
Abstract
Go is a member of the pertussis toxin-sensitive Gi/o family. Despite its abundance in the central nervous system, the precise role of Go remains largely unknown compared to other G proteins. In the present study, we explored the functions of Go in the developing cerebellar cortex by deleting its gene, Gnao. We performed a histological analysis with cerebellar sections of adult mice by cresyl violet- and immunostaining. Global deletion of Gnao induced cerebellar hypoplasia, reduced arborization of Purkinje cell dendrites, and atrophied Purkinje cell dendritic spines and the terminal boutons of climbing fibers from the inferior olivary nucleus. These results indicate that Go-mediated signaling pathway regulates maturation of presynaptic parallel fibers from granule cells and climbing fibers during the cerebellar cortical development.
Similar content being viewed by others


Introduction
When G-protein coupled receptors (GPCRs) bind their cognate ligands, their respective heterotrimeric GTP binding proteins (G-proteins) are activated, inducing the dissociation of Gα from Gβγ. Specificity of GPCR signal transduction is determined by the functions of the specific Gα subunits because all G-proteins share a pool of Gβγ subunits. Gα subunits are classified into four subfamilies: Gαs, Gαi/o, Gαq/11, and Gα12/13. So far, 16 different mammalian Gα subunits have been identified [1, 2]. Go belongs to the pertussis toxin-sensitive Gi/o family, the α subunits of which share 71% amino acid sequence homology. Gαi proteins are ubiquitously expressed and well-studied. They inhibit adenylyl cyclase (AC) directly and consequently reduce intracellular cyclic AMP (cAMP) levels [3]. Gnai (the gene encoding Gαi protein) knockout (Gnai−/−) mice show several abnormalities, especially in their immune systems and blood clotting [4, 5]. In comparison, Gαo expression is restricted to the central nervous system (CNS), particularly in synapses, and to endocrine cells and cardiac myocytes [3, 6, 7]. Accordingly, Gnao (the gene encoding Gαo protein) knockout (Gnao−/−) mice show severe neurological deficits including seizures, hyperactivity, and abnormal sexual behavior accompanied by early death [8, 9]. Mutations of Gnao gene in human patients induce early infantile epileptic encephalopathy (EIEE), which is the basis for the Gnao to be called EIEE17 [10, 11]. These findings suggest Gαo plays an important role in the CNS. Still, compared to Gi, the role of Go in the CNS is largely unknown. This is likely due to the fact that no enzymatic functions have yet been associated with Gαo and to the fact that the scarcity of Gnao−/− mice due to their low birth and survival rates make Gαo difficult to study.
Gαo is abundantly found in the neurite tips and growth cones of in vitro cultured neurons where it stimulates neurite formation [7, 12, 13]. In Gnao−/− mice, the olfactory nerve layer comprised of projecting axons of olfactory sensory neurons is atrophied, which may be associated with defective Gαo signals in neurite outgrowth [9]. Gαo is highly expressed in the cerebellar cortex [14], however, the function of Go in the cerebellum is mostly unknown except for the interaction with the Purkinje cell protein 2, Pcp2 (L7) [15] and Gi/o-coupled cannabinoid receptor 1 (CB1) [16]. Pcp2(L7)−/− mice are almost normal, showing only mild hypoplasia accompanied by normal motor learning and enhanced motor function [17]. WIN55212–2, a CB1 receptor agonist predominantly activates Gαo among various G-proteins in the rat cerebellum [16].
The cerebellum is a well-defined neural system that can help clarify the correlation between the anatomical architecture of a neural network and its function [18]. The cerebellar cortex consists of three distinct layers: the molecular layer (ML), the Purkinje cell layer (PCL), and the granule cell layer (GCL). Purkinje cells (PCs) arranged in a single layer of the PCL, each sending a single, long axon to deep cerebellar nuclei (DCN) and are the only output neurons of the cerebellar cortex. Their complex dendrites extend through the ML where they receive presynaptic inputs from climbing fibers (CFs) projecting from the inferior olivary nucleus (ION) and parallel fibers (PFs) originating in granule cells (GCs). The PC dendrites are segregated into proximal and distal territories during the postnatal period [19]. Immature proximal PC dendrites are first innervated by multiple CFs during the initial 3 weeks of the postnatal period. During the subsequent 3 weeks, a single dominant CF moves toward the pial surface while the other redundant CFs are pruned [20]. Simultaneously, the immature PCs undergo a transformation from multi- to mono-planarity as they acquire the fan-like shape that is typical for mature PCs [21]. These morphogenetic processes are controlled cell-autonomously by intrinsic PC factors in the early phases of postnatal development and by extrinsic synaptic connections with ION-CFs in the later phases [22].
