
Abstract
Neuroinflammation by activated microglia and astrocytes plays a critical role in progression of amyotrophic lateral sclerosis (ALS). Interleukin-19 (IL-19) is a negative-feedback regulator that limits pro-inflammatory responses of microglia in an autocrine and paracrine manner, but it remains unclear how IL-19 contributes to ALS pathogenesis. We investigated the role of IL-19 in ALS using transgenic mice carrying human superoxide dismutase 1 with the G93A mutation (SOD1G93A Tg mice). We generated IL-19–deficient SOD1G93A Tg (IL-19−/−/SOD1G93A Tg) mice by crossing SOD1G93A Tg mice with IL-19−/− mice, and then evaluated disease progression, motor function, survival rate, and pathological and biochemical alternations in the resultant mice. In addition, we assessed the effect of IL-19 on glial cells using primary microglia and astrocyte cultures from the embryonic brains of SOD1G93A Tg mice and IL-19−/−/SOD1G93A Tg mice. Expression of IL-19 in primary microglia and lumbar spinal cord was higher in SOD1G93A Tg mice than in wild-type mice. Unexpectedly, IL-19−/−/SOD1G93A Tg mice exhibited significant improvement of motor function. Ablation of IL-19 in SOD1G93A Tg mice increased expression of both neurotoxic and neuroprotective factors, including tumor necrosis factor-α (TNF-α), IL-1β, glial cell line–derived neurotrophic factor (GDNF), and transforming growth factor β1, in lumbar spinal cord. Primary microglia and astrocytes from IL-19−/−/SOD1G93A Tg mice expressed higher levels of TNF-α, resulting in release of GDNF from astrocytes. Inhibition of IL-19 signaling may alleviate ALS symptoms.
Similar content being viewed by others
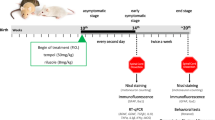

Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease characterized by motor neuron degeneration, which leads to progressive muscle weakness, amyotrophy, and death from respiratory paralysis within 3–5 years from onset [1]. ALS is thought to be caused by a multi-factorial mechanism, including glial neuroinflammation, oxidative stress, glutamate-mediated excitotoxicity, mitochondrial abnormalities, and impaired axonal transport [2]. In particular, recent evidence indicates that non-neuronal cells play a pivotal role in ALS pathogenesis [3]. Restricted expression of superoxide dismutase 1 (SOD1) mutation in neurons is not sufficient to induce ALS pathology [4], but wild-type neurons exhibit an ALS phenotype when their surroundings are altered by glial cells carrying the SOD1 mutation [5].
Interleukin-19 (IL-19) is a member of the IL-10 family of cytokines, which includes IL-20, IL-22, IL-26, IL-28A, IL-28B, and IL-29 [6]. Recently, IL-19 was grouped into the IL-20 subfamily, which includes IL-20, IL-22, and IL-24, because these cytokines share common receptor subunits (IL-20 receptor α/β heterodimer) and signaling pathways [7, 8]. IL-19 is an anti-inflammatory cytokine, produced by activated microglia and macrophages, that acts as a negative-feedback regulator to limit pro-inflammatory responses by these cells in an autocrine and paracrine manner [9, 10]. In the central nervous system (CNS), microglia and astrocytes are thought to be the main source of IL-19 production [9, 11]. In regard to its anti-inflammatory role, IL-19 inhibits the symptoms of animal models of inflammatory bowel disease [10, 12], suppresses hapten-dependent skin inflammation in contact hypersensitivity [13], decreases brain infarction area in a mouse model of cerebral ischemia [14], and improves locomotor function in a mouse model of spinal cord injury [15]. On the other hand, IL-19 also acts as a pro-inflammatory cytokine in T helper 2 cell (Th2)-mediated diseases such as asthma and rheumatoid arthritis. In fact, IL-19 stimulation induces the production of TNF-α, IL-6, and reactive oxygen species in monocytes [16], TNF-α and IL-6 in synovial fibroblasts [17], and IL-1β, IL-6, IL-8, CCL5, and CXCL9 in lung epithelial cells [18]. Therefore, IL-19 exerts either anti-inflammatory or pro-inflammatory effects in accordance with immunological conditions.
However, it remains uncertain whether IL-19 is involved in the pathomechanism of neurodegenerative diseases. In this study, we used a mouse model to investigate how IL-19 contributes to the pathogenesis of ALS.
Methods
Animals
All animal experiments were conducted under protocols approved by the Animal Experiment Committee of Yokohama City University (approved number: F-A-19–036). C57BL/6 (B6) mice were purchased from Japan SLC (Hamamatsu, Japan). IL-19−/− mice (B6 background) were obtained from Regeneron Pharmaceuticals (Tarrytown, NY, USA) [10]. Transgenic mice carrying 23 copies of a transgene encoding the G93A mutant of human SOD1 (designated as SOD1G93A Tg mice) were purchased from the Jackson Laboratory (B6.Cg-Tg(SOD1-G93A)1Gur/J; #004435, Jackson Laboratory, Bar Harbor, ME, USA) [19]. We obtained IL-19–deficient SOD1G93A Tg mice (designated as IL-19−/−/SOD1G93A Tg mice) by crossing SOD1G93A Tg mice with IL-19−/− mice.
