
Abstract
Activation of nociceptin opioid peptide receptors (NOP, a.k.a. opioid-like receptor-1, ORL-1) by the ligand nociceptin/orphanin FQ, leads to G protein-dependent regulation of Cav2.2 (N-type) voltage-gated calcium channels (VGCCs). This typically causes a reduction in calcium currents, triggering changes in presynaptic calcium levels and thus neurotransmission. Because of the widespread expression patterns of NOP and VGCCs across multiple brain regions, the dorsal horn of the spinal cord, and the dorsal root ganglia, this results in the alteration of numerous neurophysiological features. Here we review the regulation of N-type calcium channels by the NOP-nociceptin system in the context of neurological conditions such as anxiety, addiction, and pain.
Similar content being viewed by others
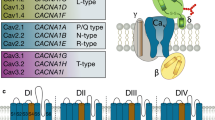

Structure and physiological roles of N-type calcium channels
Voltage-gated calcium channels (VGCCs) mediate calcium influx upon the membrane depolarization, regulating many physiological processes, such as neurotransmitter release, hormone secretion, muscle contraction, and gene expression [1]. The dysfunction of these channels has been linked to a wide range of pathological conditions including pain, epilepsy, migraine, night blindness, cardiac arrhythmias, and congenital deafness among many others [2, 3], making them important pharmacological targets [4]. VGCCs have been classified based on their electrophysiological and pharmacological profiles. They can be separated into low-voltage activated (LVA) calcium channels, which include the T-type calcium channels (Cav3.1, Cav3.2, and Cav3.3), or high-voltage activated ones (HVA), including N-type (Cav2.2), R-type (Cav2.3), P/Q-type (Cav2.1), and L-type (Cav1.1, Cav1.2, Cav1.3, and Cav1.4) channels [5, 6]. With the exception of Cav1.1 which is skeletal muscle-specific, these channels are all expressed in the brain, but show varying albeit overlapping distributions and support specific neurophysiological roles [1]. Cav2 channels operate predominantly at presynaptic terminals where they are responsible for fast synaptic transmission [7], whereas Cav1.3 and Cav1.4 channels can both be expressed at ribbon synapses (where they support features such as auditory and visual synaptic transmission [8, 9]; with Cav1.2 and Cav1.3 being expressed as well on cell bodies and dendrites of many types of central nervous system neurons [10]. All calcium channels contain a pore forming Cavα1 subunit that is obligatory to obtain a functional channel and which are comprised of four transmembrane domains that are connected by cytoplasmic linker regions and flanked by cytoplasmic N- and C-terminal regions [1, 5]. HVA channel Cavα1 subunits co-assemble with ancillary Cavα2δ and Cavβ subunits in a 1:1:1 stoichiometry [11]. These subunits help promote plasma membrane expression and regulate the biophysical and pharmacological features of the channel [12, 13]. The mammalian genome expresses four different types of Cavβ and four types of Cavα2δ subunits, with the latter being post-translationally cleaved, relinked via a disulfide bond, and then attached to the outer leaflet of the plasma membrane via a GPI anchor [14, 15]. In contrast, Cavβ subunits are cytoplasmic and attach to the Cavα1 subunit at the linker between transmembrane domains I and II. With additional diversity arising from alternative splicing events, the existence of multiple ancillary subunits gives rise to a wide range of different calcium channel complexes [16, 17]. The cryo-EM structure of Cav2.2 (and other) calcium channels has been solved [18,19,20,21] and has provided insights into the structural basis of channels function and pharmacology.
Cav2.2 calcium channels are expressed predominantly at presynaptic nerve terminals where they (along with Cav2.1 channels) are critically involved in the release of neurotransmitters [22, 23]. Their expression is primarily restricted to neurons, with wide distribution in the brain as well as in peripheral sensory afferents. Cav2.2 channels are potently inhibited by peptide toxins isolated from different types of marine fish hunting cone snails such as ω-conotoxin MVIIA (Conus magus) and ω-conotoxin GVIA (Conus geographus) whose selectivity of action on these channels has helped elucidate key roles of these channels in neurophysiological processes [24, 25]. Intrathecal delivery of such toxins mediates potent analgesic effects [26, 27], consistent with a key role of these channels in neurotransmission between sensory afferents and neurons in the spinal dorsal horn. Case in point, a synthetic version of ω-conotoxin MVIIA (a.k.a. Ziconotide or Prialt) is FDA approved for the treatment of refractory cancer pain [28]. Additional insights into the physiological roles of Cav2.2 channels have been gained from genetic models such as knockout mice lacking these channels, from small organic inhibitors of Cav2.2 channel activity, and from molecules that modulate post-translational modification of these channels. Mice deficient in Cav2.2 show hyposensitivity to different pain modalities [29,30,31], consistent with the data obtained with pharmacological inhibition. These mice also show increased vigilance [32], hyper-aggression [33], reduced anxiety levels [29], and appear to exhibit a reduction in the symptoms of alcohol withdrawal [34]. The latter is also consistent with the action of small organic Cav2.2 inhibitors [35,36,37]. A link between Cav2.2 channels and reward-seeking was highlighted by studies that target the interaction of Cav2.2 with collapsin mediator protein 2 (CRMP2) which is involved in regulating Cav2.2 channel expression at the plasma membrane [38, 39]—interfering with this regulatory pathway alters reinstatement of cocaine seeking [40]. Pharmacological inhibition of Cav2.2 has been shown to have anxiolytic effects [41, 42], and this may be related to an inhibition of GABAergic synaptic transmission in the basolateral amygdala and prefrontal cortex [43]. On the other hand, it was shown that alternative splicing of exon 37 of Cav2.2 enhances responses to aversive stimuli via alteration of glutamatergic signaling [17]. Deletion of Cav2.2 channels either directly, or by depletion of its ancillary Cavβ3 subunit has been linked to impairment of memory [44, 45]. Finally, delivery of the Cav2.2 inhibitor ω-conotoxin GVIA directly into the brain has been shown to cause depression-like behavior in rodents [46], although it should be noted that depression is more commonly associated with altered L-type calcium channel activity. Overall, these findings indicate that Cav2.2 channels mediate important physiological roles and that modulation of the activity of these channels is expected to have consequences that affect behaviors such as anxiety and pain responses.
Modulation of Cav2.2 channels by NOP receptors
Cav2.2 channels are subject to modulation by a wide range of G protein coupled receptors (GPCRs) [47]. This includes the nociceptin opioid receptor (NOP, previously called the opioid receptor-like 1 receptor (ORL-1), which was first described by Mollereau et al. [48] and added as a fourth member of the opioid receptor family of µ-, κ-, and δ-opioid receptors [49, 50]. However, the NOP receptor is not activated by any of the known endogenous opioids that act on classical opioid receptors, but instead by the endogenous agonist nociceptin/orphanin FQ [51, 52]. The crystal structure of the NOP receptor reveals a typical 7 transmembrane helix topology with an additional short helix contained within the proximal C-terminus [53].
Like other G protein-coupled receptors (GPCRs), the activation of NOP by nociceptin triggers the exchange of GDP for GTP on the Gα subunit, causing its dissociation from the Gβγ dimer. The dissociated subunits then proceed to act on different signaling pathways and effector systems. The NOP receptor couples primarily to Gαi/o, inhibiting adenylyl cyclase and thus cyclic adenosine monophosphate (cAMP) production [54]. In addition, activation of NOP receptors activates phospholipase C and MAP kinase activity via coupling to Gα14 and Gα16 [55]. The dissociated Gβγ subunits, in addition to regulating various intracellular signaling cascades, activate G protein-coupled inwardly rectifying potassium (GIRK) channels [56], and inhibit VGCCs [47], thus reducing neuronal excitability and neurotransmitter release (Fig. 1A).

Key aspects of NOP receptor signalling. A. NOP receptors at synaptic terminals. Activation of NOP by N/OFQ triggers several G protein dependent signalling pathways. B. Activation of NOP receptors triggers Gβγ mediated voltage-dependent inhibition of Cav2.2 channels, as well as Gα/tyrosine kinase dependent voltage independent modulation. Due to the formation of signalling complexes, prolonged agonist application triggers the co-internalization of NOP receptor Cav2.2 channel complexes
Specifically, Gβγ subunits inhibit members of the Cav2 calcium channel family (Fig. 1B) by directly interacting with the Cavα1 subunit at its cytoplasmic N-terminal and domain I-II linker regions [57,58,59], with Cav2.2 channels being inhibited more potently than Cav2.1 [60,61,62]. Direct Gβγ modulation of Cav2.2 is sensitive to the membrane potential and can be reversed by the application of a depolarizing pre-pulse or by trains of action potentials—it is therefore referred to as voltage-dependent inhibition [63,64,65]. The extent of G protein modulation is also dependent on the Cavβ subunit [66,67,68] and critically dependent on the Gβ isoform [61, 69], thus allowing receptors to mediate a wide range of inhibitory voltage-dependent processes. Many G protein-coupled receptors also cause voltage-independent modulation of channel activity, most likely via the activation of protein kinases and receptor tyrosine kinases (Fig. 1B). This type of inhibition is insensitive to membrane depolarizations and has also been observed with NOP receptors [70].