Various types of Gα proteins are expressed in PCs such as Gαq, Gαo, Gαi2, and Gαz. In PC dendrites, Gαz is constantly increased over development, whereas Gαq and Gαo show different temporal peaks of gene expression patterns during development stages [23], which suggested that the specificity of Gαq and Gαo in the developmental changes of PCs. During the cerebellar development, Gαq-mediated signaling cascades [metabotropic glutamate receptor type 1 (mGluR1)-Gαq-phospholipase Cβ4 (PLCβ4)-protein kinase Cγ (PKCγ)] are well-studied. Deletion of each component evokes the similar phenotypes including multiple CF innervation, motor discoordination, and ataxia [24,25,26].
In this study, we investigate possible roles of Gαo in cerebellar cortical development using Gnao−/− mice. These results showed that the defective Gαo signals prevent the proper development of presynaptic PFs and CFs as well as maturation of postsynaptic PC dendrites. Our results indicate that Gαo is essential for full PC differentiation and the refinement of presynaptic CFs during the postnatal development.
Methods
Mouse genetics
Gnao−/− mice were generated by breeding Gnao+/− mice. Hemizygotes were created via the insertion of a neomycin selection cassette at Gnao exon 6 [8]. Tail-genomic DNA was used for genotyping by polymerase chain reaction (PCR) using the following primers that allowed us to verify the disruption of the Gnao gene: int6R1 (5′- ACC TGG CCT CCC TTG GGA ATA CAG − 3′) and ex6F1 (5′- CAG CGA TCT GAA CGC AAG AAG TGG − 3′) for wild type, and int6R1 and pol2R1 (5′- TGT GCT CTA GTA GCT TTA CGG AGC − 3′) for mutant. The number of animals used for each quantitative analysis is described in each figure legend. All experimental procedures were reviewed and approved by the Institutional Animal Research Ethics Committee at Ajou University Medical Center (Suwon, South Korea). All mutant mice were compared to the wild type littermate control mice.
Cerebellar neuron culture
PCs were cultivated according to previous reports [27, 28] with slight modifications. Briefly, the extracted cerebella from C57BL/6 N at postnatal day (P) 0 were minced with a surgical knife, washed with Hank’s balanced salt solution (HBSS; Sigma-Aldrich, #H2387) containing gentamicin (50 μg/ml, Sigma-Aldrich, #G1914), and incubated in Accumax solution (Sigma-Aldrich, #A7089) for 15 min at 37 °C in a 5% CO2 incubator. After washing with HBSS, the tissues were triturated by repetitive pipetting and the debris was removed by passing the suspension through a nylon mesh. After centrifugation of the cell suspension, the cell pellet was resuspended in plating medium containing 10% (vol/vol) fetal bovine serum (FBS; Hyclone, #SH30084.03) and 50 μg/ml gentamicin in Dulbecco’s modified Eagle medium/F12 (DMEM/F12; Gibco, #11330–032) and then plated onto coverslips coated with poly-D-lysine (PDL; 0.1 mg/ml, Sigma-Aldrich, #P6407) and laminin (10 μg/ml, Gibco, #23017–015). After 2 h, the cells were washed with HBSS and the medium was replaced with Neurobasal-A medium (Gibco, #10888–022) containing B27 (Gibco, #17504–044), Glutamax (Gibco, #35050–061), and Penicillin-Streptomycin (10,000 U/ml, Gibco, #151–40,122). To culture GCs, C57BL/6 N mice were sacrificed at P7 when external granule cells extensively proliferate [29, 30]. Cerebellar cells were isolated and purified as described [31].
In situ hybridization
The template DNA for in vitro transcription-Gnao exons 4 and 5 (nt 767–914, NM_010308.3)-was chosen for the preparation of RNA probes to avoid any regions of homology with Gnai2. The cDNA encompassing exons 4 and 5 (148 bp) was amplified with a forward primer (5′-CTT TGG GCG TGG AGT ATG GTG-3′) and a reverse primer (5′-CTC CTG GAT CCC CGA GTC GCC C-3′). The PCR product was then sub-cloned into the pGEM-T easy plasmid (Promega, #A1360). This plasmid was linearized with Nco I (Roche, #10835315001) or Sal I (Roche, #10348783001) for preparing the antisense- or sense-stranded RNAs, respectively. The antisense RNA was in vitro transcribed using SP6 RNA polymerase and a UTP-11-fluorescein isothiocyanate (FITC) labeling kit (Roche, #11685619910). Cryostat brain sections (40 μm-thick) were dried at room temperature (23 ± 2 °C) for 30 min and fixed in 10% neutral buffered formalin (NBF; BBC biochemical, #0151) for 10 min. After washing with diethylpyrocarbonate (Sigma-Aldrich, #D5758)-treated phosphate buffered saline (DEPC-PBS), the sections were acetylated with 0.1 M triethylamine (TEA, pH 8.0; Sigma-Aldrich, #T0886) and 2.5% acetic anhydride (Sigma-Aldrich, #A6404) in DEPC-PBS for 10 min at room temperature. The sections were then dehydrated and defatted with a 5-min chloroform (Sigma-Aldrich, #36919) treatment. Then, the