi SM, Simjee SU. Glioblastoma multiforme: a review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac J cancer Prevention: APJCP. 2017;18(1):3.
Nicholas MK. Glioblastoma multiforme: evidence-based approach to therapy. Expert Rev Anticancer Ther. 2007;7(sup1):S23–7.
Borasi G, Nahum A, Paulides MM, Powathil G, Russo G, Fariselli L, et al. Fast and high temperature hyperthermia coupled with radiotherapy as a possible new treatment for glioblastoma. J Ther Ultrasound. 2016;4:1–10.
Ohgaki H, Kleihues P. The definition of primary and secondary glioblastoma. Clin Cancer Res. 2013;19(4):764–72.
Fan C, Liu W, Cao H, Wen C, Chen L, Jiang G. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. 2013;4(10):e876–e.
Messaoudi K, Clavreul A, Lagarce F. Toward an effective strategy in glioblastoma treatment. Part I: resistance mechanisms and strategies to overcome resistance of glioblastoma to temozolomide. Drug Discovery Today. 2015;20(7):899–905.
Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev Med. 2015;66:129–43.
Yoshimoto K, Mizoguchi M, Hata N, Murata H, Hatae R, Amano T, et al. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front Oncol. 2012;2:38784.
Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31(6):955–60.
Clarke MJ, Mulligan EA, Grogan PT, Mladek AC, Carlson BL, Schroeder MA, et al. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol Cancer Ther. 2009;8(2):407–14.
Villano JL, Seery TE, Bressler LR. Temozolomide in malignant gliomas: current use and future targets. Cancer Chemother Pharmacol. 2009;64:647–55.
Stupp R, Hegi ME, Gilbert MR, Chakravarti A. Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol. 2007;25(26):4127–36.
Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–710.
Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. Cancer J Clin. 2010;60(3):166–93.
Zangouei AS, Moghbeli M. MicroRNAs as the critical regulators of cisplatin resistance in gastric tumor cells. Genes Environ. 2021;43(1):21.
Maharati A, Zanguei AS, Khalili-Tanha G, Moghbeli M. MicroRNAs as the critical regulators of tyrosine kinase inhibitors resistance in lung tumor cells. Cell Commun Signal. 2022;20(1):27.
Hamidi AA, Taghehchian N, Basirat Z, Zangouei AS, Moghbeli M. MicroRNAs as the critical regulators of cell migration and invasion in thyroid cancer. Biomark Res. 2022;10(1):40.
Luo J, Wang X, Yang Y, Mao Q. Role of micro-RNA (miRNA) in pathogenesis of glioblastoma. Eur Rev Med Pharmacol Sci. 2015;19(9).
Moghbeli M, Zangouei AS, Nasrpour Navaii Z, Taghehchian N. Molecular mechanisms of the microRNA-132 during tumor progressions. Cancer Cell Int. 2021;21(1):439.
Tolue Ghasaban F, Maharati A, Zangouei AS, Zangooie A, Moghbeli M. MicroRNAs as the pivotal regulators of cisplatin resistance in head and neck cancers. Cancer Cell Int. 2023;23(1):170.
Moghbeli M. MicroRNAs as the pivotal regulators of cisplatin resistance in osteosarcoma. Pathol Res Pract. 2023;249:154743.
Ahmed SP, Castresana JS, Shahi MH. Glioblastoma and miRNAs. Cancers. 2021;13(7):1581.
Beylerli O, Gareev I, Sufianov A, Ilyasova T, Zhang F. The role of microRNA in the pathogenesis of glial brain tumors. Non-coding RNA Res. 2022;7(2):71–6.
Gareev I, Beylerli O, Liang Y, Xiang H, Liu C, Xu X, et al. The role of MicroRNAs in therapeutic resistance of malignant primary brain tumors. Front Cell Dev Biology. 2021;9:740303.
Wan Y, Sun G, Zhang S, Wang Z, Shi L. MicroRNA-125b inhibitor sensitizes human primary glioblastoma cells to chemotherapeutic drug temozolomide on invasion. Vitro Cell Dev Biology-Animal. 2013;49:599–607.
Yang JK, Yang JP, Tong J, Jing SY, Fan B, Wang F, et al. Exosomal miR-221 targets DNM3 to induce tumor progression and temozolomide resistance in glioma. J Neurooncol. 2017;131(2):255–65.