Inhibition of Cav2.2 calcium channels by NOP receptors has been described in both neurons and heterologous expression systems [70,71,72,73,74,75,76,77]. In addition to the classical G protein inhibition, the C-terminus of the NOP receptor physically interacts with the Cav2.2 C-terminus, allowing it to control channel density at the plasma membrane through enhanced forward trafficking by possibly promoting ER export, and via channel/receptor co-internalization after prolonged agonist exposure [70, 78, 79] (Fig. 1B). This physical interaction also allows for a tonic inhibition of Cav2.2 channels by Gβγ in the absence of an agonist, presumably due to the receptor’s constitutive activity. Another potential avenue by which the NOP can modulate Cav2.2 channels is via heterodimerization with other opioid receptor family members. NOPs form heterodimer complexes with all of the other three opioid receptors, and the prolonged activation of µ-opioid receptors triggers the internalization of Cav2.2 channels, but only in the presence of NOP receptors [80].
A number of different ligands of NOP receptors have been identified and they are not considered to be opioids. As already noted above, the archetypal agonist of the NOP receptor is nociception—a 17-amino acid peptide that is structurally related to dynorphin A [54]. A wide range of synthetic NOP receptor agonists have been described (for review, see [81]) and have been examined in preclinical and clinical studies of anxiety and pain. Conversely, a wide range of NOP receptor antagonists have been developed and tested in animals and human subjects in phase I and II studies for the treatment of cognitive impairment, depression, and addiction [81, 82]. This includes LY2940094 (aka BTRX-246040) which appears to be effective in the treatment of depression and well tolerated [83]. Mixed agonists of NOP and opioid receptors such as AT-121 and cepranopol have also been explored as potential systemically active pain therapeutics including in phase III clinical testing [84, 85]. The NOP receptor is widely distributed in the central and peripheral nervous systems [86]. In the brain, it is expressed in regions that are involved in the processing of pain, learning, memory, food intake, and fear [86,87,88], consistent with the above pharmacological studies. Given the widespread expression of VGCCs, they are co-expressed with these receptors in many areas of the brain. Below, we will highlight the role of the NOP-N/OFQ system in neuro-physiological processes that are known to rely on Cav2.2 channels.
NOP receptors as modulators of pain
NOP receptors are expressed in the dorsal and ventral horns of the spinal cord which integrate sensory processing [89]. NOP receptor expression is enhanced in the spinal cord in rodent models of persistent inflammatory and neuropathic pain [90,91,92] and there is an upregulation of plasma concentrations of nociceptin in chronic pain patients [93]. Intrathecal delivery of nociceptin mediates analgesia in rodent models of pain [94,95,96,97], as do non-peptide agonists [98,99,100]. In non-human primates, systemic or intrathecal injection of NOP agonists causes antinociception [101,102,103,104,105]. These analgesic effects are likely mediated by an inhibitory action on presynaptic Cav2.2 channels expressed in sensory afferents (note that NOP receptors are expressed on both A and C fibers [79, 86, 106], along with possible activation of postsynaptic GIRK channels). In addition, NOP receptor activity has been shown to modulate the excitability of spinal interneurons and superficial layer neurons projecting to the brain [107,108,109].
Mice lacking the NOP receptors show normal nociceptive thresholds, only slightly reduced locomotor activity, and normal immune responses, but compromised hearing ability [110]. These mice however also show alterations in morphine tolerance. Like NOP receptors, activation of μ-opioid receptors by morphine or DAMGO also inhibits Cav2.2 channel activity [111]. While NOP receptors are intrinsically resistant to the μ-opioid specific agonist morphine, they appear to affect morphine tolerance via crosstalk with μ-opioid receptors. It was shown that mice lacking NOP receptors exhibit resistance to morphine tolerance without alterations in the acute effects of morphine [112, 113], whereas mice with morphine tolerance exhibit upregulation of NOP receptors [113]. Along these lines, NOP receptor activation diminishes the development of morphine tolerance in rats and blocks morphine-induced place preference [114, 115], although this concept has been challenged [116]. Other studies have reported an antagonistic effect of NOP receptor activation on opioid-induced analgesia [117, 118]. In addition, increased NOP expression is seen after prolonged morphine exposure [113, 119, 120]. Finally, bi-functional ligands of NOP and μ-opioid receptors help prevent adverse effects associated with μ-opioid receptor activation [121,122,123]. To what extent this crosstalk is mediated by selective actions via Cav2.2 calcium channels remains to be determined, but the notion that μ-opioid receptors, NOP receptors, and Cav2.2 channels can form signaling complexes [80] hints at the possibility that converging signaling by these two receptors on Cav2.2 as a common target may play a role.
While NOP receptors mediate antinociceptive actions at the spinal level likely by inhibition of spinal excitatory neurons and synaptic transmission at peripheral afferent terminals [124], they appear to exhibit pronociceptive effects at the supraspinal level, as observed after injection of nociceptin into the brain [51, 52]. It is not entirely clear how these pronociceptive effects occur at the cellular and molecular level. One possibility may be an inhibitory effect on stress-induced analgesia that is known to be modulated by NOP receptors [125,126,127]. Indeed, there is evidence that NOP receptor activation contributes to the occurrence of allodynia and hyperalgesia in post-traumatic stress disorders (PTSD) [128], with NOP antagonists reversing this type of pain behavior [129]. It has been suggested that at supraspinal sites, the activation of NOP receptors may inhibit neuronal activity via activation of GIRK channels in brain loci that are relevant for nociceptive transmission such as the periaqueductal gray region and dorsal raphe nucleus [124], but exactly how this modulation causes pro-nociceptive effects remains to be worked out.
Role of NOP receptors in anxiety
Anxiety is marked by excessive fear (and avoidance) in response to perceived threatening situations and even in the absence of true danger [130]. NOP receptors are highly expressed in neurons and synaptic terminals of brain areas that are implicated in anxiety and depression [131]. This includes the amygdala, the nucleus accumbens (NAc), the hippocampus, the hypothalamus, the periaqueductal gray, the ventral tegmental area (VTA), and some of the prefrontal regions among others [132,133,134]. The medial prefrontal cortex (mPFC) exerts inhibitory control over amygdala activity, preventing an improper expression of emotion, but in anxiety disorders, this control becomes imperfect and consequently triggers aberrant amygdala activation through dysfunction of GABAergic and glutamatergic transmission [135,136,137,138]. NOP activation produces presynaptic inhibition of VGCCs, and thus the release of glutamate in the lateral amygdala [139, 140]. NOP agonists delivered orally or via intraperitoneal injections have revealed anxiolytic effects in rodents [141,142,143,144]. Little is known about the biochemical pathways by which NOP agonists induce anxiolytic effects but they may involve serotonin availability and changes in firing rates in the dorsal raphe nucleus (DRN, which projects to serotonergic axons of the amygdala), and concomitantly, by counteracting anxiogenic CRF actions into the bed nucleus of stria terminalis [145]. It should be noted that some studies have, however, reported anxiogenic responses to nociceptin, and the reason for these discordant findings is unclear [146, 147].
Prenatal ethanol exposure (PEE) induces an anxiety-like phenotype in adult and adolescent rats [148], and this is frequently used as an animal model to study anxiety. This results in the induction of neuronal hyper-excitability by augmenting glutamatergic transmission, which does not appear to be attenuated by GABAergic inhibition in the basolateral amygdala [149, 150]. NOP activation via intracerebroventricular administration of nociceptin reportedly inhibits associated anxiety behaviors [151]. In this model, there is an alteration of dopaminergic neuron morphology [152], along with a reduction of NOP gene expression [153], and an increase in the expression of the precursor of nociceptin (ppN/OFQ) [151]. Although the authors did not find any alterations of the NOP-nociceptin pathway in the amygdala, alterations of upstream inputs may lead to altered amygdala activity, thus perhaps contributing to the anxiety phenotype.
Altogether, anxiolytic-like effects can be observed with the delivery of nociceptin and synthetic agonists into the brain. Cav2.2 channels are also known to play a role in anxiety and are a potential pharmacological target for anxiolytics [41, 154, 155]. Indeed, as noted earlier, inhibition or depletion of Cav2.2 channels has been shown to mediate anxiolytic effects in mice [29, 156], and therefore a putative inhibition of these channels by activation of NOP would be expected to result in similar outcomes. It is however unclear precisely in which brain structures and neuronal subtypes of NOP receptor regulation of Cav2.2 channels may take place in the context of anxiety.
NOP receptors and stress
The fear/anxiety neuro-circuitry overlaps with the neuro-circuitry that regulates stress responses [130]. The amygdala integrates information from cortical and thalamic sensory inputs, to generate fear and anxiety-related behavioral outputs [157], with fear conditioning models demonstrating an increase in synaptic strength in the lateral amygdala (LA) and the PFC [158,159,160]. PTSD involves an over-reactivity of the HPA axis, decreased inhibitory signaling by GABA, and increased excitatory neurotransmission. This is associated with increased activity in emotion-processing brain regions, such as the limbic system (hippocampus, amygdala, PFC). Overall, a major characteristic of PTSD is impaired fear extinction, following exposure to a traumatic event [161]. These responses against chronic stress lead to altered NOP receptor signaling which may contribute to an increase in corticosterone levels [145]. In PTSD experimental models, corticosterone levels, as well as nociceptin and NOP expression are increased in different brain regions and the cerebrospinal fluid, and consequently, NOP antagonists protect against molecular and behavioral deficits [162, 163]. Much of the underlying mechanisms appear to involve alterations in L-type calcium channel activity [156, 164,165,166,167,168,169,170]. However, given that Cav2.2 channels are present at synaptic junctions in the HPA axis, and are typically inhibited by NOP receptor agonists, it is possible that these channels play a detrimental role in fear extinction during PTSD, in addition to their potential pronociceptive effects that were discussed earlier. On the other hand, it is interesting to note that compounds such as topiramate which block R-type and N-type channels appear to show some promise in treating PTSD [171]. Clearly, more work is needed to elucidate the potential role of Cav2.2 channels in PTSD and its modulation by the NOP receptors.