sections were rinsed with ethanol, air dried, and stored at − 70 °C until used. The sections were pre-warmed at 55 °C for 15 min and pretreated at 37 °C for 2 h in hybridization solutions (Sigma-Aldrich, #H7782) supplemented with 0.2 g/ml dextran sulfate (Sigma-Aldrich, #D8906), 50% (vol/vol) formamide (Sigma-Aldrich, #F9037), 1 mg/ml Herring’s sperm DNA (Sigma-Aldrich, #D7290), and 1 mg/ml Ribonucleic acid from torula yeast (Sigma-Aldrich, #R6625) in 1.5× saline-sodium citrate (SSC; Invitrogen, #AM9763). Hybridization was performed in the presence of 0.1 μg/μl FITC-labeled RNA probe for 40 h at 37 °C. Then, unbound the probe was removed by sequential washing with 2× SSC for 5 min at room temp, 1× SSC for 1 min at room temp, 0.5× SSC containing 0.1% sodium dodecyl sulfate (SDS; Sigma-Aldrich, #L4390) for 20 min at 55 °C, 0.1× SSC containing 0.1% SDS for 20 min at 55 °C, 0.1× SSC containing 0.1% SDS for 20 min at room temp twice, and finally Tris-buffered saline (TBS, pH 7.5) without SDS for 5 min at room temperature 3 times. The sections were counterstained with bisbenzamide (Hoechst 33258; Invitrogen, #H3569) and mounted using Fluoromount-G® mounting solution (Southern Biotech, #0100–01). Images were acquired using an LSM710 confocal microscope (Carl Zeiss). Sense probes were generated with T7 RNA polymerase and used to verify Gnao-specific signals.
Immunofluorescence analysis
Mice were deeply anesthetized by i.p. injection of 2,2,2-tribromoethanol (0.02 ml/g, Sigma-Aldrich, #T48402) and perfused transcardially with 10% NBF. Extracted brains were incubated overnight in 10% NBF at 4 °C for post-fixation. For frozen sections, the brain was placed in 30% sucrose (Sigma-Aldrich, #S7903) in 0.1 M phosphate buffer (pH 7.4) at room temp for 48 h and embedded in O.C.T compound (Tissue-Tek, Sakura Finetek, #4583) to cut into 30 μm-thick slices. For paraffin sections, the brain was embedded in paraffin (Merck, #1.15161.2504) following standard procedures and cut into 7 μm-thick slices. To perform the staining, epitopes were unmasked by microwave (Daewoo Electronics, South Korea)-heating in 10 mM sodium citrate (pH 6.0; Sigma-Aldrich, #S4641) buffer including 0.05% (vol/vol) Tween 20 (Anatrace, #T1003) at 95 °C for 15 min. The samples were incubated with blocking solution [10% (vol/vol) normal goat serum (Gibco, # 16210–072), 1% bovine serum albumin (BSA; Sigma-Aldrich, #A2153) and 0.1% (vol/vol) Triton X-100 (Sigma-Aldrich, #T8787) in PBS (T-PBS)] for 1 h at room temp. The sections were incubated with primary antibodies overnight at 4 °C. The antibodies used were specific for Gαo (1:200, rabbit, Santa Cruz, #SC-387), Calbindin-D28K (Calb, 1:100, mouse, Sigma-Aldrich, #C9848 and 1:200, rabbit, Swant, #CB38a), Pcp2 (1:200, mouse, Santa Cruz, #SC-137064), vesicular glutamate transporter 1 (vGluT1, 1:100, rabbit, Invitrogen, #48–2400), vesicular glutamate transporter 2 (vGluT2, 1:100, mouse, Millipore, #MAB5504), and Tubulin β-III (Tubb3; 1:500, mouse, Biolegend, #801201). After washing with T-PBS, sections were incubated with secondary antibodies conjugated with Alexa 488 or 568 (Invitrogen) for 1 h at room temperature. The sections were counterstained with bisbenzamide and mounted as described above.
For immunocytochemistry, the coverslips with live cells attached were fixed with 10% NBF for 10 min at room temperature. After washing, the coverslips were incubated with blocking solution to block non-specific signals for 1 h at room temp and then incubated with primary antibodies overnight at 4 °C. The samples were incubated in the presence of secondary antibodies and mounted as described above. All fluorescence images were acquired using an LSM710 confocal microscope (Carl Zeiss) or a slide scanner Axio-Scan.Z1 slide scanner (Carl Zeiss). Isotype-specific IgG or normal serum were used instead of the primary antibody to validate the specificity of the immunoassays.
Western blot analysis
Approximately 100 mg of brain tissue was homogenized in 1 ml RIPA buffer (50 mM Tris-Cl, pH 8.0, 1% (vol/vol) IGEPAL® CA-630 (Sigma-Aldrich, #I8896), 0.1% SDS, 0.5% sodium deoxycholate (Sigma-Aldrich, #D6750), 150 mM Sodium chloride (Affymetrix/USB, #21618). The homogenate was centrifuged at 14,000 x g for 10 min at 4 °C and the supernatant was used for western blot analysis with anti- Gαo (1:1000, Santa Cruz), anti- Gαi1/2/3 (1:1000, Santa Cruz, #SC-26761), anti- Gαq (1:500, Santa Cruz, #SC-136181) and anti-Pcp2 (1:100, Santa Cruz, #SC-137064) as previously described [32]. The specific immunoreactivity was visualized using a secondary antibody conjugated with horseradish peroxidase (1:5000, Zymed) and an ECL kit (Pierce, #32106).