Li Y, Liu Y, Ren J, Deng S, Yi G, Guo M, et al. miR-1268a regulates ABCC1 expression to mediate temozolomide resistance in glioblastoma. J Neurooncol. 2018;138(3):499–508.
Chen J, Fu X, Wan Y, Wang Z, Jiang D, Shi L. miR-125b inhibitor enhance the chemosensitivity of glioblastoma stem cells to temozolomide by targeting Bak1. Tumour Biol. 2014;35(7):6293–302.
Wang X, Li X, Zhou Y, Huang X, Jiang X. Long non-coding RNA OIP5-AS1 inhibition upregulates microRNA-129-5p to repress resistance to temozolomide in glioblastoma cells via downregulating IGF2BP2. Cell Biol Toxicol. 2022;38(6):963–77.
Montazer M, Taghehchian N, Mojarrad M, Moghbeli M. Role of microRNAs in regulation of WNT signaling pathway in urothelial and prostate cancers. Egypt J Med Hum Genet. 2022;23(1):1–12.
Zhang C, Yang X, Fu C, Liu X. Combination with TMZ and miR-505 inhibits the development of glioblastoma by regulating the WNT7B/Wnt/β-catenin signaling pathway. Gene. 2018;672:172–9.
Kannoji A, Phukan S, Sudher Babu V, Balaji VN. GSK3beta: a master switch and a promising target. Expert Opin Ther Targets. 2008;12(11):1443–55.
Luo J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009;273(2):194–200.
Phukan S, Babu VS, Kannoji A, Hariharan R, Balaji VN. GSK3beta: role in therapeutic landscape and development of modulators. Br J Pharmacol. 2010;160(1):1–19.
Tian T, Mingyi M, Qiu X, Qiu Y. MicroRNA-101 reverses temozolomide resistance by inhibition of GSK3β in glioblastoma. Oncotarget. 2016;7(48):79584–95.
Ghahhari NM, Babashah S. Interplay between microRNAs and WNT/β-catenin signalling pathway regulates epithelial–mesenchymal transition in cancer. Eur J Cancer. 2015;51(12):1638–49.
Yang S, Liu Y, Li M-Y, Ng CS, Yang S-l, Wang S, et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/β-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. 2017;16(1):1–12.
Mahmoudian RA, Akhlaghipour I, Lotfi M, Shahidsales S, Moghbeli M. Circular RNAs as the pivotal regulators of epithelial-mesenchymal transition in gastrointestinal tumor cells. Pathol Res Pract. 2023;245:154472.
Li D-M, Chen Q-D, Wei G-N, Wei J, Yin J-X, He J-H, et al. Hypoxia-induced miR-137 inhibition increased glioblastoma multiforme growth and chemoresistance through LRP6. Front Oncol. 2021;10:611699.
Lu Y, Tian M, Liu J, Wang K. LINC00511 facilitates Temozolomide resistance of glioblastoma cells via sponging mir-126-5p and activating Wnt/β-catenin signaling. J Biochem Mol Toxicol. 2021;35(9):e22848.
Asem MS, Buechler S, Wates RB, Miller DL, Stack MS. Wnt5a signaling in cancer. Cancers. 2016;8(9):79.
Endo M, Nishita M, Fujii M, Minami Y. Insight into the role of Wnt5a-induced signaling in normal and cancer cells. Int Rev cell Mol Biology. 2015;314:117–48.
Pu P, Zhang Z, Kang C, Jiang R, Jia Z, Wang G, et al. Downregulation of Wnt2 and β-catenin by siRNA suppresses malignant glioma cell growth. Cancer Gene Ther. 2009;16(4):351–61.
Zeng A, Yin J, Li Y, Li R, Wang Z, Zhou X, et al. Mir-129-5p targets Wnt5a to block PKC/ERK/NF-κB and JNK pathways in glioblastoma. Cell Death Dis. 2018;9(3):394.
Weina K, Utikal J. SOX2 and cancer: current research and its implications in the clinic. Clin Translational Med. 2014;3(1):1–10.
Luo G, Luo W, Sun X, Lin J, Wang M, Zhang Y, et al. MicroRNA–21 promotes migration and invasion of glioma cells via activation of Sox2 and β–catenin signaling. Mol Med Rep. 2017;15(1):187–93.
Zhou K, Zhang C, Yao H, Zhang X, Zhou Y, Che Y, et al. Knockdown of long non-coding RNA NEAT1 inhibits glioma cell migration and invasion via modulation of SOX2 targeted by miR-132. Mol Cancer. 2018;17:1–11.