NOP receptors as targets for addiction
Dopamine participates in drug-seeking reward behavior by modulating the activity of the mesolimbic pathway [172]. The dopaminergic axons in the ventral tegmental area (VTA) project to downstream regions such as the nucleus accumbens, the nucleus of the stria terminalis, and the lateral hypothalamus [173]. Nociceptin and NOP receptors are densely expressed in the VTA where they regulate dopaminergic transmission [174, 175]. This occurs by disinhibition of GABAergic control over dopaminergic neurons, such that NOP agonists mediate suppression of dopamine release [115, 176, 177]. Genetic deletion or pharmacological inhibition of NOP receptors in rats was shown to inhibit nicotine-motivated behaviors [178]. Along these lines, alcohol reward-seeking behavior was shown to be inhibited upon NOP receptor antagonism or deletion [179,180,181,182,183,184]. This fits with observations that nociceptin and NOP receptor expression are aberrantly increased in many brain regions of Marchigian Sardinian alcohol-preferring rats, including the central amygdala [185]. However, as with the role of NOP receptors in anxiety, there are also several studies that show that NOP agonists may in fact inhibit reward-seeking behavior related to alcohol and cocaine [186,187,188]. The reason for this discrepancy is not entirely clear, and a recent review article suggested the possibility of some NOP agonists acting as functional antagonists [163]. This then makes it difficult to predict how NOP receptors modulate the reward circuitry by putative actions on calcium channels.
L-type channels have been extensively implicated in reward-seeking behavior and are known to be dysregulated during states of addiction [155, 189,190,191,192]. But there is also evidence that N-type calcium channels play a role, as Cav2.2 knockout mice exhibit reduced ethanol consumption [34]. Along these lines, the mixed N-type/T-type calcium channel inhibitor NP078585 reduced alcohol-induced intoxication and reinstatement, but failed to do so in mice lacking Cav2.2. This indicates that its primary target was indeed the N-type channel [35] and altogether suggests that the Cav2.2 channel is a potential target for the treatment of alcoholism [36]. While an inhibitory action of NOP agonists on Cav2.2 channels would be consistent with some of the results discussed above, the fact that NOP antagonists can also attenuate reward-seeking behavior suggests that Cav2.2 channels are not the exclusive effector of NOP receptors in the context of addiction.
Role of NOP receptors in other neuropsychiatric conditions
Khan et al. [193] suggested a potential role of the NOP system in schizophrenia. Some indications that support this concept are derived from experiments involving a resident-intruder test—an animal model of aggression that is thought to reflect certain symptoms of bipolar disorder, schizophrenia, or PTSD. In this model, systemic injection of NOP antagonists had little effect, but the aggressive behavior of mice that were treated with NOP agonists was greatly increased [194]. The observed effects were similar to those seen with those of para-chlorophenylalanine, an inhibitor of serotonin synthesis [194]. An equivalent increase in aggressive behavior was seen in Cav2.2 null mice, or upon microinjection of a Cav2.2 channel inhibitor into the dorsal raphe nucleus of wild-type mice [33]. Hence, it is tempting to speculate that the aggressive behavior observed with NOP receptor agonists may have involved the inhibition of Cav2.2 channels. This might fit with the idea that Cav2.2 null mice also presented with an increase in serotonin levels in the hypothalamus. It is important to note that a recent study has shown that P/Q-type calcium channels may also play a role in aggressive behavior [195]—a Cav2 channel subtype that is also inhibited by NOP receptors.
Modeling bipolar-like behavior through the administration of psychostimulants affects several neurotransmitter systems, and thus it is difficult to attribute precise cellular and molecular mechanisms that underlie the resultant behavioral phenotype [196]. In mouse models of methylphenidate-induced manic-like behavior, the synthetic full NOP agonist Ro 65-6570 attenuated drug-induced hyperlocomotion, but did not affect spontaneous locomotor activity, whereas NOP receptor antagonists had no effect [197]. Interestingly, methylphenidate mediated similar effects in WT and NOP receptor null mice, indicating that the establishment of the hyperlocomotion does not per se involve a dysregulation of the NOP receptor system [197]. It has been shown that N-type and P/Q-type channel inhibitors prevent methylphenidate-induced locomotion [198, 199], potentially by inhibiting dopamine release. This is thus consistent with a mechanism by which NOP receptor activation mediates stimulant-induced hyperlocomotion by inhibition of Cav2.2 channels.
NOP receptor function in movement disorders
Central vestibular neurons are important for processing motion-related multisensory signals and their transformation into motor commands [200]. Participation of NOP receptors and nociceptin in vestibular afferent excitability has been demonstrated via the locomotor performance of rats after traumatic head injury. Such rats exhibit motor deficits and increased levels of nociceptin in the afferent vestibular nuclei, as well as an increase in NOP receptor expression in brain regions that are associated with the vestibular function [201, 202]. The behavioral and functional deficits that occur following a traumatic brain injury also correlate with the elevated NOP expression in brain regions that are associated with vestibular function, including the motor cortex, the dorsal striatum, and the vestibular nuclei [76]. In cultured rat vestibular afferent neurons, nociceptin inhibits HVA calcium currents (principally, those carried by Cav2.2), without modifying LVA channels, and this inhibitory action occurs via voltage-dependent Gβγ modulation [76]. This fits with the idea that blockers of Cav2.2 channels are neuroprotective during traumatic brain injury [203].
In Parkinson's disease, the degeneration of the dopaminergic neurons of the substantia nigra (SN) pars compacta affects the entire basal ganglia network [204, 205], leading to hyperactivity that is sustained by enhanced glutamatergic inputs [206, 207]. In addition to the basal ganglia, dopamine depletion in the cortical and subcortical motor areas results in the upregulation of the nociceptin-NOP system in the SN, and a down-regulation in the striatum, and this may contribute to associated motor dysfunction [208, 209]. 6-hydroxydopamine (6-OHDA) hemilesioned rats exhibit an elevated expression and release of nociceptin into the SN [210] thereby causing motor deficits. There are conflicting results from pharmacological studies in Parkinson’s disease models. On one hand, exogenous nociceptin or synthetic NOP agonists, when administered systemically, attenuated dyskinesias [211, 212]. However, there was a biphasic effect on motor function, where low doses improved Parkinson-like symptoms, and higher doses disrupted motor coordination and global behavior [212]. Conversely, NOP antagonists also produced antiparkinsonian effects [213, 214] by ameliorating akinesia, bradykinesia, and overall gait problems at low doses, but they inhibited motor activity at intermediate concentrations. Interestingly, these different doses differentially regulated glutamatergic and GABAergic signaling in the SN and the ventromedial thalamus [215]. While the precise contributions of VGCCs to this NOP-dependent regulation of excitatory and inhibitory neurons remain to be determined, there is evidence that 6-OHDA mediates a PKA-dependent upregulation of Cav2.2 channels in the SN [216]. Hence, a putative inhibition of Cav2.2 channels would be consistent with a protective effect of NOP agonists.
Conclusion
The nociceptin-NOP pathway is expressed widely in the peripheral and central nervous system and dysregulated in a number of neurological and neuropsychiatric conditions. A key target of NOP receptor activation are Cav2.2 channels which are typically inhibited upon receptor activation, but whose forward and reverse trafficking is also regulated by NOP receptor association with the channel complex. In the afferent pain pathway, there is a tight correlation between Cav2.2 channel activity and NOP receptor activation in the context of pain signaling (Fig. 2). In conditions such as anxiety, reward-seeking, and motor control the importance of Cav2.2 channels as downstream targets of NOP receptors is more tenuous (Fig. 2). While actions on Cav2.2 channels are consistent with what is known about this VGCC subtype, NOP receptors activate a plethora of downstream signaling cascades that are expected to have complex effects on neurons, glial cells, and neuronal circuits. Future studies involving the activation of these receptors in Cav2.2 null mice will ultimately provide deeper insights into putative linkages.

Graphical overview of key physiological roles of NOP receptors vis a vis a putative action on Cav2.2 channels
Availability of data and materials
Not applicable.
Abbreviations
- NOP:
-
Nociceptin opioid peptide
- GABA:
-
Gamma-aminobutyric acid
- PKA:
-
Protein kinase A
- HVA:
-
High voltage-activated
- LVA:
-
Low voltage-activated
- VTA:
-
Ventral tegmental area
- SN:
-
Substantia nigra
- PTSD:
-
Post-traumatic stress syndrome
- PFC:
-
Prefrontal cortex
- GIRK channel:
-
G protein-coupled inwardly rectifying potassium channel
- VGCC:
-
Voltage-gated calcium channel
- CRMP2:
-
Collapsin mediator protein 2
- SN:
-
Substantia nigra
- 6-OHDA:
-
6-Hydroxydopamine
References
Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev. 2015;67(4):821–70.
Lory P, Mezghrani A. Calcium channelopathies in inherited neurological disorders: relevance to drug screening for acquired channel disorders. IDrugs. 2010;13(7):467–71.
Striessnig J. Voltage-gated Ca(2+)-channel α1-subunit de novo missense mutations: gain or loss of function—implications for potential therapies. Front Synaptic Neurosci. 2021;13: 634760.
Dixon RE, Navedo MF, Binder MD, Santana LF. Mechanisms and physiological implications of cooperative gating of clustered ion channels. Physiol Rev. 2022;102(3):1159–210.
Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3(8): a003947.