Behavioral assessments
After the age of 8 weeks, mice were assessed weekly by body weight scaling and Rotarod test; assessments were performed in randomized order by an observer blinded to the genotype [20]. Time of disease onset was defined as the first day when the mice exhibited hindlimb motor deficits such as abnormal reflexes or tremor, which are early symptoms observed in SOD1G93A Tg mice [20, 21]. For the Rotarod test, the time that the mouse remained on a rotating cylinder (Ugo-Basile, Monvalle, Varese, Italy) at a constant speed of 15 rpm was measured. Each mouse was evaluated three times, and the longest latency to fall was recorded; 180 s was chosen as the arbitrary cut-off time.
Cells
Primary cultures were prepared from SOD1G93A Tg mice or IL-19−/−/SOD1G93A Tg mice. Primary microglia cultures were isolated from primary mixed glial-cell cultures prepared from newborn mice using the “shaking off” method after 14 days in vitro, as described previously [3, 22]. The purity of the cultures, as determined by anti-CD11b immunostaining, was > 99%. Cultures were maintained in Dulbecco’s modified Eagle’s minimum essential medium (DMEM) (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (FBS) (Equitech-Bio, Kerrville, TX, USA), 5 μg/ml bovine insulin (Sigma-Aldrich), and 0.2% glucose. Primary astrocyte cultures were purified from primary mixed glial cultures by three to four repetitions of trypsinization and replating, as described previously [4, 23]. Astrocyte purity, as determined by glial fibrillary acidic protein (GFAP)-specific immunostaining, was > 95%.
Quantitative reverse transcription PCR analysis
Lumbar spinal cords were collected from 20-week-old mice. Microglia and astrocytes were seeded at a density of 1 × 105 cells/well (500 µl/well) in 48-well plates. Microglia were treated with 0–1000 ng/ml lipopolysaccharide (LPS) for 24 h. Astrocytes were treated with 0–1000 ng/ml LPS for 24 h. Total RNA was extracted from cultured microglia, astrocytes, and lumbar spinal cords using the miRNeasy Mini Kit (Qiagen, Valencia, CA, USA). For quantitative reverse transcription PCR (qPCR) experiments, RNA was reverse transcribed into cDNA using SuperScript III (Life Technologies, Carlsbad, CA, USA). Levels of mRNAs encoding IL-19, IL-6, IL-10, inducible nitric oxide synthase (iNOS), tumor necrosis factor α (TNF-α), IL-1β, glial cell line–derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF), TGF-β1, IL-20 receptor α (IL-20Rα), IL-20 receptor β (IL-20Rβ), and hypoxanthine phosphoribosyltransferase 1 (HPRT1) were measured using quantitative PCR (qPCR), which was performed on a LightCycler using the KAPA SYBR FAST qPCR Master Mix kit (Sigma-Aldrich). Relative expression levels were determined using the ΔΔCT method; the genes of interest were normalized against the geometric mean of HPRT1. Primers for qPCR are indicated in Table1.
Histological analysis
Frozen sections (20 µm thick) of mouse lumbar spinal cords were prepared using a previously described method [20, 24] at each disease stage (early stage, 12 weeks; middle stage, 16 weeks; late stage, 20 weeks; end stage, 24 weeks). The sections were permeabilized with 1% Triton-X-100 in PBS for 30 min, blocked with 10% FBS for 1 h, and incubated overnight with rabbit anti–mouse Iba1 polyclonal antibodies (1:300; Wako, Tokyo, Japan), mouse anti-NeuN monoclonal antibody (CI: A60, 1:500; Chemicon, Temecula, CA, USA), rabbit anti–mouse GFAP polyclonal antibodies (1:1000; Dako, Glostrup, Denmark), and rat anti-CD68 monoclonal antibody (CI: FA-11, 1:500; Bio-Rad, Hercules, CA, USA), followed by incubation with Alexa Fluor 488 or Alexa Fluor 546–conjugated secondary antibodies (Life Technologies). The stained cells were analyzed in six random fields per section using a deconvolution fluorescence microscope system (BZ-X800; Keyence, Osaka, Japan) as described previously [20, 24]. Data were collected from six sections per mouse and four mice per group.
Statistical analysis
Statistical significance was analyzed using Student’s t-test or one-way analysis of variance (ANOVA) followed by post hoc Tukey’s test. Survival time and onset time were analyzed by log-rank test. All statistical analyses were performed using GraphPad Prism version 8 (Graph Pad Software, La Jolla, CA, USA).