Piva M, Domenici G, Iriondo O, Rábano M, Simoes BM, Comaills V, et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol Med. 2014;6(1):66–79.
Li D, Zhao L-N, Zheng X-L, Lin P, Lin F, Li Y, et al. Sox2 is involved in paclitaxel resistance of the prostate cancer cell line PC-3 via the PI3K/Akt pathway. Mol Med Rep. 2014;10(6):3169–76.
Zeng H, Wang L, Wang J, Chen T, Li H, Zhang K, et al. microRNA-129-5p suppresses adriamycin resistance in breast cancer by targeting SOX2. Arch Biochem Biophys. 2018;651:52–60.
He J, Shi J, Zhang K, Xue J, Li J, Yang J, et al. Sox2 inhibits wnt-beta-catenin signaling and metastatic potency of cisplatin-resistant lung adenocarcinoma cells. Mol Med Rep. 2017;15(4):1693–701.
Luo W, Yan D, Song Z, Zhu X, Liu X, Li X, et al. Mir-126-3p sensitizes glioblastoma cells to temozolomide by inactivating Wnt/β-catenin signaling via targeting SOX2. Life Sci. 2019;226:98–106.
Lopez-Bertoni H, Kotchetkov IS, Mihelson N, Lal B, Rui Y, Ames H, et al. A Sox2:mir-486-5p Axis regulates survival of GBM cells by inhibiting tumor suppressor networks. Cancer Res. 2020;80(8):1644–55.
Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab Invest. 2017;97(5):498–518.
Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neurooncology. 2006;8(3):261–79.
Mao H, LeBrun DG, Yang J, Zhu VF, Li M. Deregulated signaling pathways in glioblastoma multiforme: molecular mechanisms and therapeutic targets. Cancer Invest. 2012;30(1):48–56.
Matias D, Predes D, Niemeyer Filho P, Lopes M, Abreu J, Lima F, et al. Microglia-glioblastoma interactions: new role for wnt signaling. Biochim et Biophys Acta (BBA)-Reviews Cancer. 2017;1868(1):333–40.
Li Z, Zhang J, Zheng H, Li C, Xiong J, Wang W, et al. Modulating lncRNA SNHG15/CDK6/miR-627 circuit by palbociclib, overcomes temozolomide resistance and reduces M2-polarization of glioma associated microglia in glioblastoma multiforme. J Experimental Clin Cancer Res. 2019;38(1):1–13.
Navaei ZN, Khalili-Tanha G, Zangouei AS, Abbaszadegan MR, Moghbeli M. PI3K/AKT signaling pathway as a critical regulator of cisplatin response in tumor cells. Oncol Res. 2021;29(4):235–50.
Maharati A, Moghbeli M. PI3K/AKT signaling pathway as a critical regulator of epithelial-mesenchymal transition in colorectal tumor cells. Cell Commun Signal. 2023;21(1):201.
Bouras A, Kaluzova M, Hadjipanayis CG. Radiosensitivity enhancement of radioresistant glioblastoma by epidermal growth factor receptor antibody-conjugated iron-oxide nanoparticles. J Neurooncol. 2015;124:13–22.
Meng X, Zhao Y, Han B, Zha C, Zhang Y, Li Z, et al. Dual functionalized brain-targeting nanoinhibitors restrain temozolomide-resistant glioma via attenuating EGFR and MET signaling pathways. Nat Commun. 2020;11(1):594.
Mukherjee B, McEllin B, Camacho CV, Tomimatsu N, Sirasanagandala S, Nannepaga S, et al. EGFRvIII and DNA double-strand break repair: a molecular mechanism for radioresistance in glioblastoma. Cancer Res. 2009;69(10):4252–9.
Dancey JE, Freidlin B. Targeting epidermal growth factor receptor—are we missing the mark? Lancet. 2003;362(9377):62–4.
Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor–resistant disease. J Clin Oncol. 2013;31(8):1070.
Blobel CP. ADAMs: key components in EGFR signalling and development. Nat Rev Mol Cell Biol. 2005;6(1):32–43.
Ciardiello F, Tortora G. EGFR antagonists in cancer treatment. N Engl J Med. 2008;358(11):1160–74.