Simms BA, Zamponi GW. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron. 2014;82(1):24–45.
Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron. 1992;9(6):1099–115.
Zhang G, Liu JB, Yuan HL, Chen SY, Singer JH, Ke JB. Multiple calcium channel types with unique expression patterns mediate retinal signaling at bipolar cell ribbon synapses. J Neurosci. 2022;42(34):6487–505.
Pangrsic T, Singer JH, Koschak A. Voltage-gated calcium channels: key players in sensory coding in the retina and the inner ear. Physiol Rev. 2018;98(4):2063–96.
Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, et al. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123(4):949–62.
Catterall WA, Wisedchaisri G, Zheng N. The conformational cycle of a prototypical voltage-gated sodium channel. Nat Chem Biol. 2020;16(12):1314–20.
Buraei Z, Yang J. The ß subunit of voltage-gated Ca2+ channels. Physiol Rev. 2010;90(4):1461–506.
Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. 2016;594(19):5369–90.
Davies A, Kadurin I, Alvarez-Laviada A, Douglas L, Nieto-Rostro M, Bauer CS, et al. The alpha2delta subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci U S A. 2010;107(4):1654–9.
Kadurin I, Ferron L, Rothwell SW, Meyer JO, Douglas LR, Bauer CS, et al. Proteolytic maturation of α. Elife. 2016;5:e21143.
Lipscombe D, Lopez-Soto EJ. Epigenetic control of ion channel expression and cell-specific splicing in nociceptors: chronic pain mechanisms and potential therapeutic targets. Channels (Austin). 2021;15(1):156–64.
Bunda A, LaCarubba B, Bertolino M, Akiki M, Bath K, Lopez-Soto J, et al. Cacna1b alternative splicing impacts excitatory neurotransmission and is linked to behavioral responses to aversive stimuli. Mol Brain. 2019;12(1):81.
Gao S, Yan N. Structural basis of the modulation of the voltage-gated calcium ion channel Ca. Angew Chem Int Ed Engl. 2021;60(6):3131–7.
Dong Y, Gao Y, Xu S, Wang Y, Yu Z, Li Y, et al. Closed-state inactivation and pore-blocker modulation mechanisms of human Ca. Cell Rep. 2021;37(5): 109931.
Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, et al. Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 Å resolution. Nature. 2016;537(7619):191–6.
Zhao Y, Huang G, Wu Q, Wu K, Li R, Lei J, et al. Cryo-EM structures of apo and antagonist-bound human Ca. Nature. 2019;576(7787):492–7.
Wheeler DB, Sather WA, Randall A, Tsien RW. Distinctive properties of a neuronal calcium channel and its contribution to excitatory synaptic transmission in the central nervous system. Adv Second Messenger Phosphoprotein Res. 1994;29:155–71.
Ishikawa T, Kaneko M, Shin HS, Takahashi T. Presynaptic N-type and P/Q-type Ca2+ channels mediating synaptic transmission at the calyx of Held of mice. J Physiol. 2005;568(Pt 1):199–209.
Adams ME, Myers RA, Imperial JS, Olivera BM. Toxityping rat brain calcium channels with omega-toxins from spider and cone snail venoms. Biochemistry. 1993;32(47):12566–70.
Olivera BM, Imperial JS, Cruz LJ, Bindokas VP, Venema VJ, Adams ME. Calcium channel-targeted polypeptide toxins. Ann N Y Acad Sci. 1991;635:114–22.
Wang YX, Gao D, Pettus M, Phillips C, Bowersox SS. Interactions of intrathecally administered ziconotide, a selective blocker of neuronal N-type voltage-sensitive calcium channels, with morphine on nociception in rats. Pain. 2000;84(2–3):271–81.
Scott DA, Wright CE, Angus JA. Actions of intrathecal omega-conotoxins CVID, GVIA, MVIIA, and morphine in acute and neuropathic pain in the rat. Eur J Pharmacol. 2002;451(3):279–86.
McGivern JG. Ziconotide: a review of its pharmacology and use in the treatment of pain. Neuropsychiatr Dis Treat. 2007;3(1):69–85.
Saegusa H, Kurihara T, Zong S, Kazuno A, Matsuda Y, Nonaka T, et al. Suppression of inflammatory and neuropathic pain symptoms in mice lacking the N-type Ca2+ channel. Embo j. 2001;20(10):2349–56.
Kim C, Jun K, Lee T, Kim SS, McEnery MW, Chin H, et al. Altered nociceptive response in mice deficient in the alpha(1B) subunit of the voltage-dependent calcium channel. Mol Cell Neurosci. 2001;18(2):235–45.
Hatakeyama S, Wakamori M, Ino M, Miyamoto N, Takahashi E, Yoshinaga T, et al. Differential nociceptive responses in mice lacking the alpha(1B) subunit of N-type Ca(2+) channels. NeuroReport. 2001;12(11):2423–7.
Beuckmann CT, Sinton CM, Miyamoto N, Ino M, Yanagisawa M. N-type calcium channel alpha1B subunit (Cav2.2) knock-out mice display hyperactivity and vigilance state differences. J Neurosci. 2003;23(17):6793–7.
Kim C, Jeon D, Kim YH, Lee CJ, Kim H, Shin HS. Deletion of N-type Ca(2+) channel Ca(v)2.2 results in hyperaggressive behaviors in mice. J Biol Chem. 2009;284(5):2738–45.
Newton PM, Orr CJ, Wallace MJ, Kim C, Shin HS, Messing RO. Deletion of N-type calcium channels alters ethanol reward and reduces ethanol consumption in mice. J Neurosci. 2004;24(44):9862–9.
Newton PM, Zeng L, Wang V, Connolly J, Wallace MJ, Kim C, et al. A blocker of N- and T-type voltage-gated calcium channels attenuates ethanol-induced intoxication, place preference, self-administration, and reinstatement. J Neurosci. 2008;28(45):11712–9.
Newton PM, Messing RO. The N-type calcium channel is a novel target for treating alcohol use disorders. Channels (Austin). 2009;3(2):77–81.
Belardetti F. Evolving therapeutic indications for N-type calcium channel blockers: from chronic pain to alcohol abuse. Future Med Chem. 2010;2(5):791–802.
Brittain JM, Piekarz AD, Wang Y, Kondo T, Cummins TR, Khanna R. An atypical role for collapsin response mediator protein 2 (CRMP-2) in neurotransmitter release via interaction with presynaptic voltage-gated calcium channels. J Biol Chem. 2009;284(45):31375–90.
Brittain JM, Duarte DB, Wilson SM, Zhu W, Ballard C, Johnson PL, et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat Med. 2011;17(7):822–9.
Buchta WC, Moutal A, Hines B, Garcia-Keller C, Smith ACW, Kalivas P, et al. Dynamic CRMP2 regulation of CaV2.2 in the prefrontal cortex contributes to the reinstatement of cocaine seeking. Mol Neurobiol. 2020;57(1):346–57.
Schuwald AM, Nöldner M, Wilmes T, Klugbauer N, Leuner K, Müller WE. Lavender oil-potent anxiolytic properties via modulating voltage dependent calcium channels. PLoS ONE. 2013;8(4): e59998.
Zamani M, Budde T, Bozorgi H. Intracerebroventricular administration of N-type calcium channel blocker ziconotide displays anticonvulsant, anxiolytic, and sedative effects in rats: a preclinical and pilot study. Epilepsy Behav. 2020;111: 107251.
Blazon M, LaCarubba B, Bunda A, Czepiel N, Mallat S, Londrigan L, et al. N-type calcium channels control GABAergic transmission in brain areas related to fear and anxiety. OBM Neurobiol. 2021;5(1):10.21926/obm.neurobiol.2101083.
Jeon D, Kim C, Yang YM, Rhim H, Yim E, Oh U, et al. Impaired long-term memory and long-term potentiation in N-type Ca2+ channel-deficient mice. Genes Brain Behav. 2007;6(4):375–88.
Murakami M, Nakagawasai O, Yanai K, Nunoki K, Tan-No K, Tadano T, et al. Modified behavioral characteristics following ablation of the voltage-dependent calcium channel beta3 subunit. Brain Res. 2007;1160:102–12.
Zhou Y NK, Li W, Takahashi E. Role of Cav2.2-mediated signaling in depressive behaviors. Integr Mol Med. 2015. https://doi.org/10.15761/IMM.1000170.
Tedford HW, Zamponi GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol Rev. 2006;58(4):837–62.
Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, et al. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994;341(1):33–8.
Snyder SH, Pasternak GW. Historical review: opioid receptors. Trends Pharmacol Sci. 2003;24(4):198–205.
Stein C. Opioid receptors. Annu Rev Med. 2016;67:433–51.
Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377(6549):532–5.
Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Orphanin FQ, et al. A neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270(5237):792–4.
Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485(7398):395–9.
Toll L, Bruchas MR, Calo G, Cox BM, Zaveri NT. Nociceptin/orphanin FQ receptor structure, signaling, ligands, functions, and interactions with opioid systems. Pharmacol Rev. 2016;68(2):419–57.
New DC, Wong YH. The ORL1 receptor: molecular pharmacology and signalling mechanisms. Neurosignals. 2002;11(4):197–212.
Liao YY, Trapella C, Chiou LC. 1-Benzyl-N-[3-[spiroisobenzofuran-1(3H),4′-piperidin-1-yl]propyl]pyrrolidine-2-carboxamide (Compound 24) antagonizes NOP receptor-mediated potassium channel activation in rat periaqueductal gray slices. Eur J Pharmacol. 2009;606(1–3):84–9.
Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380(6571):258–62.
Agler HL, Evans J, Tay LH, Anderson MJ, Colecraft HM, Yue DT. G protein-gated inhibitory module of N-type (ca(v)2.2) ca2+ channels. Neuron. 2005;46(6):891–904.
Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385(6615):442–6.
Bourinet E, Zamponi GW, Stea A, Soong TW, Lewis BA, Jones LP, et al. The alpha 1E calcium channel exhibits permeation properties similar to low-voltage-activated calcium channels. J Neurosci. 1996;16(16):4983–93.
Arnot MI, Stotz SC, Jarvis SE, Zamponi GW. Differential modulation of N-type 1B and P/Q-type 1A calcium channels by different G protein subunit isoforms. J Physiol. 2000;527 Pt 2(2(Pt 2)):203–12.
Agler HL, Evans J, Colecraft HM, Yue DT. Custom distinctions in the interaction of G-protein beta subunits with N-type (CaV2.2) versus P/Q-type (CaV2.1) calcium channels. J Gen Physiol. 2003;121(6):495–510.
Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature. 1989;340(6229):153–6.
Brody DL, Patil PG, Mulle JG, Snutch TP, Yue DT. Bursts of action potential waveforms relieve G-protein inhibition of recombinant P/Q-type Ca2+ channels in HEK 293 cells. J Physiol. 1997;499(Pt 3):637–44.
Zamponi GW, Snutch TP. Modulation of voltage-dependent calcium channels by G proteins. Curr Opin Neurobiol. 1998;8(3):351–6.
Meir A, Bell DC, Stephens GJ, Page KM, Dolphin AC. Calcium channel beta subunit promotes voltage-dependent modulation of alpha 1 B by G beta gamma. Biophys J. 2000;79(2):731–46.
Feng ZP, Arnot MI, Doering CJ, Zamponi GW. Calcium channel beta subunits differentially regulate the inhibition of N-type channels by individual Gbeta isoforms. J Biol Chem. 2001;276(48):45051–8.
Meir A, Dolphin AC. Kinetics and Gbetagamma modulation of Ca(v)2.2 channels with different auxiliary beta subunits. Pflugers Arch. 2002;444(1–2):263–75.
Jeong SW, Ikeda SR. Effect of G protein heterotrimer composition on coupling of neurotransmitter receptors to N-type Ca(2+) channel modulation in sympathetic neurons. Proc Natl Acad Sci USA. 2000;97(2):907–12.
Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C, et al. Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci. 2004;7(2):118–25.
Knoflach F, Reinscheid RK, Civelli O, Kemp JA. Modulation of voltage-gated calcium channels by orphanin FQ in freshly dissociated hippocampal neurons. J Neurosci. 1996;16(21):6657–64.
Larsson KP, Olsen UB, Hansen AJ. Nociceptin is a potent inhibitor of N-type Ca(2+) channels in rat sympathetic ganglion neurons. Neurosci Lett. 2000;296(2–3):121–4.
Borgland SL, Connor M, Christie MJ. Nociceptin inhibits calcium channel currents in a subpopulation of small nociceptive trigeminal ganglion neurons in mouse. J Physiol. 2001;536(Pt 1):35–47.
Connor M, Yeo A, Henderson G. The effect of nociceptin on Ca2+ channel current and intracellular Ca2+ in the SH-SY5Y human neuroblastoma cell line. Br J Pharmacol. 1996;118(2):205–7.
Chin JH, Harris K, MacTavish D, Jhamandas JH. Nociceptin/orphanin FQ modulation of ionic conductances in rat basal forebrain neurons. J Pharmacol Exp Ther. 2002;303(1):188–95.
Seseña E, Soto E, Bueno J, Vega R. Nociceptin/orphanin FQ peptide receptor mediates inhibition of N-type calcium currents in vestibular afferent neurons of the rat. J Neurophysiol. 2020;124(6):1605–14.
Ruiz-Velasco V, Puhl HL, Fuller BC, Sumner AD. Modulation of Ca2+ channels by opioid receptor-like 1 receptors natively expressed in rat stellate ganglion neurons innervating cardiac muscle. J Pharmacol Exp Ther. 2005;314(3):987–94.
Altier C, Khosravani H, Evans RM, Hameed S, Peloquin JB, Vartian BA, et al. ORL1 receptor-mediated internalization of N-type calcium channels. Nat Neurosci. 2006;9(1):31–40.
Murali SS, Napier IA, Rycroft BK, Christie MJ. Opioid-related (ORL1) receptors are enriched in a subpopulation of sensory neurons and prolonged activation produces no functional loss of surface N-type calcium channels. J Physiol. 2012;590(7).
Evans RM, You H, Hameed S, Altier C, Mezghrani A, Bourinet E, et al. Heterodimerization of ORL1 and opioid receptors and its consequences for N-type calcium channel regulation. J Biol Chem. 2010;285(2):1032–40.
Zaveri NT. Nociceptin opioid receptor (NOP) as a therapeutic target: progress in translation from preclinical research to clinical utility. J Med Chem. 2016;59(15):7011–28.
Witkin JM, Rorick-Kehn LM, Benvenga MJ, Adams BL, Gleason SD, Knitowski KM, et al. Preclinical findings predicting efficacy and side-effect profile of LY2940094, an antagonist of nociceptin receptors. Pharmacol Res Perspect. 2016;4(6): e00275.
Post A, Smart TS, Krikke-Workel J, Dawson GR, Harmer CJ, Browning M, et al. A selective nociceptin receptor antagonist to treat depression: evidence from preclinical and clinical studies. Neuropsychopharmacology. 2016;41(7):1803–12.
Lambert DG. Mixed mu-nociceptin/orphanin FQ opioid receptor agonists and the search for the analgesic holy grail. Br J Anaesth. 2019;122(6):e95–7.
Ding H, Trapella C, Kiguchi N, Hsu FC, Caló G, Ko MC. Functional profile of systemic and intrathecal cebranopadol in nonhuman primates. Anesthesiology. 135: Copyright© 2021, the American Society of Anesthesiologists. All Rights Reserved.; 2021. p. 482–93.
Ozawa A, Brunori G, Mercatelli D, Wu J, Cippitelli A, Zou B, et al. Knock-in mice with NOP-eGFP receptors identify receptor cellular and regional localization. J Neurosci. 2015;35(33):11682–93.
Neal CR Jr, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Akil H, et al. Opioid receptor-like (ORL1) receptor distribution in the rat central nervous system: comparison of ORL1 receptor mRNA expression with (125)I-[(14)Tyr]-orphanin FQ binding. J Comp Neurol. 1999;412(4):563–605.
Neal CR Jr, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Watson SJ Jr. Localization of orphanin FQ (nociceptin) peptide and messenger RNA in the central nervous system of the rat. J Comp Neurol. 1999;406(4):503–47.
Ozawa A, Brunori G, Cippitelli A, Toll N, Schoch J, Kieffer BL, et al. Analysis of the distribution of spinal NOP receptors in a chronic pain model using NOP-eGFP knock-in mice. Br J Pharmacol. 2018;175(13):2662–75.
Jia Y, Linden DR, Serie JR, Seybold VS. Nociceptin/orphanin FQ binding increases in superficial laminae of the rat spinal cord during persistent peripheral inflammation. Neurosci Lett. 1998;250(1):21–4.
Briscini L, Corradini L, Ongini E, Bertorelli R. Up-regulation of ORL-1 receptors in spinal tissue of allodynic rats after sciatic nerve injury. Eur J Pharmacol. 2002;447(1):59–65.
Mika J, Schäfer MK, Obara I, Weihe E, Przewlocka B. Morphine and endomorphin-1 differently influence pronociceptin/orphanin FQ system in neuropathic rats. Pharmacol Biochem Behav. 2004;78(1):171–8.
Ko MH, Kim YH, Woo RS, Kim KW. Quantitative analysis of nociceptin in blood of patients with acute and chronic pain. NeuroReport. 2002;13(13):1631–3.
Courteix C, Coudoré-Civiale MA, Privat AM, Pélissier T, Eschalier A, Fialip J. Evidence for an exclusive antinociceptive effect of nociceptin/orphanin FQ, an endogenous ligand for the ORL1 receptor, in two animal models of neuropathic pain. Pain. 2004;110(1–2):236–45.
Yamamoto T, Nozaki-Taguchi N, Kimura S. Analgesic effect of intrathecally administered nociceptin, an opioid receptor-like1 receptor agonist, in the rat formalin test. Neuroscience. 1997;81(1):249–54.
Wu Q, Liu L. ORL(1) activation mediates a novel ORL(1) receptor agonist SCH221510 analgesia in neuropathic pain in rats. J Mol Neurosci. 2018;66(1):10–6.
Chen Y, Sommer C. Activation of the nociceptin opioid system in rat sensory neurons produces antinociceptive effects in inflammatory pain: involvement of inflammatory mediators. J Neurosci Res. 2007;85(7):1478–88.
Reiss D, Wichmann J, Tekeshima H, Kieffer BL, Ouagazzal AM. Effects of nociceptin/orphanin FQ receptor (NOP) agonist, Ro64-6198, on reactivity to acute pain in mice: comparison to morphine. Eur J Pharmacol. 2008;579(1–3):141–8.
Hayashi S, Nakata E, Morita A, Mizuno K, Yamamura K, Kato A, et al. Discovery of {1-[4-(2-{hexahydropyrrolo[3,4-c]pyrrol-2(1H)-yl}-1H-benzimidazol-1-yl)piperidin-1-yl]cyclooctyl}methanol, systemically potent novel non-peptide agonist of nociceptin/orphanin FQ receptor as analgesic for the treatment of neuropathic pain: design, synthesis, and structure-activity relationships. Bioorg Med Chem. 2010;18(21):7675–99.