Results
IL-19 is upregulated in primary microglia and lumbar spinal cord of SOD1G93A Tg mice
First, we examined IL-19 expression levels in primary microglia and lumbar spinal cords of wild-type and SOD1G93A Tg mice. qPCR analyses revealed that IL-19 expression in primary microglia was significantly higher in SOD1G93A Tg mice than in wild-type mice (Fig. 1a). SOD1G93A Tg mice also exhibited elevated expression of IL-19 in the lumbar spinal cords as the disease progressed (Fig. 1b). In addition, expression levels of IL-19 receptor (IL-20 receptor α/β heterodimer) were higher in lumbar spinal cords of SOD1G93A Tg mice than in wild-type mice. (Fig. 1c, d).

Expression levels of IL-19 and IL-19 receptor are elevated in primary microglia and lumbar spinal cord of SOD1Tg mice. a qPCR data for IL-19 in primary microglia of wild-type and SOD1G93ATg mice. b qPCR data for IL-19 in lumbar spinal cord of wild-type and SOD1G93ATg mice. c qPCR data for IL-20Rα in lumbar spinal cord of wild-type and SOD1G93ATg mice. d qPCR data for IL-20Rβ in lumbar spinal cord of wild-type and SOD1G93ATg mice. Values are means ± SD (n = 4). *p < 0.05
Ablation of IL-19 in SOD1G93A Tg mice improves motor function, but not lifespan
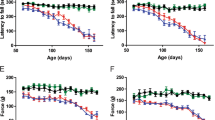
Next, we investigated the role of IL-19 in ALS by comparing SOD1G93A Tg mice with IL-19−/−/SOD1G93A Tg mice. IL-19 deficiency did not cause a significant difference in mean survival time (Fig. 2a; IL-19−/−/ SOD1G93A Tg, 178.4 ± 11 days vs. SOD1G93A Tg, 173.6 ± 12 days), disease onset (Fig. 2b; IL-19−/−/ SOD1G93A Tg, 68.1 ± 1.6 days vs. SOD1G93A Tg, 66.8 ± 2.0 days), or body weight (Fig. 2c), but it did significantly improve motor function at the late stage of the disease (Fig. 2d, 20–22 weeks). These results indicated that IL-19 influences disease progression in SOD1G93A Tg mice.

Interleukin-19 deficiency improves motor function at the late stage in the SOD1G93ATg mice. a Kaplan–Meier survival curves for SOD1G93ATg and IL-19−/−/SOD1G93ATg mice. b Kaplan–Meier curves for time to onset of SOD1G93ATg and IL-19−/−/SOD1G93ATg mice. c Body weight. Significant differences were not observed between the two types of model mice. d Rotarod test. IL-19−/−/SOD1G93ATg mice exhibited a significant improvement in motor function at the late stage (20–22 weeks) relative to SOD1G93ATg mice. Values are means ± SD (SOD1G93ATg, n = 29; IL-19−/−/SOD1G93ATg, n = 30). *, p < 0.05
Ablation of IL-19 in SOD1G93A Tg mice alters microglial phenotype in lumbar spinal cords
Next, we examined the effect of IL-19 on glial cells in lumbar spinal cord at the late (20 weeks) and end (24 weeks) stages. Immunohistochemical analyses of microglia, astrocytes, and neurons did not reveal significant differences in the extent of gliosis or neuronal loss between SOD1G93A Tg mice and IL-19−/−/SOD1G93A Tg mice at either disease stage (Fig. 3a–f). However, ablation of IL-19 decreased positivity for CD68 (neurotoxic microglial marker; Fig. 4a, b, e) and increased positivity for arginase1 (neuroprotective microglial marker; Fig. 4c, d, f) in lumbar spinal cord microglia of 20-week-old SOD1G93ATg mice. These data indicated that IL-19 modulates microglial phenotype in SOD1G93A Tg mice.

Ablation of IL-19 does not affect gliosis and neuronal loss in SOD1G93A Tg. a–c Images of lumbar spinal cord of SOD1G93A Tg and IL-19−/−/ SOD1G93A Tg mice at the late stage (20 weeks) and end stage (24 weeks), immunostained for (a) Iba1, (b) GFAP, and (c) NeuN. d Percentage of Iba1-positive areas in the lumbar spinal cord sections. e Percentage of GFAP-positive areas in the lumbar spinal cord sections. f Numbers of NeuN-positive cells in the ventral horn of lumbar spinal cord sections. Scale bar, 200 µm. Values are means ± SD

Ablation of IL-19 modulates microglial phenotype in SOD1G93A Tg at the late stage. Images of lumbar spinal cord of 20-week-old (a) SOD1G93ATg mice and (b) IL-19−/−/SOD1G93A Tg mice immunostained for CD68 (green) and Iba1 (red). Images of lumbar spinal cord of 20-week-old (c) SOD1G93A Tg mice and (d) IL-19−/−/SOD1G93A Tg mice immunostained for arginase 1 (green) and Iba1 (red). e Percentage of CD68 positive cells in Iba1 positive cells in lumbar spinal cord sections. f Percentage of arginase 1 positive cells in Iba1 positive cells in the lumbar spinal cord sections. Scale bars: 200 µm in merged images and 50 µm in enlarged images. Values are means ± SD. *p < 0.05
Ablation of IL-19 upregulates both neurotoxic and neuroprotective factors in lumbar spinal cord of SOD1G93A Tg mice
Next, we evaluated the expression levels of neurotoxic and neuroprotective factors in the lumbar spinal cords of SOD1G93A Tg and IL-19−/−/SOD1G93A Tg mice at the late stage (20 weeks) when motor function began to show a significant difference. qPCR analyses for pro-inflammatory factors revealed that expression levels of TNF-α and IL-1β were significantly upregulated in IL-19−/−/SOD1G93A Tg mice compared to SOD1G93A Tg mice (Fig. 5a–d). As for neurotrophic factors and anti-inflammatory factors, GDNF and TGF-β were significantly upregulated in IL-19−/−/SOD1G93A Tg mice compared to SOD1G93A Tg mice (Fig. 5e–h). Interestingly, these data imply that IL-19 affects expression levels of both neurotoxic and neuroprotective factors in lumbar spinal cord of SOD1G93A Tg mice.