Chen Y, Li R, Pan M, Shi Z, Yan W, Liu N, et al. MiR-181b modulates chemosensitivity of glioblastoma multiforme cells to temozolomide by targeting the epidermal growth factor receptor. J Neurooncol. 2017;133(3):477–85.
Areeb Z, Stuart SF, West AJ, Gomez J, Nguyen HP, Paradiso L, et al. Reduced EGFR and increased miR-221 is associated with increased resistance to temozolomide and radiotherapy in glioblastoma. Sci Rep. 2020;10(1):17768.
Liu X, Guo Q, Gao G, Cao Z, Guan Z, Jia B, et al. Exosome-transmitted circCABIN1 promotes temozolomide resistance in glioblastoma via sustaining ErbB downstream signaling. J Nanobiotechnol. 2023;21(1):45.
Qi X-c, Xie D-j, Yan Q-f, Wang Y-r, Zhu Y-x, Qian C, et al. LRIG1 dictates the chemo-sensitivity of temozolomide (TMZ) in U251 glioblastoma cells via down-regulation of EGFR/topoisomerase-2/Bcl-2. Biochem Biophys Res Commun. 2013;437(4):565–72.
Wei J, Qi X, Zhan Q, Zhou D, Yan Q, Wang Y, et al. miR-20a mediates temozolomide-resistance in glioblastoma cells via negatively regulating LRIG1 expression. Biomed Pharmacother. 2015;71:112–8.
Rashed WM. C-MET as a potential target therapy toward personalized therapy in some pediatric tumors: an overview. Crit Rev Oncol/Hematol. 2018;131:7–15.
Piao Y, Park SY, Henry V, Smith BD, Tiao N, Flynn DL, et al. Novel MET/TIE2/VEGFR2 inhibitor altiratinib inhibits tumor growth and invasiveness in bevacizumab-resistant glioblastoma mouse models. Neurooncology. 2016;18(9):1230–41.
Fornari F, Milazzo M, Chieco P, Negrini M, Calin GA, Grazi GL, et al. MiR-199a-3p regulates mTOR and c-Met to influence the doxorubicin sensitivity of human hepatocarcinoma cells. Cancer Res. 2010;70(12):5184–93.
Kong DS, Song SY, Kim DH, Joo KM, Yoo JS, Koh JS, et al. Prognostic significance of c-Met expression in glioblastomas. Cancer. 2009;115(1):140–8.
Zhao C, Guo R, Guan F, Ma S, Li M, Wu J, et al. MicroRNA-128-3p enhances the chemosensitivity of temozolomide in glioblastoma by targeting c-Met and EMT. Sci Rep. 2020;10(1):9471.
Cheung M, Testa R. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013;13(3):234–44.
Zhang J, Zhang J, Zhang J, Qiu W, Xu S, Yu Q, et al. MicroRNA-625 inhibits the proliferation and increases the chemosensitivity of glioma by directly targeting AKT2. Am J cancer Res. 2017;7(9):1835.
Liu J, Han G, Liu H, Qin C. Suppression of cholangiocarcinoma cell growth by human umbilical cord mesenchymal stem cells: a possible role of wnt and Akt signaling. PLoS ONE. 2013;8(4):e62844.
Baxter PA, Lin Q, Mao H, Kogiso M, Zhao X, Liu Z, et al. Silencing BMI1 eliminates tumor formation of pediatric glioma CD133 + cells not by affecting known targets but by down-regulating a novel set of core genes. Acta Neuropathol Commun. 2014;2:1–14.
Shi L, Fei X, Wang Z, You Y. PI3K inhibitor combined with miR-125b inhibitor sensitize TMZ-induced anti-glioma stem cancer effects through inactivation of Wnt/β-catenin signaling pathway. Vitro Cell Dev Biology-Animal. 2015;51:1047–55.
Cai T, Liu Y, Xiao J. Long noncoding RNA MALAT 1 knockdown reverses chemoresistance to temozolomide via promoting micro RNA-101 in glioblastoma. Cancer Med. 2018;7(4):1404–15.
Trojan J, Cloix J-F, Ardourel M-Y, Chatel M, Anthony D. Insulin-like growth factor type I biology and targeting in malignant gliomas. Neuroscience. 2007;145(3):795–811.
Schlenska-Lange A. Cell proliferation and migration in glioblastoma multiforma cell lines are influenced by insulin-like growth factor 1 in vitro (28, Pg 1059, 2008). Anticancer Res. 2008;28(3 B):1965.