Cerebellar surface area measurement
The cerebella of mice older than P21 were considered mature. 1 mm-thick midsagittal mouse brain sections were obtained using a mouse brain matrix (ASI-Instruments, RBM-2000C) and used to make paraffin blocks. Paraffin-embedded cerebellar sections (7 μm-thick) were de-paraffinized via standard procedures and stained with cresyl violet (Sigma-Aldrich, #C5042) or nuclear fast red solution (Sigma-Aldrich, #N3020) for 1–5 min at room temperature. The sections were mounted using Cytoseal™ XYL (Thermo Scientific, #8312–4) and scanned with Aperio Scanscope XT scanner (Aperio Technologies). Cresyl violet-stained images of the whole cerebellar sections and the GCL were obtained, converted to black-and-white, and used to measure the occupancy of the GCL and ML via the ImageJ 1.50 program (NIH) [33]. The area covered by the ML was measured by subtracting the area covered by the GCL and white matter from the area of the whole cerebellum.
Measurement of PC dendrites and CF boutons
To measure the thickness of PC dendrites, paraffin sections (7 μm-thick) from Gnao−/− mice were stained with an anti-Calbindin antibody. Fluorescent images were taken with an LSM710 confocal microscope using a 63X-oil immersion objective. 50 μm-long dendrites were selected from the confocal images. The thickness of each PC dendritic trunk was measured at a distance of one cell-body diameter from the cell body as described [34].
To measure spine length and density, all types of spines (i.e., protrusions, stubby, filopodia, and mushroom-type spines) were counted within a 10 μm stretch of distal dendrite in Gnao−/− mice and their wild-type littermates, Gnao+/+. According to the typical criteria for classifying dendritic spines, only spines separated from the dendritic shaft by at least 0.5 μm were counted [35]. vGluT2-positive CF boutons were counted on 50 μm stretches of the main dendritic trunks from Gnao−/− mice and Gnao+/+ mice. The size of the vGluT2-positive fluorescent boutons was measured in a 200 μm-wide area of the ML from lobule II/III of each genotype. All quantifications for these results were performed semi-automatically using the ImageJ 1.50 software package (NIH).
Statistical analysis
Statistical analyses were performed to determine any significant differences between two groups by two-tailed unpaired t-tests using the GraphPad Prism 8 (GraphPad Software Inc.) or SigmaPlot 12.0 software (Systat Software, Inc.). All quantitative values are presented as means ± Standard error of mean (SEM). For all tests, p < 0.05 is considered significant and p values are presented in the results and/or figure legends.
Results
Gαo is required for cerebellar cortex development
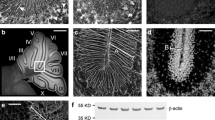
The anatomical development of the cerebellar lobules is completed around P21 [36]. Although the survival rates of Gnao−/− mice is extremely low, we were able to analyze the cerebella of the few Gnao−/− mice that survived to P180. To avoid lobular differences, we used sagittal sections of the vermal region in all experiments. Cresyl violet staining showed that the lack of Gαo does not alter the overall lobulation of the cerebellar cortex; the only significant difference we observed in Gnao−/− mice was a 62% reduction in the depth of the intercrural fissure between lobules VI and VII [37] (0.25 ± 0.02 mm in Gnao+/+ mice; 0.10 ± 0.04 mm in Gnao−/− mice; n = 3 mice for each genotype≥P25; *p < 0.05; arrows in Fig. 1a-b, and g). The overall size of the cerebellum was also reduced, with the cerebellar surface area in the midsagittal sections of the vermis being 25% smaller in Gnao−/− mice compared to Gnao+/+ mice (5.9 ± 0.39 mm2 in Gnao+/+ mice; 4.4 ± 0.23 mm2 in Gnao−/− mice; n = 4 mice for each genotype≥P25; *p < 0.05; Fig. 1h and Additional file 1). This hypoplasia was evident in the ML (Fig. 1c-d), the thickness of which in each folium was reduced by 11% in Gnao−/− mice (138.6 ± 2.27 μm in Gnao+/+ mice; 123.7 ± 2.16 μm in Gnao−/− mice; n = 4 mice for each genotype≥P25; ***p < 0.001; Fig. 1i). Since the thickness and shape of the GCL varies among the folia, we compared the total area occupied by the GCL in Gnao−/− mice to that of their wild-type littermates (Additional file 1). The GCL was reduced to an extent similar to that of the ML (2.4 ± 0.14 mm2 in Gnao+/+ mice; 1.9 ± 0.01 mm2 in Gnao−/− mice; n = 3 mice for each genotype≥P25; *p < 0.05; Fig. 1j), and a total number of GCs were reduced by 27% in Gnao−/− mice compared to their wild-type littermates (155.0*103 ± 2.67*103 cells in Gnao+/+ mice; 113.7*103 ± 5.77*103 cells in Gnao−/− mice; n = 3 mice for each genotype≥P25; **p < 0.005; Fig. 1k). Thus, we did not find a significant difference in the occupancy ratios of the ML to the GCL in Gnao−/− mice compared to their wild-type littermates (n = 4 for each genotype≥P25; Fig. 1l).