qPCR analyses of pro-inflammatory cytokines, anti-inflammatory cytokines, and growth factors in the lumbar spinal cords of 20-week-old SOD1G93A Tg mice and IL-19−/−/ SOD1G93A Tg mice. qPCR data are shown for a iNOS mRNA, b IL-1β mRNA, c IL-6 mRNA, d TNF-α mRNA, e BDNF mRNA, f GDNF mRNA, g IL-10 mRNA, and h TGF-β1 mRNA. Values are means ± SD (n = 4). *p < 0.05
Ablation of IL-19 increases TNF-α release from microglia and astrocytes, thereby promoting GDNF production in astrocytes, in SOD1G93A Tg mice
Because IL-19 deficiency increased the expression of TNF-α and GDNF in lumbar spinal cord of SOD1G93A Tg mice (Fig. 5d, f), and in light of a previous observation that TNF-α induces GDNF release from astrocytes [25], we investigated the interaction between TNF-α and GDNF in microglia and astrocytes from ALS mice.
First, we sought to determine whether IL-19 deficiency would alter the expression levels of TNF-α and GDNF in microglia and astrocytes from ALS mice. We used LPS to stimulate glial cell cultures because previous studies revealed that mutant SOD1 induces microglial activation via TLR4 and CD14, which serve as the receptor complex for LPS [26, 27]. TNF-α expression was higher in 24-h LPS-stimulated primary microglia or astrocytes from IL-19−/−/SOD1G93A Tg mice than in those from SOD1G93A Tg mice (Fig. 6a, c), whereas levels of GDNF were not affected (Fig. 6b, d).

Ablation of IL-19 upregulates TNF-α expression in primary microglia and astrocytes from SOD1G93A Tg. a, b qPCR data for TNF-α and GDNF expression in LPS (1000 ng/ml)-stimulated primary microglia from SOD1G93A Tg and IL-19−/−/SOD1G93A Tg. c, d qPCR data for TNF-α and GDNF expression in LPS (1000 ng/ml)-stimulated primary astrocytes from SOD1G93A Tg and IL-19−/−/SOD1G93A Tg. Values are means ± SD (n = 4). *p < 0.05
Next, we investigated whether IL-19 deficiency alters the expression level of TNF-α–induced GDNF in microglia and astrocytes from ALS mice. For this purpose, we stimulated primary microglia and astrocytes from SOD1G93ATg and IL-19−/−/SOD1G93ATg mice with TNF-α (0–100 ng/ml) for 24 h. TNF-α stimulation dose-dependently upregulated GDNF expression level in astrocytes (Fig. 7b), but not in microglia (Fig. 7a). Moreover, primary astrocytes from IL-19−/−/SOD1G93A Tg mice exhibited a significantly greater increase in GDNF expression level than those from SOD1G93ATg mice in response to TNF-α stimulation (Fig. 7b). These results suggested that astrocytes secrete GDNF upon stimulation with TNF-α released from microglia and astrocytes, which acts in an autocrine and paracrine manner.

Ablation of IL-19 enhanced TNF-α–induced upregulation of GDNF in primary astrocytes and lumbar spinal cord from SOD1G93A Tg mice. a qPCR for GDNF mRNA in primary microglia stimulated with TNF-α (0–100 ng/ml). Values are means ± SD (n = 4). *p < 0.05. b qPCR for GDNF mRNA in primary astrocytes stimulated by TNF-α (0–100 ng/ml). Values are means ± SD (n = 4). *p < 0.05. c qPCR data for GDNF expression in lumbar spinal cord from SOD1G93A Tg and IL-19−/−/SOD1G93A Tg. Values are means ± SD (n = 6). *p < 0.05. d qPCR data for TNF-α expression in lumbar spinal cord from SOD1G93A Tg and IL-19−/−/SOD1G93A Tg. Values are means ± SD (n = 6). *p < 0.05 when data of 12–20-week-old mice were analyzed. **p < 0.05 when data of 12–24-week-old mice were analyzed
In addition, we examined the levels of TNF-α and GDNF in the lumbar spinal cord over a time course. GDNF expression was significantly higher in IL-19−/−/SOD1G93A Tg mice after the late stage (20–24 weeks) than in SOD1G93ATg mice as the TNF-α upregulation (Fig. 7c, d). The chronological expression data in lumbar spinal cord are consistent with our in vitro observations of astrocytic release of GDNF in response to TNF-α stimulation.