Yin D, Tamaki N, Parent AD, Zhang JH. Insulin-like growth factor-I decreased etoposide-induced apoptosis in glioma cells by increasing bcl-2 expression and decreasing CPP32 activity. Neurol Res. 2005;27(1):27–35.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93.
Li X, Wu C, Chen N, Gu H, Yen A, Cao L, et al. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget. 2016;7(22):33440.
Duzgun Z, Eroglu Z, Avci CB. Role of mTOR in glioblastoma. Gene. 2016;575(2):187–90.
Chen P-H, Cheng C-H, Shih C-M, Ho K-H, Lin C-W, Lee C-C, et al. The inhibition of microRNA-128 on IGF-1-activating mTOR signaling involves in temozolomide-induced glioma cell apoptotic death. PLoS ONE. 2016;11(11):e0167096.
Lin L, Lin D, Jin L, Wang J, Lin Z, Zhang S et al. LncRNA HOXA-AS2 Promotes Temozolomide Resistance in Glioblastoma by Regulated miR-302a-3p/IGF1 Axis. Genetics Research. 2022;2022.
Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385(6618):729–33.
Moss ML, Jin S-LC, Milla ME, Burkhart W, Carter HL, Chen W-J, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature. 1997;385(6618):733–6.
Grötzinger J, Lorenzen I, Düsterhöft S. Molecular insights into the multilayered regulation of ADAM17: the role of the extracellular region. Biochim et Biophys Acta (BBA)-Molecular Cell Res. 2017;1864(11):2088–95.
Saad MI, Rose-John S, Jenkins BJ. ADAM17: an emerging therapeutic target for lung cancer. Cancers. 2019;11(9):1218.
Yang J-T, Lee I-N, Huang C, Huang H-C, Wu Y-P, Chong Z-Y, et al. ADAM17 confers Temozolomide Resistance in Human Glioblastoma cells and miR-145 regulates its expression. Int J Mol Sci. 2023;24(9):7703.
Colak S, Ten Dijke P. Targeting TGF-β signaling in Cancer. Trends cancer. 2017;3(1):56–71.
Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30.
Han J, Alvarez-Breckenridge CA, Wang QE, Yu J. TGF-β signaling and its targeting for glioma treatment. Am J cancer Res. 2015;5(3):945–55.
Mao J, Sun Z, Cui Y, Du N, Guo H, Wei J, et al. PCBP2 promotes the development of glioma by regulating FHL3/TGF-β/Smad signaling pathway. J Cell Physiol. 2020;235(4):3280–91.
Nie E, Jin X, Miao F, Yu T, Zhi T, Shi Z, et al. TGF-β1 modulates temozolomide resistance in glioblastoma via altered microRNA processing and elevated MGMT. Neurooncology. 2021;23(3):435–46.
Shree B, Tripathi S, Sharma V. Transforming growth factor-Beta-regulated LncRNA-MUF promotes Invasion by modulating the miR-34a Snail1 Axis in Glioblastoma Multiforme. Front Oncol. 2021;11:788755.
Hegi ME, Liu L, Herman JG, Stupp R, Wick W, Weller M, et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncology: Official J Am Soc Clin Oncol. 2008;26(25):4189–99.
Sarkaria JN, Kitange GJ, James CD, Plummer R, Calvert H, Weller M, et al. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin cancer Research: Official J Am Association Cancer Res. 2008;14(10):2900–8.
Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003.
Cabrini G, Fabbri E, Lo Nigro C, Dechecchi MC, Gambari R. Regulation of expression of O6-methylguanine-DNA methyltransferase and the treatment of glioblastoma (review). Int J Oncol. 2015;47(2):417–28.
Nie E, Jin X, Wu W, Yu T, Zhou X, Shi Z, et al. MiR-198 enhances temozolomide sensitivity in glioblastoma by targeting MGMT. J Neurooncol. 2017;133:59–68.
Nakanishi C, Toi M. Nuclear factor-kappab inhibitors as sensitizers to anticancer drugs. Nat Rev Cancer. 2005;5(4):297–309.
Wang CY, Cusack JC Jr., Liu R, Baldwin AS. Jr. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med. 1999;5(4):412–7.
Hymowitz SG, Wertz IE. A20: from ubiquitin editing to tumour suppression. Nat Rev Cancer. 2010;10(5):332–41.
Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Sci (New York NY). 2010;327(5969):1135–9.