Gαo is required for cerebellar cortex development. a-b Overall cerebellum size of Gnao−/− mice is smaller than that of their wild-type Gnao+/+littermates. Both Gnao+/+ and Gnao−/− mice show the ten typical cerebellar lobules, but the depth of the folia in lobules VI-VII is reduced in Gnao−/− mice (arrows). c-d Cresyl violet staining shows the normal arrangement of the three cortical layers. The ML is thinner in Gnao−/− mice than Gnao+/+ mice. e-f Gαo is abundant in the ML and GCL of Gnao+/+, but undetectable in the ML and GCL of Gnao−/− mice. The asterisks in e highlight the absence of Gαo in PC soma. Note the difference of ML thickness. Scale bars, 500 μm in a-b, 50 μm in c-f. g The intercrural fissure between lobules VI and VII is reduced in Gnao−/− mice. h Cerebellar areas in mid-sagittal sections are smaller in Gnao−/− mice. i ML thickness is reduced in Gnao−/− mice (n = 119 sections from 4 Gnao+/+ mice and 122 sections from 4 Gnao−/− mice). j GCL occupancy is 20% lower in Gnao−/− mice than Gnao+/+ mice. k The number of total GCs in the GCL area is reduced by 27% in Gnao−/− mice. l Occupancy ratios for the GCL (43.4 ± 0.73 in Gnao+/+ mice and 44.8 ± 0.96 in Gnao−/− mice; p = 0.300) and for the ML (45.3 ± 0.97 in Gnao+/+ mice and 43.1 ± 1.33 in Gnao−/− mice; p = 0.248) are similar when the smaller size of the cerebellum in Gnao−/− mice is taken into account. m upper. Genotyping shows the mutant (Gnao−) and the wild type (Gnao+) alleles as 275 and 224 bp, respectively. Lower. Western blot analysis shows the absence of Gαo, but not Gαi. Data are means ± SEM; *p < 0.05, **p < 0.005, ***p < 0.001 vs. Gnao+/+. N.S.; No significant difference, STD; standard marker
Immunostaining with an anti-Gαo antibody revealed a broad distribution of Gαo protein in the ML and GCL (Fig. 1e), but not in the PC somata within the PCL (asterisks in Fig. 1e). We verified the specificity of the anti-Gαo antibody by confirming an absence of immunoreactivity in Gnao−/− mice (Fig. 1f) as well by western analysis (Fig. 1m).
Gαo is targeted to the synaptic membranes of cerebellar neurons
The apparent absence of Gαo-immunoreactivity we observed in PC somata within the PCL was not in accordance with previous reports of Gαo expression in PCs (Table 1). To determine the precise location of Gαo protein, we performed an immunostaining experiment to detect Gαo and Pcp2 in the adult cerebellum. Pcp2 is a well-known PC-specific marker that interacts with Gαo [15]. We observed Gαo protein in the membrane compartment of PC dendrites but not in the cytosolic compartment of PC soma (Fig. 1e, asterisk), whereas we observed Pcp2 staining both in PC dendrites and soma (Fig. 2a). Importantly, Gαo was co-localized with Pcp2 in the dendritic spines (Fig. 2a). Such co-localization was evident in the sprouting dendritic tips and in pseudopodial protrusions emerging from the apical pole of early phase PCs in P7 (Fig. 2b). An in situ hybridization experiment revealed the presence of Gnao mRNA in the perikaryon of PCs, GCs, and interneurons of the ML (arrowheads in Fig. 2c1, c2, and c3, respectively). Gαo protein expression was more evident upon in vitro dissociation in cultured PCs (Fig. 2d). Unlike in the in vivo immunostaining experiment, we observed Gαo protein both in PC soma (asterisk) and dendrites (arrows in Fig. 2d). This immunoreactivity showed an overlap with that of anti-Calb, another PC-specific marker (Fig. 2d). These results indicate Gαo protein is highly mobile in the membrane compartment after being synthesized in the PC soma. This leads to the apparent absence of Gαo protein in the PCL in vivo (Table 1). Further studies with immune-electron microscopy with the Gnao+/+ and Gnao−/− mice may clarify this issue. In dissociated cultures of P7 cerebellum in which external granule layer (EGL) cells highly proliferate, we found most cells are Tubb3- and Gαo-positive (Fig. 2e). Considering that GCs greatly outnumber interneurons by 414:1 [40, 41], it is very likely most Tubb3- and Gαo-positive cells in Fig. 2e are GCs although Gnao mRNA is expressed both in GCs and interneurons in the ML (Fig. 2c). Thus, the reduced size of the ML indicates a reduced total number of GCs in the absence of Gαo (Fig. 1k).