Discussion
Previous studies showed that IL-19 acts in an autocrine and paracrine negative-feedback regulator to limit pro-inflammatory responses by microglia and macrophages [9, 10], and that it can slow the progression of microglia/macrophage-mediated CNS disorders such as stroke [14] and spinal cord injury [15]. In fact, we recently showed that IL-19 deficiency worsened microglia/macrophage-mediated experimental autoimmune encephalitis, whereas IL-19 treatment abrogated this condition by inhibiting macrophage activation [28]. In this study, we observed upregulation of IL-19 expression in lumbar spinal cord of SOD1G93A Tg mice in parallel with disease progression (Fig. 1). Therefore, we hypothesized that ablation of IL-19 would exacerbate disease progression in SOD1G93A Tg mice.
Unexpectedly, however, we observed that ablation of IL-19 improved motor function at the late stage in SOD1G93A Tg mice (Fig. 2). At this stage, immunohistochemical analyses detected that IL-19 deficiency resulted in conversion of microglia from a pro-inflammatory and neurotoxic phenotype to an anti-inflammatory and neuroprotective phenotype (Fig. 4). qPCR analyses revealed that expression levels of TNF-α and GDNF in lumbar spinal cord were higher in IL-19−/−/SOD1G93A Tg mice than in SOD1G93A Tg mice (Fig. 5). Moreover, IL-19 deficiency enhanced TNF-α upregulation in microglia and astrocytes and augmented TNF-α–induced GDNF upregulation in astrocytes from SOD1G93A Tg mice, which was corroborated by the chronological expression levels of GDNF and TNF-α in the lumbar spinal cord of these mice (Figs. 6 and 7). Due to the technical difficulties of isolating and maintaining glial cells from diseased adult mice, we used primary glial cell cultures from neonatal mouse brains to represent disease phenotypes in adult mice. In general, cell culture models recapitulated the disease progression of animal models on a compressed time scale. In ALS models, previous studies also demonstrated that neonatal primary cultures from mutant SOD1 transgenic mice underwent significant phenotypic changes corresponding to those in adult mice [29,30,31]. Therefore, we believe that our cell models at least partly recapitulated the disease pathology in ALS mice. Interestingly, previous studies documented that TNF-α promotes TNF-α release from microglia and astrocytes in an autocrine and paracrine manner, mediated by TNF-α receptor 1 (TNFR1) [25, 32,33,34], and that TNF-α induces astrocytic GDNF production [25]. Other studies reported TNF-α elevation in the spinal cord of SOD1G93A Tg mice prior to disease onset, and showed that ablation of TNFR1 but not TNFR2 enhanced motor neuron loss and accelerated disease progression in SOD1G93ATg mice [35, 36]. Microglia-derived TNF-α plays a crucial role as a paracrine signal to regulate astrocytic neuroprotective functions [37], and microglia in SOD1G93A Tg mice produce high levels of TNF-α [26]. Based on these findings, we hypothesized that the TNF-α–GDNF axis would exert neuroprotective effects in ALS pathogenesis (Fig. 8). Mutant SOD1–derived pro-inflammatory stimuli induce microglia and astrocytes to release TNF-α in an autocrine and paracrine manner. Secreted TNF-α induces astrocytic GDNF release, which exerts a neuroprotective effect. Ablation of IL-19 promotes neuroprotection by enhancing this TNF-α–GDNF neuroprotective cascade.

Model of the roles of TNF-α and GDNF in glia-mediated neuroprotection in ALS. Stimuli such as mutant SOD1 induce microglia and astrocytes to release TNF-α. Secreted TNF-α induces GDNF release from astrocytes in an autocrine and paracrine manner, resulting in enhancement of neuroprotection. Blockade of IL-19 signaling may augment this TNF-α–induced GDNF neuroprotective cascade (red arrows)
Several lines of evidence indicate that activated microglia and astrocytes are pivotal players in ALS pathogenesis, and that glial activation exerts both neurotoxic and neuroprotective effects depending on its spatiotemporal distribution [29, 38]. In particular, glial activation by cytokines often shows conflicting abilities in neurotoxicity and neuroprotection [39]. For example, overexpression of TGF-β1, an anti-inflammatory cytokine, paradoxically accelerates the disease progression of SOD1G93A Tg by astrocytic activation [40]. Furthermore, IL-10, a major anti-inflammatory cytokine, exacerbates amyloid-β plaque burden and cognitive impairment, whereas IL-10 deficiency enhanced amyloid-β clearance in Alzheimer’s disease model mice [41, 42]. We also observed that the level of IL-1β in the lumbar spinal cord was higher in IL-19−/−/SOD1G93A Tg mice than in SOD1G93A Tg mice. IL-1β is a major pro-inflammatory cytokine that enhances glial NF-κB activation and subsequent TNF-α production [43]. Therefore, it is possible that IL-1β elevation could induce and augment glial TNF-α production in IL-19−/−/SOD1G93A Tg mice.