Chen Y, Vallee S, Wu J, Vu D, Sondek J, Ghosh G. Inhibition of NF-kappaB activity by IkappaBbeta in association with kappab-ras. Mol Cell Biol. 2004;24(7):3048–56.
Fenwick C, Na SY, Voll RE, Zhong H, Im SY, Lee JW, et al. A subclass of Ras proteins that regulate the degradation of IkappaB. Volume 287. New York, NY: Science; 2000. pp. 869–73. 5454.
Lin H, Wang Y, Zhang X, Liu B, Zhang W, Cheng J. Prognostic significance of kappaB-Ras1 expression in gliomas. Med Oncol (Northwood Lond Engl). 2012;29(2):1272–9.
Tago K, Funakoshi-Tago M, Sakinawa M, Mizuno N, Itoh H. KappaB-Ras is a nuclear-cytoplasmic small GTPase that inhibits NF-kappaB activation through the suppression of transcriptional activation of p65/RelA. J Biol Chem. 2010;285(40):30622–33.
Haemmig S, Baumgartner U, Glück A, Zbinden S, Tschan MP, Kappeler A, et al. miR-125b controls apoptosis and temozolomide resistance by targeting TNFAIP3 and NKIRAS2 in glioblastomas. Cell Death Dis. 2014;5(6):e1279.
Rahnama F, Shimokawa T, Lauth M, Finta C, Kogerman P, Teglund S, et al. Inhibition of GLI1 gene activation by Patched1. Biochem J. 2006;394(Pt 1):19–26.
Thomas Z, Gibson W, Sexton J, Aird K, Ingram S, Aldrich A, et al. Targeting GLI1 expression in human inflammatory breast cancer cells enhances apoptosis and attenuates migration. Br J Cancer. 2011;104(10):1575–86.
Munoz JL, Rodriguez-Cruz V, Ramkissoon SH, Ligon KL, Greco SJ, Rameshwar P. Temozolomide resistance in glioblastoma occurs by miRNA-9-targeted PTCH1, independent of sonic hedgehog level. Oncotarget. 2015;6(2):1190.
Maharati A, Moghbeli M. Long non-coding RNAs as the critical regulators of PI3K/AKT, TGF-beta, and MAPK signaling pathways during breast tumor progression. J Transl Med. 2023;21(1):556.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim et Biophys Acta (BBA)-Molecular Cell Res. 2007;1773(8):1263–84.
Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK–RAS–RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):103–19.
Ye Y-Y, Mei J-W, Xiang S-S, Li H-F, Ma Q, Song X-L, et al. MicroRNA-30a-5p inhibits gallbladder cancer cell proliferation, migration and metastasis by targeting E2F7. Cell Death Dis. 2018;9(3):410.
Yin W, Wang B, Ding M, Huo Y, Hu H, Cai R, et al. Elevated E2F7 expression predicts poor prognosis in human patients with gliomas. J Clin Neurosci. 2016;33:187–93.
Lu C, Wei Y, Wang X, Zhang Z, Yin J, Li W, et al. DNA-methylation-mediated activating of lncRNA SNHG12 promotes temozolomide resistance in glioblastoma. Mol Cancer. 2020;19:1–19.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773(8):1263–84.
Wei Y, Lu C, Zhou P, Zhao L, Lyu X, Yin J, et al. EIF4A3-induced circular RNA ASAP1 promotes tumorigenesis and temozolomide resistance of glioblastoma via NRAS/MEK1/ERK1-2 signaling. Neuro Oncol. 2021;23(4):611–24.
Wittchen ES, Aghajanian A, Burridge K. Isoform-specific differences between Rap1A and Rap1B GTPases in the formation of endothelial cell junctions. Small GTPases. 2011;2(2):65–76.
Bertoni A, Tadokoro S, Eto K, Pampori N, Parise LV, White GC, et al. Relationships between Rap1b, affinity modulation of integrin alpha IIbbeta 3, and the actin cytoskeleton. J Biol Chem. 2002;277(28):25715–21.
Malchinkhuu E, Sato K, Maehama T, Ishiuchi S, Yoshimoto Y, Mogi C, et al. Role of Rap1B and tumor suppressor PTEN in the negative regulation of lysophosphatidic acid—induced migration by isoproterenol in glioma cells. Mol Biol Cell. 2009;20(24):5156–65.
Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582(14):2093–101.
de Toledo M, Anguille C, Roger L, Roux P, Gadea G. Cooperative anti-invasive effect of Cdc42/Rac1 activation and ROCK inhibition in SW620 colorectal cancer cells with elevated blebbing activity. PLoS ONE. 2012;7(11):e48344.
She X, Yu Z, Cui Y, Lei Q, Wang Z, Xu G, et al. miR-128 and miR-149 enhance the chemosensitivity of temozolomide by Rap1B-mediated cytoskeletal remodeling in glioblastoma. Oncol Rep. 2014;32(3):957–64.
Nishimatsu H, Suzuki E, Nagata D, Moriyama N, Satonaka H, Walsh K, et al. Adrenomedullin induces endothelium-dependent vasorelaxation via the phosphatidylinositol 3-kinase/Akt–dependent pathway in rat aorta. Circul Res. 2001;89(1):63–70.
Okumura H, Nagaya N, Itoh T, Okano I, Hino J, Mori K, et al. Adrenomedullin infusion attenuates myocardial ischemia/reperfusion injury through the phosphatidylinositol 3-kinase/Akt-dependent pathway. Circulation. 2004;109(2):242–8.
Fritz-Six KL, Dunworth WP, Li M, Caron KM. Adrenomedullin signaling is necessary for murine lymphatic vascular development. J Clin Investig. 2008;118(1):40–50.
Oehler M, Norbury C, Hague S, Rees MC, Bicknell R. Adrenomedullin inhibits hypoxic cell death by upregulation of Bcl-2 in endometrial cancer cells: a possible promotion mechanism for tumour growth. Oncogene. 2001;20(23):2937–45.
He Z, Cheng M, Hu J, Liu L, Liu P, Chen L, et al. miR-1297 sensitizes glioma cells to temozolomide (TMZ) treatment through targeting adrenomedullin (ADM). J Translational Med. 2022;20(1):443.
Sakurai T, Kudo M, Umemura A, He G, Elsharkawy AM, Seki E, et al. p38α inhibits liver fibrogenesis and consequent hepatocarcinogenesis by curtailing accumulation of reactive oxygen species. Cancer Res. 2013;73(1):215–24.
Kumphune S, Chattipakorn S, Chattipakorn N. Roles of p38-MAPK in insulin resistant heart: evidence from bench to future bedside application. Curr Pharm Design. 2013;19(32):5742–54.
Liu Q, Zou R, Zhou R, Gong C, Wang Z, Cai T, et al. miR-155 regulates glioma cells invasion and chemosensitivity by p38 isforms in vitro. J Cell Biochem. 2015;116(7):1213–21.
Su Z, Yang Z, Xu Y, Chen Y, Yu Q. MicroRNAs in apoptosis, autophagy and necroptosis. Oncotarget. 2015;6(11):8474.
Tolue Ghasaban F, Maharati A, Akhlaghipour I, Moghbeli M. MicroRNAs as the critical regulators of autophagy-mediated cisplatin response in tumor cells. Cancer Cell Int. 2023;23(1):80.
He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
Mijaljica D, Prescott M, Devenish RJ. The intriguing life of autophagosomes. Int J Mol Sci. 2012;13(3):3618–35.
Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–56.
Kong Q, Xu L-H, Xu W, Fang J-P, Xu H-G. HMGB1 translocation is involved in the transformation of autophagy complexes and promotes chemoresistance in leukaemia. Int J Oncol. 2015;47(1):161–70.
Sehgal A, Konig H, Johnson D, Tang D, Amaravadi R, Boyiadzis M, et al. You eat what you are: autophagy inhibition as a therapeutic strategy in leukemia. Leukemia. 2015;29(3):517–25.
Yang M-C, Loh J-K, Li Y-Y, Huang W-S, Chou C-H, Cheng J-T, et al. Bcl2L12 with a BH3-like domain in regulating apoptosis and TMZ-induced autophagy: a prospective combination of ABT-737 and TMZ for treating glioma. Int J Oncol. 2015;46(3):1304–16.
Bhoopathi P, Chetty C, Gujrati M, Dinh DH, Rao JS, Lakka S. Cathepsin B facilitates autophagy-mediated apoptosis in SPARC overexpressed primitive neuroectodermal tumor cells. Cell Death Differ. 2010;17(10):1529–39.
Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64(12):4286–93.
Franzetti E, Huang Z-J, Shi Y-X, Xie K, Deng X-J, Li J-P, et al. Autophagy precedes apoptosis during the remodeling of silkworm larval midgut. Apoptosis. 2012;17:305–24.