Gαo is highly targeted to the synaptic terminal regions of cerebellar neurons in wild-type mice. a Gαo-immunoreactivity appears at high levels throughout PC dendrites, especially in Pcp2-positive spines (Arrows in a1-a3, magnified views of a box shown in a). Scale bars, 50 μm in a, 10 μm in a1-a3. b PCs start to differentiate with many protrusions and neurites in a monolayer. Gαo is co-localized with Calb, another PC-specific marker at every PC boundary (yellow, arrows in b2 and b3) except in PC soma devoid of Gαo (asterisks in b2). Scale bars, 20 μm in b-b3. c Strong Gnao mRNA signals appear in the perinuclear regions of PC soma (c1), GCs (c2), and ML interneurons (c3). Scale bars, 20 μm in c, 10 μm in c1-c3. d In vitro culture of PCs shows Gαo and calbindin-D28K (Calb) expression at 21 days in vitro (DIV). The cultured PCs show significant Gαo signal in the soma (asterisk, d2) and in the dendrites (arrows in d1-d2). e In GCs at DIV7, Gαo protein appears in the soma (asterisks, e2) and in the neurites (arrows in e1-e2) of Tubb3-positive GCs. Scale bars, 20 μm in d-e. Bisbenzimide (Hoechst 33342) staining (blue) was used to visualize cell nuclei in a-e
Gαo is required for the maturation of PC dendrites
After immunostaining PFs in Gnao−/− mice with an anti-vGluT1 antibody, we found a generalized reduction in the thickness of the ML, but no more specific abnormalities (Additional file 2). Immunostaining of Gnao−/− mice with an anti-Calb antibody revealed no difference in the arrangement and number of PC somata in the PCL in Gnao−/− mice (n = 4 for each genotype; Fig. 3a-b, and g). We estimated PC monoplanarity by counting the number of PCs with primary dendrites stretching longer than 30 μm. About 70.3 ± 6.20% of the wild-type PCs retained primary dendrites longer than 30 μm (Fig. 3c, h), which is consistent with a previous report that roughly 70–80% PCs are monoplanar after P25 [21]. This suggests our semi-quantitative approach is relevant and indirectly indicates planarity as was previously noted [34]. In comparison, Gnao−/− mice show only 47.1 ± 4.85% monoplanar PCs, suggesting that roughly 33% PCs in Gnao−/− mice do not acquire monoplanarity during the early postnatal period (n = 3 mice for each genotype≥P25; *p < 0.05; Fig. 3d, h). Importantly, we observed a reduction in the thickness of PC dendritic trunks by 25% (3.2 ± 0.06 μm in Gnao+/+ mice and 2.4 ± 0.06 μm in Gnao−/− mice; n = 3 mice for each genotype≥P25; ***p < 0.001; Fig. 3e-f, and i) and in the number of spines by 16% (1.01 ± 0.01 /μm in Gnao+/+ mice; 0.85 ± 0.01 /μm in Gnao−/− mice; n = 3 mice for each genotype≥P180; ***p < 0.001; Fig. 3j) accompanied by an increase in average spine length by 9% (0.7 ± 0.01 μm in Gnao+/+ mice; 0.8 ± 0.02 μm in Gnao−/− mice; n = 3 mice for each genotype≥P180; ***p < 0.001; Fig. 3k). These data suggest Gαo deficiency disrupts the development of the monoplanarity of typical fan-shaped PCs, their dendritic arborization, and their spine formation without affecting their migration or alignment. Interestingly, when we stained Gnao−/− mice and visualized the vGluT2-positive terminal boutons of CFs with vertical extensions along the main dendrite trunks of PCs (Fig. 4a-b), we found a 32% reduction in the number of CF boutons on PCs (23.3 ± 2.02 in Gnao+/+ mice; 16.0 ± 1.38 in Gnao−/− mice; n = 4 mice for each genotype≥P25; *p < 0.05; Fig. 4c) and an 11% reduction in the average size of CF boutons (2.1 ± 0.05 μm2 in Gnao+/+ mice; 1.9 ± 0.06 μm2 in Gnao−/− mice; n = 4 mice for each genotype≥P25; **p < 0.005; Fig. 4d). This atrophy of CF boutons and PC spines may indicate an essential role for Gαo in full functional maturation of synapses between presynaptic ION neurons and postsynaptic PCs.