El Daibani A, Che T. Spotlight on nociceptin/orphanin FQ receptor in the treatment of pain. Molecules. 2022;27(3):595.
Ko MC, Wei H, Woods JH, Kennedy RT. Effects of intrathecally administered nociceptin/orphanin FQ in monkeys: behavioral and mass spectrometric studies. J Pharmacol Exp Ther. 2006;318(3):1257–64.
Ko MC, Naughton NN. Antinociceptive effects of nociceptin/orphanin FQ administered intrathecally in monkeys. J Pain. 2009;10(5):509–16.
Ko MC, Woods JH, Fantegrossi WE, Galuska CM, Wichmann J, Prinssen EP. Behavioral effects of a synthetic agonist selective for nociceptin/orphanin FQ peptide receptors in monkeys. Neuropsychopharmacology. 2009;34(9):2088–96.
Schröder W, Lambert DG, Ko MC, Koch T. Functional plasticity of the N/OFQ-NOP receptor system determines analgesic properties of NOP receptor agonists. Br J Pharmacol. 2014;171(16):3777–800.
Kiguchi N, Ko MC. Effects of NOP-related ligands in nonhuman primates. Handb Exp Pharmacol. 2019;254:323–43.
Pettersson LM, Sundler F, Danielsen N. Expression of orphanin FQ/nociceptin and its receptor in rat peripheral ganglia and spinal cord. Brain Res. 2002;945(2):266–75.
Ahmadi S, Liebel JT, Zeilhofer HU. The role of the ORL1 receptor in the modulation of spinal neurotransmission by nociceptin/orphanin FQ and nocistatin. Eur J Pharmacol. 2001;412(1):39–44.
Zeilhofer HU, Selbach UM, Guhring H, Erb K, Ahmadi S. Selective suppression of inhibitory synaptic transmission by nocistatin in the rat spinal cord dorsal horn. J Neurosci. 2000;20(13):4922–9.
Luo C, Kumamoto E, Furue H, Chen J, Yoshimura M. Nociceptin inhibits excitatory but not inhibitory transmission to substantia gelatinosa neurones of adult rat spinal cord. Neuroscience. 2002;109(2):349–58.
Nishi M, Houtani T, Noda Y, Mamiya T, Sato K, Doi T, et al. Unrestrained nociceptive response and disregulation of hearing ability in mice lacking the nociceptin/orphaninFQ receptor. Embo J. 1997;16(8):1858–64.
Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc Natl Acad Sci USA. 1996;93(4):1486–91.
Ueda H, Yamaguchi T, Tokuyama S, Inoue M, Nishi M, Takeshima H. Partial loss of tolerance liability to morphine analgesia in mice lacking the nociceptin receptor gene. Neurosci Lett. 1997;237(2–3):136–8.
Ueda H, Inoue M, Takeshima H, Iwasawa Y. Enhanced spinal nociceptin receptor expression develops morphine tolerance and dependence. J Neurosci. 2000;20(20):7640–7.
Lutfy K, Hossain SM, Khaliq I, Maidment NT. Orphanin FQ/nociceptin attenuates the development of morphine tolerance in rats. Br J Pharmacol. 2001;134(3):529–34.
Murphy NP, Ly HT, Maidment NT. Intracerebroventricular orphanin FQ/nociceptin suppresses dopamine release in the nucleus accumbens of anaesthetized rats. Neuroscience. 1996;75(1):1–4.
Zaratin PF, Petrone G, Sbacchi M, Garnier M, Fossati C, Petrillo P, et al. Modification of nociception and morphine tolerance by the selective opiate receptor-like orphan receptor antagonist (-)-cis-1-methyl-7-[[4-(2,6-dichlorophenyl)piperidin-1-yl]methyl]-6,7,8,9-tetrahydro-5H-benzocyclohepten-5-ol (SB-612111). J Pharmacol Exp Ther. 2004;308(2):454–61.
Mogil JS, Grisel JE, Reinscheid RK, Civelli O, Belknap JK, Grandy DK. Orphanin FQ is a functional anti-opioid peptide. Neuroscience. 1996;75(2):333–7.
Mogil JS, Grisel JE, Zhangs G, Belknap JK, Grandy DK. Functional antagonism of mu-, delta- and kappa-opioid antinociception by orphanin FQ. Neurosci Lett. 1996;214(2–3):131–4.
Gouardères C, Tafani JA, Meunier JC, Jhamandas K, Zajac JM. Nociceptin receptors in the rat spinal cord during morphine tolerance. Brain Res. 1999;838(1–2):85–94.
Ray SB, Gupta YK, Wadhwa S. Expression of opioid receptor-like 1 (ORL1) & mu opioid receptors in the spinal cord of morphine tolerant mice. Indian J Med Res. 2005;121(3):194–202.
Khroyan TV, Zaveri NT, Polgar WE, Orduna J, Olsen C, Jiang F, et al. SR 16435 [1-(1-(bicyclo[3.3.1]nonan-9-yl)piperidin-4-yl)indolin-2-one], a novel mixed nociceptin/orphanin FQ/mu-opioid receptor partial agonist: analgesic and rewarding properties in mice. J Pharmacol Exp Ther. 2007;320(2):934–43.
Bird MF, McDonald J, Horley B, O’Doherty JP, Fraser B, Gibson CL, et al. MOP and NOP receptor interaction: studies with a dual expression system and bivalent peptide ligands. PLoS ONE. 2022;17(1): e0260880.
Sukhtankar DD, Zaveri NT, Husbands SM, Ko MC. Effects of spinally administered bifunctional nociceptin/orphanin FQ peptide receptor/μ-opioid receptor ligands in mouse models of neuropathic and inflammatory pain. J Pharmacol Exp Ther. 2013;346(1):11–22.
Rizzi A, Nazzaro C, Marzola GG, Zucchini S, Trapella C, Guerrini R, et al. Endogenous nociceptin/orphanin FQ signalling produces opposite spinal antinociceptive and supraspinal pronociceptive effects in the mouse formalin test: pharmacological and genetic evidences. Pain. 2006;124(1–2):100–8.
Rizzi A, Marzola G, Bigoni R, Guerrini R, Salvadori S, Mogil JS, et al. Endogenous nociceptin signaling and stress-induced analgesia. NeuroReport. 2001;12(14):3009–13.
Xie X, Wisor JP, Hara J, Crowder TL, LeWinter R, Khroyan TV, et al. Hypocretin/orexin and nociceptin/orphanin FQ coordinately regulate analgesia in a mouse model of stress-induced analgesia. J Clin Invest. 2008;118(7):2471–81.
Gerashchenko D, Horvath TL, Xie XS. Direct inhibition of hypocretin/orexin neurons in the lateral hypothalamus by nociceptin/orphanin FQ blocks stress-induced analgesia in rats. Neuropharmacology. 2011;60(4):543–9.
Zhang Y, Gandhi PR, Standifer KM. Increased nociceptive sensitivity and nociceptin/orphanin FQ levels in a rat model of PTSD. Mol Pain. 2012;8:76.
Zhang Y, Simpson-Durand CD, Standifer KM. Nociceptin/orphanin FQ peptide receptor antagonist JTC-801 reverses pain and anxiety symptoms in a rat model of post-traumatic stress disorder. Br J Pharmacol. 2015;172(2):571–82.
Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010;35(1):169–91.
Park JY, Chae S, Kim CS, Kim YJ, Yi HJ, Han E, et al. Role of nociceptin/orphanin FQ and nociceptin opioid peptide receptor in depression and antidepressant effects of nociceptin opioid peptide receptor antagonists. Korean J Physiol Pharmacol. 2019;23(6):427–48.
LM S, I L. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology. 2010;35(1):169-91.
Tovote P, Fadok JP, Lüthi A. Neuronal circuits for fear and anxiety. Nat Rev Neurosci. 2015;16(6):317–31.
Calhoon GG, Tye KM. Resolving the neural circuits of anxiety. Nat Neurosci. 2015;18(10):1394–404.
Nuss P. Anxiety disorders and GABA neurotransmission: a disturbance of modulation. Neuropsychiatr Dis Treat. 2015;11:165–75.
Kaur S, Singh R. Role of different neurotransmitters in anxiety: a systematic review. Int J Pharmaceut Sci Res. 2017;8(2):411–21.
Liu WZ, Zhang WH, Zheng ZH, Zou JX, Liu XX, Huang SH, et al. Identification of a prefrontal cortex-to-amygdala pathway for chronic stress-induced anxiety. Nat Commun. 2020;11(1):2221.
Möhler H. The GABA system in anxiety and depression and its therapeutic potential. Neuropharmacology. 2012;62(1):42–53.
Donica CL, Awwad HO, Thakker DR, Standifer KM. Cellular mechanisms of nociceptin/orphanin FQ (N/OFQ) peptide (NOP) receptor regulation and heterologous regulation by N/OFQ. Mol Pharmacol. 2013;83(5):907–18.
Meis S, Pape HC. Control of glutamate and GABA release by nociceptin/orphanin FQ in the rat lateral amygdala. J Physiol. 2001;532(Pt 3):701–12.
Jenck F, Moreau JL, Martin JR, Kilpatrick GJ, Reinscheid RK, Monsma FJ Jr, et al. Orphanin FQ acts as an anxiolytic to attenuate behavioral responses to stress. Proc Natl Acad Sci USA. 1997;94(26):14854–8.