Lorente M, Torres S, Salazar M, Carracedo A, Hernández-Tiedra S, Rodríguez-Fornés F, et al. Stimulation of the midkine/ALK axis renders glioma cells resistant to cannabinoid antitumoral action. Cell Death Differ. 2011;18(6):959–73.
Zhang N, Chen Y, Jiang R, Li E, Chen X, Xi Z, et al. PARP and RIP 1 are required for autophagy induced by 11’-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy. 2011;7(6):598–612.
You L, Wang Z, Li H, Shou J, Jing Z, Xie J, et al. The role of STAT3 in autophagy. Autophagy. 2015;11(5):729–39.
Li H, Chen L, Li J-j, Zhou Q, Huang A, Liu W-w, et al. miR-519a enhances chemosensitivity and promotes autophagy in glioblastoma by targeting STAT3/Bcl2 signaling pathway. J Hematol Oncol. 2018;11:1–16.
Comincini S, Allavena G, Palumbo S, Morini M, Durando F, Angeletti F, et al. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol Ther. 2013;14(7):574–86.
Xu J, Huang H, Peng R, Ding X, Jiang B, Yuan X, et al. MicroRNA-30a increases the chemosensitivity of U251 glioblastoma cells to temozolomide by directly targeting beclin 1 and inhibiting autophagy. Exp Ther Med. 2018;15(6):4798–804.
Shukla SK, Purohit V, Mehla K, Gunda V, Chaika NV, Vernucci E, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32(1):71–87. e7.
Tabatabai G, Frank B, Möhle R, Weller M, Wick W. Irradiation and hypoxia promote homing of haematopoietic progenitor cells towards gliomas by TGF-β-dependent HIF-1α-mediated induction of CXCL12. Brain. 2006;129(9):2426–35.
Renfrow JJ, Soike MH, Debinski W, Ramkissoon SH, Mott RT, Frenkel MB, et al. Hypoxia-inducible factor 2α: a novel target in gliomas. Future Med Chem. 2018;10(18):2227–36.
Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625–34.
Shi Y, Guryanova OA, Zhou W, Liu C, Huang Z, Fang X et al. Ibrutinib inactivates BMX-STAT3 in glioma stem cells to impair malignant growth and radioresistance. Sci Transl Med. 2018;10(443).
Ma Y, Gong Y, Cheng Z, Loganathan S, Kao C, Sarkaria JN, et al. Critical functions of RhoB in support of glioblastoma tumorigenesis. Neurooncology. 2015;17(4):516–25.
Prendergast GC. Actin’up: RhoB in cancer and apoptosis. Nat Rev Cancer. 2001;1(2):162–8.
Skuli N, Monferran S, Delmas C, Lajoie-Mazenc I, Favre G, Toulas C, et al. Activation of RhoB by hypoxia controls hypoxia-inducible factor-1α stabilization through glycogen synthase kinase-3 in U87 glioblastoma cells. Cancer Res. 2006;66(1):482–9.
Yin J, Ge X, Shi Z, Yu C, Lu C, Wei Y, et al. Extracellular vesicles derived from hypoxic glioma stem-like cells confer temozolomide resistance on glioblastoma by delivering miR-30b-3p. Theranostics. 2021;11(4):1763.
Matheson CJ, Backos DS, Reigan P. Targeting WEE1 kinase in cancer. Trends Pharmacol Sci. 2016;37(10):872–81.
Ling Z, Zhang J, Liu Q. Oncogenic forkhead box D3 antisense RNA 1 promotes cell survival and confers temozolomide resistance in glioblastoma cells through the miR-128-3p/WEE1 G2 checkpoint kinase axis. Bioengineered. 2022;13(3):6012–23.
Shi L, Wan Y, Sun G, Zhang S, Wang Z, Zeng Y. miR-125b inhibitor may enhance the invasion-prevention activity of temozolomide in glioblastoma stem cells by targeting PIAS3. BioDrugs. 2014;28:41–54.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MPY and AB were involved in search strategy and drafting. MM revised, structured, and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Palizkaran Yazdi, M., Barjasteh, A. & Moghbeli, M. MicroRNAs as the pivotal regulators of Temozolomide resistance in glioblastoma. Mol Brain 17, 42 (2024). https://doi.org/10.1186/s13041-024-01113-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-024-01113-6