Gαo is required for PC maturation. a-b The thickness of the ML is reduced (Bars in a, b) without changing PCL alignment. Scale bar, 100 μm. c-d Calbindin-D28k (Calb) staining reveals whole PC dendritic trees throughout the ML of the adult cerebellum. Scale bar, 20 μm in c-d. e-f Images of dendritic spines detected with an anti-Calb antibody. Arrows indicate spines longer than 1.5 μm in Gnao−/− mice. Scale bar, 5 μm in e-f. g The average number of PCs per 200 μm is similar between Gnao+/+ mice and Gnao−/− mice (11.5 ± 0.23 in Gnao+/+ mice and 11.0 ± 0.30 in Gnao−/− mice; p = 0.232 vs. Gnao+/+). h 70.3 ± 6.20% of total PCs in Gnao+/+ mice and 47.1 ± 4.85% of total PCs in Gnao−/− mice extend primary dendrites longer than 30 μm (n = 61 PCs from 3 Gnao+/+ mice and 76 PCs from 3 Gnao−/− mice). i Dendritic trunk widths are decreased in Gnao−/− mice (n = 41 primary dendrites from 3 Gnao+/+ mice and 39 primary dendrites from 3 Gnao−/− mice). j Gnao−/− mice exhibit a significant reduction of spine density (n = 123 segments of 122 distal dendrites from 3 Gnao+/+ mice and 130 segments of 106 distal dendrites from 3 Gnao−/− mice). k Average spine length is longer in Gnao−/− mice (n = 210 dendrite spines from 3 Gnao+/+ mice and 195 dendrite spines from 3 Gnao−/− mice). Data are means ± SEM; *p < 0.05, ***p < 0.001 vs. Gnao+/+. N.S.; No significant difference

Gαo is required for the maturation of CF-PC synapses. a-b Gnao−/− mice show atrophied vGluT2-positive morphology of CF synaptic boutons (open arrowheads in b1) and PC dendrites in the ML relative to Gnao+/+ mice (arrowheads in a1). Scale bar, 10 μm. c Gnao−/− mice show fewer vGluT2-positive dendritic boutons (n = 9 main dendrites from 4 Gnao+/+ mice and n = 7 main dendrites from 4 Gnao−/− mice). d Bouton size is smaller in Gnao−/− mice (P25) (n = 655 vGluT2-positive boutons from 4 Gnao+/+ mice and 432 vGluT2-positive boutons from 4 Gnao−/− mice). Data are means ± SEM; *p < 0.05, **p < 0.005 vs. Gnao+/+
Discussion
The cerebellum is a useful model utilized for studying many aspects of neural development because of its typical cytoarchitecture and developmental program [42]. In this study, we show that Go plays a crucial role in the postnatal development of the cerebellar cortex. Specifically, Gnao−/− mice exhibit hypoplasia of the cerebellum (Fig. 1 and Additional file 1) with overall defects in cerebellar cortex development. In Gnao−/− mice, the thickness of the ML and GCL are significantly reduced (Fig. 1) without more specific abnormalities in vGluT1-expression (Additional file 2), the monoplanarity, dendritic arborization and spinogenesis of PCs are reduced (Fig. 3), and the maturation of CF boutons are atrophied (Fig. 4).
The intensive remodeling and maturation of PC dendrites that occurs during the postnatal period is controlled by intrinsic factors early in development and by a careful orchestration of intrinsic and extrinsic factors later in development [22]. The abnormalities we observed in the PC dendrites and spines of Gnao−/− mice may therefore be either a consequence of the lack of Go-mediated signaling in the PCs themselves (intrinsic factors) or a consequence of their defective formation of synapses with CF boutons (extrinsic interactions).
Intrinsic factors that regulate Ca2+ homeostasis, such as mGluR1 and its downstream signaling pathways, are critical in PCs for the selection of the winning CF and the pruning of redundant CFs that originate in the ION [43]. Mice lacking even a single component of the mGluR1-Gαq-PLCβ4-PKCγ signaling cascade in PCs show multiple CF effects. Although such mice show normal PC dendritic growth, they have motor defects that include ataxia and impaired motor learning [24,25,26, 44]. Activation of PLCβ occurs when Gβγ dimers are released from Gq and Gi/o, but the binding sites and activation kinetics differ between Gαq and Gβγ [45]. Western analyses indicated that the expression levels of Gαi and Gαq were not altered by Gnao-deletion (Fig. 1m and Additional file 3). Furthermore, the loss of Gαo does not increase free Gβγ dimers in Gnao−/− mice [8]. Thus, the phenotypes of Gnao−/− mice are more likely due to the loss of some of Gαo specific unique functions rather than being due to the activation of the Gβγ-PLCβ pathway.