IL-19 also exerts a potent anti-inflammatory effect on CNS-infiltrating monocytes and macrophages under Th1-prone condition [14, 15], whereas it exerts a pro-inflammatory effect on tissue-infiltrating monocytes/macrophages and fibroblasts in Th2-prone conditions [16,17,18, 44]. Previous studies also reported conflicting observations of IL-19 effects (pro-inflammatory vs. anti-inflammatory) even in similar mouse models of colitis [10, 45], suggesting that the effects of IL-19 depend strongly on the condition and distribution of inflammation. In addition to the unexpected pro-inflammatory effect of IL-19 in ALS mice, we observed another small discrepancy in the trends of GDNF and TNF-α expression levels in lumbar spinal cord in the end stage (24 weeks) (Fig. 7c, d). Although differential expression levels of TNF-α were observed between SOD1G93A Tg and IL-19−/−/SOD1G93A Tg mice in the late stage (20 weeks), rapidly increasing TNF-α expression in the end stage reached similar levels in both Tg mice (Fig. 7d). By contrast, GDNF expression was significantly higher in IL-19−/−/SOD1G93A Tg mice than in SOD1G93A Tg mice from the late to the end stage (Fig. 7c). Nevertheless, these mice had similar lifespans. Strong upregulation of TNF-α in the end stage may have exacerbated neuroinflammation, disrupted CNS homeostasis, and overwhelmed the neuroprotective effect of GDNF. Further investigations are needed to elucidate these issues.
Our observations reveal a complex neuro–glia–immune interaction under inflamed conditions in ALS mice. Spatiotemporal manipulation of glial activation might facilitate the development of novel therapeutic strategies for neurodegenerative diseases such as ALS.
Conclusions
In this study, we demonstrated that IL-19 deficiency delayed disease onset and improved motor function at the late stage in the SOD1G93ATg mice. Ablation of IL-19 slowed the disease progression of ALS mice by enhancing glial neuroprotection through such glia-secreted factors as TNF-α and GDNF. Our results suggest that blockade of IL-19 signaling represents a potential therapeutic strategy for ameliorating neurodegenerative diseases including ALS.
Availability of data and materials
The datasets used and analyzed during this study are available from the corresponding author on reasonable request.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- BDNF:
-
Brain-derived neurotrophic factor
- CNS:
-
Central nervous system
- DMEM:
-
Dulbecco’s modified Eagle’s minimum essential medium
- FBS:
-
Fetal bovine serum
- GDNF:
-
Glial cell–derived neurotrophic factor
- GFAP:
-
Glial fibrillary acidic protein
- Hprt1:
-
Hypoxanthine phosphoribosyltransferase 1
- IL-19:
-
Interleukin-19
- Iba1:
-
Ionized calcium-binding adapter molecule 1
- IL-1β:
-
Interleukin-1 beta
- SOD1:
-
Superoxide dismutase 1
- IL-20Rα:
-
IL-20 receptor alpha subunit
- IL-20Rβ:
-
IL-20 receptor beta subunit
- LPS:
-
Lipopolysaccharide
- iNOS:
-
Inducible nitric oxide synthase
- PBS:
-
Phosphate-buffered saline
- qPCR:
-
Quantitative reverse transcription PCR
- TNF-α:
-
Tumor necrosis factor alpha
- TGF-β1:
-
Transforming growth factor beta 1
References
Traxinger K, Kelly C, Johnson BA, Lyles RH, Glass JD. Prognosis and epidemiology of amyotrophic lateral sclerosis: Analysis of a clinic population, 1997–2011. Neurol Clin Pract. 2013;3(4):313–20.
Gasco S, Zaragoza P, Garcia-Redondo A, Calvo AC, Osta R. Inflammatory and non-inflammatory monocytes as novel prognostic biomarkers of survival in SOD1G93A mouse model of Amyotrophic Lateral Sclerosis. PLoS ONE. 2017;12(9):e0184626.
Musaro A. State of the art and the dark side of amyotrophic lateral sclerosis. World J Biol Chem. 2010;1(5):62–8.
Pramatarova A, Laganiere J, Roussel J, Brisebois K, Rouleau GA. Neuron-specific expression of mutant superoxide dismutase 1 in transgenic mice does not lead to motor impairment. J Neurosci. 2001;21(10):3369–74.
Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302(5642):113–7.
Gallagher G, Dickensheets H, Eskdale J, Izotova LS, Mirochnitchenko OV, Peat JD, Vazquez N, Pestka S, Donnelly RP, Kotenko SV. Cloning, expression and initial characterization of interleukin-19 (IL-19), a novel homologue of human interleukin-10 (IL-10). Genes Immun. 2000;1(7):442–50.
Sabat R, Wallace E, Endesfelder S, Wolk K. IL-19 and IL-20: two novel cytokines with importance in inflammatory diseases. Expert Opin Ther Targets. 2007;11(5):601–12.