Hirao A, Imai A, Sugie Y, Tamura T, Shimokawa H, Toide K. Pharmacological properties of a novel nociceptin/orphanin FQ receptor agonist, 2-(3,5-dimethylpiperazin-1-yl)-1-[1-(1-methylcyclooctyl)piperidin-4-yl]-1H-benzimidazole, with anxiolytic potential. Eur J Pharmacol. 2008;579(1–3):189–95.
Varty GB, Lu SX, Morgan CA, Cohen-Williams ME, Hodgson RA, Smith-Torhan A, et al. The anxiolytic-like effects of the novel, orally active nociceptin opioid receptor agonist 8-[bis(2-methylphenyl)methyl]-3-phenyl-8-azabicyclo[3.2.1]octan-3-ol (SCH 221510). J Pharmacol Exp Ther. 2008;326(2):672–82.
Raffaele M, Kovacovicova K, Biagini T, Lo Re O, Frohlich J, Giallongo S, et al. Nociceptin/orphanin FQ opioid receptor (NOP) selective ligand MCOPPB links anxiolytic and senolytic effects. Geroscience. 2022;44(1):463–83.
Gavioli EC, Holanda VAD, Ruzza C. NOP ligands for the treatment of anxiety and mood disorders. Handb Exp Pharmacol. 2019;254:233–57.
Fernandez F, Misilmeri MA, Felger JC, Devine DP. Nociceptin/orphanin FQ increases anxiety-related behavior and circulating levels of corticosterone during neophobic tests of anxiety. Neuropsychopharmacology. 2004;29(1):59–71.
Green MK, Barbieri EV, Brown BD, Chen KW, Devine DP. Roles of the bed nucleus of stria terminalis and of the amygdala in N/OFQ-mediated anxiety and HPA axis activation. Neuropeptides. 2007;41(6):399–410.
Cullen CL, Burne TH, Lavidis NA, Moritz KM. Low dose prenatal ethanol exposure induces anxiety-like behaviour and alters dendritic morphology in the basolateral amygdala of rat offspring. PLoS ONE. 2013;8(1): e54924.
Zhou R, Wang S, Zhu X. Prenatal ethanol exposure attenuates GABAergic inhibition in basolateral amygdala leading to neuronal hyperexcitability and anxiety-like behavior of adult rat offspring. Neuroscience. 2010;170(3):749–57.
Baculis BC, Diaz MR, Valenzuela CF. Third trimester-equivalent ethanol exposure increases anxiety-like behavior and glutamatergic transmission in the basolateral amygdala. Pharmacol Biochem Behav. 2015;137:78–85.
Wille-Bille A, Miranda-Morales RS, Pucci M, Bellia F, D’Addario C, Pautassi RM. Prenatal ethanol induces an anxiety phenotype and alters expression of dynorphin & nociceptin/orphanin FQ genes. Prog Neuropsychopharmacol Biol Psychiatry. 2018;85:77–88.
Aghaie CI, Hausknecht KA, Wang R, Dezfuli PH, Haj-Dahmane S, Kane CJM, et al. Prenatal ethanol exposure and postnatal environmental intervention alter dopaminergic neuron and microglia morphology in the ventral tegmental area during adulthood. Alcohol Clin Exp Res. 2020;44(2):435–44.
Caputi FF, Stopponi S, Rullo L, Palmisano M, Ubaldi M, Candeletti S, et al. Dysregulation of nociceptin/orphanin FQ and dynorphin systems in the extended amygdala of alcohol preferring marchigian sardinian (msP) Rats. Int J Mol Sci. 2021;22(5):2448.
Takahashi E, Niimi K. Modulators of voltage-dependent calcium channels for the treatment of nervous system diseases. Recent Pat CNS Drug Discov. 2009;4(2):96–111.
Zamponi GW. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov. 2016;15(1):19–34.
Shinnick-Gallagher P, McKernan MG, Xie J, Zinebi F. L-type voltage-gated calcium channels are involved in the in vivo and in vitro expression of fear conditioning. Ann N Y Acad Sci. 2003;985:135–49.
Babaev O, PilettiChatain C, Krueger-Burg D. Inhibition in the amygdala anxiety circuitry. Exp Mol Med. 2018;50(4):1–16.
Schroeder BW, Shinnick-Gallagher P. Fear memories induce a switch in stimulus response and signaling mechanisms for long-term potentiation in the lateral amygdala. Eur J Neurosci. 2004;20(2):549–56.
Steimer T. Animal models of anxiety disorders in rats and mice: some conceptual issues. Dialogues Clin Neurosci. 2011;13(4):495–506.
Indovina I, Robbins TW, Núñez-Elizalde AO, Dunn BD, Bishop SJ. Fear-conditioning mechanisms associated with trait vulnerability to anxiety in humans. Neuron. 2011;69(3):563–71.
Sherin JE, Nemeroff CB. Post-traumatic stress disorder: the neurobiological impact of psychological trauma. Dialogues Clin Neurosci. 2011;13(3):263–78.
Al Yacoub ON, Awwad HO, Zhang Y, Standifer KM. Therapeutic potential of nociceptin/orphanin FQ peptide (NOP) receptor modulators for treatment of traumatic brain injury, traumatic stress, and their co-morbidities. Pharmacol Ther. 2022;231: 107982.
Ubaldi M, Cannella N, Borruto AM, Petrella M, Micioni Di Bonaventura MV, Soverchia L, et al. Role of nociceptin/orphanin FQ-NOP receptor system in the regulation of stress-related disorders. Int J Mol Sci. 2021;22(23):12956.
Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22(12):5239–49.
Busquet P, Hetzenauer A, Sinnegger-Brauns MJ, Striessnig J, Singewald N. Role of L-type Ca2+ channel isoforms in the extinction of conditioned fear. Learn Mem. 2008;15(5):378–86.
Davis SE, Bauer EP. L-type voltage-gated calcium channels in the basolateral amygdala are necessary for fear extinction. J Neurosci. 2012;32(39):13582–6.
Temme SJ, Murphy GG. The L-type voltage-gated calcium channel CaV1.2 mediates fear extinction and modulates synaptic tone in the lateral amygdala. Learn Memory (Cold Spring Harbor, NY). 2017;24(11):580–8.
Marks WN, Zabder NK, Snutch TP, Howland JG. T-type calcium channels regulate the acquisition and recall of conditioned fear in male, Wistar rats. Behav Brain Res. 2020;393: 112747.
Moon AL, Brydges NM, Wilkinson LS, Hall J, Thomas KL. Cacna1c hemizygosity results in aberrant fear conditioning to neutral stimuli. Schizophr Bull. 2020;46(5):1231–8.
Popik B, Amorim FE, Amaral OB, De Oliveira Alvares L. Shifting from fear to safety through deconditioning-update. Elife. 2020;9:e51207.
Sofuoglu M, Rosenheck R, Petrakis I. Pharmacological treatment of comorbid PTSD and substance use disorder: recent progress. Addict Behav. 2014;39(2):428–33.
Arias-Carrión O, Stamelou M, Murillo-Rodríguez E, Menéndez-González M, Pöppel E. Dopaminergic reward system: a short integrative review. Int Arch Med. 2010;3:24.
Gardner EL, Ashby CR Jr. Heterogeneity of the mesotelencephalic dopamine fibers: physiology and pharmacology. Neurosci Biobehav Rev. 2000;24(1):115–8.
Kallupi M, Varodayan FP, Oleata CS, Correia D, Luu G, Roberto M. Nociceptin/orphanin FQ decreases glutamate transmission and blocks ethanol-induced effects in the central amygdala of naive and ethanol-dependent rats. Neuropsychopharmacology. 2014;39(5):1081–92.
Roberto M, Siggins GR. Nociceptin/orphanin FQ presynaptically decreases GABAergic transmission and blocks the ethanol-induced increase of GABA release in central amygdala. Proc Natl Acad Sci USA. 2006;103(25):9715–20.
Murphy NP, Maidment NT. Orphanin FQ/nociceptin modulation of mesolimbic dopamine transmission determined by microdialysis. J Neurochem. 1999;73(1):179–86.
Zheng F, Grandy DK, Johnson SW. Actions of orphanin FQ/nociceptin on rat ventral tegmental area neurons in vitro. Br J Pharmacol. 2002;136(7):1065–71.
Domi A, Lunerti V, Petrella M, Domi E, Borruto AM, Ubaldi M, et al. Genetic deletion or pharmacological blockade of nociceptin/orphanin FQ receptors in the ventral tegmental area attenuates nicotine-motivated behaviour. Br J Pharmacol. 2022;179(11):2647–58.
Ciccocioppo R, Economidou D, Fedeli A, Angeletti S, Weiss F, Heilig M, et al. Attenuation of ethanol self-administration and of conditioned reinstatement of alcohol-seeking behaviour by the antiopioid peptide nociceptin/orphanin FQ in alcohol-preferring rats. Psychopharmacology. 2004;172(2):170–8.
Borruto AM, Fotio Y, Stopponi S, Petrella M, De Carlo S, Domi A, et al. NOP receptor antagonism attenuates reinstatement of alcohol-seeking through modulation of the mesolimbic circuitry in male and female alcohol-preferring rats. Neuropsychopharmacology. 2021;46(12):2121–31.
Borruto AM, Fotio Y, Stopponi S, Brunori G, Petrella M, Caputi FF, et al. NOP receptor antagonism reduces alcohol drinking in male and female rats through mechanisms involving the central amygdala and ventral tegmental area. Br J Pharmacol. 2020;177(7):1525–37.