Parrish-Novak J, Xu W, Brender T, Yao L, Jones C, West J, Brandt C, Jelinek L, Madden K, McKernan PA, et al. Interleukins 19, 20, and 24 signal through two distinct receptor complexes. Differences in receptor-ligand interactions mediate unique biological functions. J Biol Chem. 2002;277(49):47517–23.
Horiuchi H, Parajuli B, Wang Y, Azuma YT, Mizuno T, Takeuchi H, Suzumura A. Interleukin-19 acts as a negative autocrine regulator of activated microglia. PLoS ONE. 2015;10(3):e0118640.
Azuma YT, Matsuo Y, Kuwamura M, Yancopoulos GD, Valenzuela DM, Murphy AJ, Nakajima H, Karow M, Takeuchi T. Interleukin-19 protects mice from innate-mediated colonic inflammation. Inflamm Bowel Dis. 2010;16(6):1017–28.
Cooley ID, Chauhan VS, Donneyz MA, Marriott I. Astrocytes produce IL-19 in response to bacterial challenge and are sensitive to the immunosuppressive effects of this IL-10 family member. Glia. 2014;62(5):818–28.
Matsuo Y, Azuma YT, Kuwamura M, Kuramoto N, Nishiyama K, Yoshida N, Ikeda Y, Fujimoto Y, Nakajima H, Takeuchi T. Interleukin 19 reduces inflammation in chemically induced experimental colitis. Int Immunopharmacol. 2015;29(2):468–75.
Fujimoto Y, Fujita T, Kuramoto N, Kuwamura M, Izawa T, Nishiyama K, Yoshida N, Nakajima H, Takeuchi T, Azuma YT. The role of interleukin-19 in contact hypersensitivity. Biol Pharm Bull. 2018;41(2):182–9.
Xie W, Fang L, Gan S, Xuan H. Interleukin-19 alleviates brain injury by anti-inflammatory effects in a mice model of focal cerebral ischemia. Brain Res. 2016;1650:172–7.
Guo J, Wang H, Li L, Yuan Y, Shi X, Hou S. Treatment with IL-19 improves locomotor functional recovery after contusion trauma to the spinal cord. Br J Pharmacol. 2018;175(13):2611–21.
Liao YC, Liang WG, Chen FW, Hsu JH, Yang JJ, Chang MS. IL-19 induces production of IL-6 and TNF-alpha and results in cell apoptosis through TNF-alpha. J Immunol. 2002;169(8):4288–97.
Hsu YH, Hsieh PP, Chang MS. Interleukin-19 blockade attenuates collagen-induced arthritis in rats. Rheumatology (Oxford). 2012;51(3):434–42.
Zhong H, Wu Y, Belardinelli L, Zeng D. A2B adenosine receptors induce IL-19 from bronchial epithelial cells, resulting in TNF-alpha increase. Am J Respir Cell Mol Biol. 2006;35(5):587–92.
Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–5.
Takeuchi H, Mizoguchi H, Doi Y, Jin S, Noda M, Liang J, Li H, Zhou Y, Mori R, Yasuoka S, et al. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer’s disease. PLoS ONE. 2011;6(6):e21108.
Hayworth CR, Gonzalez-Lima F. Pre-symptomatic detection of chronic motor deficits and genotype prediction in congenic B6.SOD1(G93A) ALS mouse model. Neuroscience. 2009;164(3):975–85.
Suzumura A, Mezitis SG, Gonatas NK, Silberberg DH. MHC antigen expression on bulk isolated macrophage-microglia from newborn mouse brain: induction of Ia antigen expression by gamma-interferon. J Neuroimmunol. 1987;15(3):263–78.
Liang J, Takeuchi H, Doi Y, Kawanokuchi J, Sonobe Y, Jin S, Yawata I, Li H, Yasuoka S, Mizuno T, et al. Excitatory amino acid transporter expression by astrocytes is neuroprotective against microglial excitotoxicity. Brain Res. 2008;1210:11–9.
Komiya H, Takeuchi H, Ogawa Y, Hatooka Y, Takahashi K, Katsumoto A, Kubota S, Nakamura H, Kunii M, Tada M, et al. CCR2 is localized in microglia and neurons, as well as infiltrating monocytes, in the lumbar spinal cord of ALS mice. Mol Brain. 2020;13(1):64.
Kuno R, Yoshida Y, Nitta A, Nabeshima T, Wang J, Sonobe Y, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A. The role of TNF-alpha and its receptors in the production of NGF and GDNF by astrocytes. Brain Res. 2006;1116(1):12–8.
Zhao W, Beers DR, Henkel JS, Zhang W, Urushitani M, Julien JP, Appel SH. Extracellular mutant SOD1 induces microglial-mediated motoneuron injury. Glia. 2010;58(2):231–43.
Lee JY, Lee JD, Phipps S, Noakes PG, Woodruff TM. Absence of toll-like receptor 4 (TLR4) extends survival in the hSOD1 G93A mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2015;12:90.
Horiuchi H, Parajuli B, Komiya H, Ogawa Y, Jin S, Takahashi K, Azuma YT, Tanaka F, Suzumura A, Takeuchi H. Interleukin-19 abrogates experimental autoimmune encephalomyelitis by attenuating antigen-presenting cell activation. Front Immunol. 2021;12:615898.
Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615–22.
Hensley K, Abdel-Moaty H, Hunter J, Mhatre M, Mou S, Nguyen K, Potapova T, Pye QN, Qi M, Rice H, et al. Primary glia expressing the G93A-SOD1 mutation present a neuroinflammatory phenotype and provide a cellular system for studies of glial inflammation. J Neuroinflammation. 2006;3:2.
Kawaguchi-Niida M, Yamamoto T, Kato Y, Inose Y, Shibata N. MCP-1/CCR2 signaling-mediated astrocytosis is accelerated in a transgenic mouse model of SOD1-mutated familial ALS. Acta Neuropathol Commun. 2013;1:21.
Kuno R, Wang J, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A. Autocrine activation of microglia by tumor necrosis factor-alpha. J Neuroimmunol. 2005;162(1–2):89–96.
Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281(30):21362–8.
Horiuchi H, Parajuli B, Kawanokuchi J, Jin S, Mizuno T, Takeuchi H, Suzumura A. Oligomeric amyloid β facilitates microglial excitotoxicity by upregulating tumor necrosis factor-α and downregulating excitatory amino acid transporter 2 in astrocytes. Clin Exp Neuroimmunol. 2015;6(2):183–90.
Brambilla L, Guidotti G, Martorana F, Iyer AM, Aronica E, Valori CF, Rossi D. Disruption of the astrocytic TNFR1-GDNF axis accelerates motor neuron degeneration and disease progression in amyotrophic lateral sclerosis. Hum Mol Genet. 2016;25(14):3080–95.
Tortarolo M, Vallarola A, Lidonnici D, Battaglia E, Gensano F, Spaltro G, Fiordaliso F, Corbelli A, Garetto S, Martini E, et al. Lack of TNF-alpha receptor type 2 protects motor neurons in a cellular model of amyotrophic lateral sclerosis and in mutant SOD1 mice but does not affect disease progression. J Neurochem. 2015;135(1):109–24.
Chen SH, Oyarzabal EA, Sung YF, Chu CH, Wang Q, Chen SL, Lu RB, Hong JS. Microglial regulation of immunological and neuroprotective functions of astroglia. Glia. 2015;63(1):118–31.
Henkel JS, Beers DR, Zhao W, Appel SH. Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol. 2009;4(4):389–98.
Stoll G, Jander S, Schroeter M. Cytokines in CNS disorders: neurotoxicity versus neuroprotection. J Neural Transm Suppl. 2000;59:81–9.
Endo F, Komine O, Fujimori-Tonou N, Katsuno M, Jin S, Watanabe S, Sobue G, Dezawa M, Wyss-Coray T, Yamanaka K. Astrocyte-derived TGF-β1 accelerates disease progression in ALS mice by interfering with the neuroprotective functions of microglia and T cells. Cell Rep. 2015;11(4):592–604.
Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, DiNunno N, Rosario AM, Cruz PE, Verbeeck C, et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85(3):519–33.
Guillot-Sestier MV, Doty KR, Gate D, Rodriguez J Jr, Leung BP, Rezai-Zadeh K, Town T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015;85(3):534–48.
Moynagh PN. The interleukin-1 signalling pathway in astrocytes: a key contributor to inflammation in the brain. J Anat. 2005;207(3):265–9.
Sakurai N, Kuroiwa T, Ikeuchi H, Hiramatsu N, Maeshima A, Kaneko Y, Hiromura K, Nojima Y. Expression of IL-19 and its receptors in RA: potential role for synovial hyperplasia formation. Rheumatology (Oxford). 2008;47(6):815–20.
Steinert A, Linas I, Kaya B, Ibrahim M, Schlitzer A, Hruz P, Radulovic K, Terracciano L, Macpherson AJ, Niess JH. The stimulation of macrophages with TLR ligands supports increased IL-19 expression in inflammatory bowel disease patients and in colitis models. J Immunol. 2017;199(7):2570–84.
Acknowledgements
Not applicable
Funding
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (HT); grants from the Ministry of Health, Labour and Welfare of Japan (HT, FT); the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation (NIBIO) of Japan (HT); a Grant from the Naito Foundation (HT); and a Grant for Strategic Research Promotion from Yokohama City University (# SK2804, FT).
Author information
Authors and Affiliations
Contributions
HK and HT designed the study; HK, HT, YO, KS, AO, KT, YA, and HD performed the research; HK, HT, YO, KS, AO, HD, and FT analyzed the data; and HK, HT, and FT wrote the paper. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All animal experiments were conducted under protocols approved by the Animal Experiment Committee of Yokohama City University (approved number: F-A-19-036).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Komiya, H., Takeuchi, H., Ogawa, Y. et al. Ablation of interleukin-19 improves motor function in a mouse model of amyotrophic lateral sclerosis. Mol Brain 14, 74 (2021). https://doi.org/10.1186/s13041-021-00785-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-021-00785-8