Brunori G, Weger M, Schoch J, Targowska-Duda K, Barnes M, Borruto AM, et al. NOP receptor antagonists decrease alcohol drinking in the dark in C57BL/6J mice. Alcohol Clin Exp Res. 2019;43(10):2167–78.
Rorick-Kehn LM, Ciccocioppo R, Wong CJ, Witkin JM, Martinez-Grau MA, Stopponi S, et al. A novel, orally bioavailable nociceptin receptor antagonist, LY2940094, reduces ethanol self-administration and ethanol seeking in animal models. Alcohol Clin Exp Res. 2016;40(5):945–54.
Kallupi M, Scuppa G, de Guglielmo G, Calò G, Weiss F, Statnick MA, et al. Genetic deletion of the nociceptin/orphanin FQ receptor in the rat confers resilience to the development of drug addiction. Neuropsychopharmacology. 2017;42(3):695–706.
Economidou D, Hansson AC, Weiss F, Terasmaa A, Sommer WH, Cippitelli A, et al. Dysregulation of nociceptin/orphanin FQ activity in the amygdala is linked to excessive alcohol drinking in the rat. Biol Psychiatry. 2008;64(3):211–8.
de Guglielmo G, Martin-Fardon R, Teshima K, Ciccocioppo R, Weiss F. MT-7716, a potent NOP receptor agonist, preferentially reduces ethanol seeking and reinforcement in post-dependent rats. Addict Biol. 2015;20(4):643–51.
Li H, Scuppa G, Shen Q, Masi A, Nasuti C, Cannella N, et al. NOP receptor agonist Ro 64–6198 decreases escalation of cocaine self-administration in rats genetically selected for alcohol preference. Front Psychiatry. 2019;10:176.
Cippitelli A, Barnes M, Zaveri NT, Toll L. Potent and selective NOP receptor activation reduces cocaine self-administration in rats by lowering hedonic set point. Addict Biol. 2020;25(6): e12844.
Baliño P, Pastor R, Aragon CM. Participation of L-type calcium channels in ethanol-induced behavioral stimulation and motor incoordination: effects of diltiazem and verapamil. Behav Brain Res. 2010;209(2):196–204.
Uhrig S, Vandael D, Marcantoni A, Dedic N, Bilbao A, Vogt MA, et al. Differential roles for L-type calcium channel subtypes in alcohol dependence. Neuropsychopharmacology. 2017;42(5):1058–69.
Martínez-Rivera A, Hao J, Tropea TF, Giordano TP, Kosovsky M, Rice RC, et al. Enhancing VTA Ca(v)1.3 L-type Ca(2+) channel activity promotes cocaine and mood-related behaviors via overlapping AMPA receptor mechanisms in the nucleus accumbens. Mol Psychiatry. 2017;22(12):1735–45.
Burgdorf CE, Schierberl KC, Lee AS, Fischer DK, Van Kempen TA, Mudragel V, et al. Extinction of contextual cocaine memories requires Cav1.2 within D1R-expressing cells and recruits hippocampal Cav1.2-dependent signaling mechanisms. J Neurosci. 2017;37(49):11894–911.
Khan MS, Boileau I, Kolla N, Mizrahi R. A systematic review of the role of the nociceptin receptor system in stress, cognition, and reward: relevance to schizophrenia. Transl Psychiatry. 2018;8(1):38.
Silva EF, Silva AI, Asth L, Souza LS, Zaveri NT, Guerrini R, et al. Nociceptin/orphanin FQ receptor agonists increase aggressiveness in the mouse resident-intruder test. Behav Brain Res. 2019;356:120–6.
Bohne P, Volkmann A, Schwarz MK, Mark MD. Deletion of the P/Q-type calcium channel from serotonergic neurons drives male aggression in mice. J Neurosci. 2022;42(34):6637–53.
Freund N, Juckel G. Bipolar disorder: its etiology and how to model in rodents. Methods Mol Biol. 2019;2011:61–77.
Asth L, Tiago PRF, Costa LRF, Holanda VAD, Pacifico S, Zaveri NT, et al. Effects of non-peptide nociceptin/orphanin FQ receptor ligands on methylphenidate-induced hyperactivity in mice: implications for bipolar disorders. Neuropeptides. 2020;82: 102059.
Ogura H, Furuya Y, Teramoto T, Niidome T, Nishizawa Y, Yamanishi Y. Peptide N- and P/Q-type Ca2+ blockers inhibit stimulant-induced hyperactivity in mice. Peptides. 1998;19(6):1017–22.
Yamada K, Teraoka T, Morita S, Hasegawa T, Nabeshima T. Omega-conotoxin GVIA inhibits the methylphenidate-induced but not methamphetamine-induced behavior. Neurosci Lett. 1994;165(1–2):191–4.
Straka H, Vibert N, Vidal PP, Moore LE, Dutia MB. Intrinsic membrane properties of vertebrate vestibular neurons: function, development and plasticity. Prog Neurobiol. 2005;76(6):349–92.
Witta J, Buzas B, Cox BM. Traumatic brain injury induces nociceptin/orphanin FQ expression in neurons of the rat cerebral cortex. J Neurotrauma. 2003;20(6):523–32.
Awwad HO, Durand CD, Gonzalez LP, Tompkins P, Zhang Y, Lerner MR, et al. Post-blast treatment with Nociceptin/Orphanin FQ peptide (NOP) receptor antagonist reduces brain injury-induced hypoxia and signaling proteins in vestibulomotor-related brain regions. Behav Brain Res. 2018;340:183–94.
Gurkoff G, Shahlaie K, Lyeth B, Berman R. Voltage-gated calcium channel antagonists and traumatic brain injury. Pharmaceuticals (Basel). 2013;6(7):788–812.
Guatteo E, Cucchiaroni ML, Mercuri NB. Substantia nigra control of basal ganglia nuclei. J Neural Transm Suppl. 2009;73:91–101.
Meoni S, Cury RG, Moro E. New players in basal ganglia dysfunction in Parkinson’s disease. Prog Brain Res. 2020;252:307–27.
Blandini F, Nappi G, Tassorelli C, Martignoni E. Functional changes of the basal ganglia circuitry in Parkinson’s disease. Prog Neurobiol. 2000;62(1):63–88.
Fasano A, Mazzoni A, Falotico E. Reaching and grasping movements in Parkinson’s disease: a review. J Parkinsons Dis. 2022;12(4):1083–113.
Mercatelli D, Bezard E, Eleopra R, Zaveri NT, Morari M. Managing Parkinson’s disease: moving ON with NOP. Br J Pharmacol. 2020;177(1):28–47.
Mercatelli D, Pisanò CA, Novello S, Morari M. NOP receptor ligands and Parkinson’s disease. Handb Exp Pharmacol. 2019;254:213–32.
Marti M, Mela F, Fantin M, Zucchini S, Brown JM, Witta J, et al. Blockade of nociceptin/orphanin FQ transmission attenuates symptoms and neurodegeneration associated with Parkinson’s disease. J Neurosci. 2005;25(42):9591–601.
Marti M, Rodi D, Li Q, Guerrini R, Fasano S, Morella I, et al. Nociceptin/orphanin FQ receptor agonists attenuate L-DOPA-induced dyskinesias. J Neurosci. 2012;32(46):16106–19.
Arcuri L, Novello S, Frassineti M, Mercatelli D, Pisanò CA, Morella I, et al. Anti-Parkinsonian and anti-dyskinetic profiles of two novel potent and selective nociceptin/orphanin FQ receptor agonists. Br J Pharmacol. 2018;175(5):782–96.
Marti M, Mela F, Budri M, Volta M, Malfacini D, Molinari S, et al. Acute and chronic antiparkinsonian effects of the novel nociceptin/orphanin FQ receptor antagonist NiK-21273 in comparison with SB-612111. Br J Pharmacol. 2013;168(4):863–79.
Mabrouk OS, Viaro R, Volta M, Ledonne A, Mercuri N, Morari M. Stimulation of δ opioid receptor and blockade of nociceptin/orphanin FQ receptor synergistically attenuate parkinsonism. J Neurosci. 2014;34(39):12953–62.
Volta M, Marti M, McDonald J, Molinari S, Camarda V, Pelà M, et al. Pharmacological profile and antiparkinsonian properties of the novel nociceptin/orphanin FQ receptor antagonist 1-[1-cyclooctylmethyl-5-(1-hydroxy-1-methyl-ethyl)-1,2,3,6-tetrahydro-pyridin-4-yl]-3-ethyl-1,3-dihydro-benzoimidazol-2-one (GF-4). Peptides. 2010;31(6):1194–204.
Qu L, Wang Y, Zhang HT, Li N, Wang Q, Yang Q, et al. 6-OHDA induced calcium influx through N-type calcium channel alters membrane properties via PKA pathway in substantia nigra pars compacta dopaminergic neurons. Neurosci Lett. 2014;575:1–6.
Acknowledgements
Not applicable.
Funding
This work was supported by grants to GWZ from the Canadian Institutes of Health Research. GWZ holds a Canada Research Chair. FTTA holds an Eyes High Fellowship from the University of Calgary.
Author information
Authors and Affiliations
Contributions
FTTA and GWZ wrote the manuscript, ESC, ED, and IAS provided intellectual contributions and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Caminski, E.S., Antunes, F.T.T., Souza, I.A. et al. Regulation of N-type calcium channels by nociceptin receptors and its possible role in neurological disorders. Mol Brain 15, 95 (2022). https://doi.org/10.1186/s13041-022-00982-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-022-00982-z