Gi/o signals are known to mediate presynaptic inhibitory effects of many neurotransmitters on transmitter release in axon terminals [46]. Activation of Gi/o-coupled CB1 receptors decreases glutamate release in presynaptic PF terminals by suppressing Ca2+ entry via voltage-gated Ca2+ channels (N-, P/Q-, and R-types) or modulating vesicle-release related proteins [47,48,49]. Considering that a CB1 agonist predominantly activates Go in the cerebellum and Go exerts its inhibitory function on N- and P/Q type channels [16, 50], the Ca2+ influx in PF terminals is highly likely mediated by the Go signals. In PCs, Pcp2 stimulates GDP release from Gαo through its GoLoco domain and modulates the inhibitory functions of Go on P/Q-type (Cav2.1) channels [15, 17, 51]. Deletion of Cacna1a, the gene encoding the pore-forming α1 subunit of P/Q-type channels in the entire cerebellum or in PCs impairs the maturation of CFs [52, 53]. Pcp2 expression was not altered in our Gnao−/− mice, while the terminal boutons of CFs were atrophied (Fig. 4 and Additional file 3). It would be of great interest to determine whether the functions of voltage-gated Ca2+ channels are altered in Gnao−/− mice.
Gαo is highly enriched in the cerebellar ML, particularly at the plasma membranes of PC dendrites and in PFs of the ML. Gαo membrane targeting may be associated with lipid modifications such as myristoylation or palmitoylation [54]. The distinctive membrane compartmentalization of Gαo in PCs (Table 1) makes it well-suited to perform synapse-specific studies. Consistent with our data, Gαo appears at high levels in postsynaptic densities in vivo as well as in the neurite tips and growth cones of in vitro cultured neurons [7, 12, 13, 55]. Consistently, we show that Gαo is found in the sprouting dendrites of PCs in P7 cerebellum (Fig. 2b). It is noteworthy that migration of granule cells from EGL to GCL appear normal, suggesting that the Bergman’s glia functions properly.
In conclusion, our study suggests Go may perform critical roles in the postnatal development of the cerebellar cortex. Future studies cell type specific deletion of Gnao will reveal the correlation of intrinsic signaling pathways in cerebellar cortical neurons with PF-PC synapse and CF-PC synapse. Gabra6 (Δα6)- and Pcp2(L7)-driven Cre-expressing mice have been utilized for the study of GCs and PCs, respectively [56,57,58]. However, Gαo protein is detectable due to its high stability even after the Gnao gene is deleted by Cre enzymes in postnatal neurons (data not shown). Inducible Cre mouse lines such as Gli1-CreERT2 may be an alternative strategy to induce concomitant deletion of the Gnao gene and the existing Gαo proteins in proliferating EGL cells [59].
Availability of data and materials
All of the data generated and analyzed in this study are included in the published article.
Abbreviations
- AC:
-
Adenylyl cyclase
- Calb:
-
Calbindin-D28K
- cAMP:
-
Cyclic AMP, 3′,5′-cyclic adenosine monophosphate
- CB1:
-
Cannabinoid receptor 1
- CFs:
-
Climbing fibers
- CNS:
-
Central nervous system
- DCN:
-
Deep cerebellar nuclei
- DEPC:
-
Diethylpyrocarbonate
- DIV:
-
Days in vitro
- DMEM:
-
Dulbecco’s modified Eagle medium
- EGL:
-
External granule layer
- EIEE:
-
Early infantile epileptic encephalopathy
- FBS:
-
Fetal bovine serum
- FITC:
-
Fluorescein isothiocyanate
- GCL:
-
Granule cell layer
- GCs:
-
Granule cells
- GPCRs:
-
G-protein coupled receptors
- G-proteins:
-
GTP binding proteins
- Gαo :
-
GTP-binding Protein alpha subunit of Go
- HBSS:
-
Hank’s balanced salt solution
- ION:
-
Inferior olivary nucleus
- mGluR1:
-
Metabotropic glutamate receptor type 1
- ML:
-
Molecular layer
- N.S.:
-
No significant
- NBF:
-
Neutral buffered formalin
- ND:
-
Not Determined
- P:
-
Postnatal day
- PBS:
-
Phosphate buffered saline
- PCL:
-
Purkinje cell layer
- Pcp2:
-
Purkinje cell protein 2
- PCR:
-
Polymerase chain reaction
- PCs:
-
Purkinje cells
- PDL:
-
Poly-D-lysine
- PFs:
-
Parallel fibers
- PKCγ:
-
Protein kinase Cγ
- PLCβ4:
-
Phospholipase Cβ4
- SDS:
-
Sodium dodecyl sulfate
- SEM:
-
Standard error of mean
- SSC:
-
Saline-sodium citrate
- STD:
-
Standard marker
- TBS:
-
Tris-buffered saline
- vGluT1/2:
-
Vesicular glutamate transporter 1/2
References
Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science. 1991;252:802–8.
Neer EJ. Heterotrimeric G proteins: organizers of transmembrane signals. Cell. 1995;80:249–57.
Birnbaumer L. Expansion of signal transduction by G proteins. The second 15 years or so: from 3 to 16 alpha subunits plus betagamma dimers. Biochim Biophys Acta. 2007;1768:772–93.
Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, et al. Gi2 alpha protein deficiency: a model of inflammatory bowel disease. J Clin Immunol. 1995;15:101S–5S.
Devanathan V, Hagedorn I, Kohler D, Pexa K, Cherpokova D, Kraft P, et al. Platelet Gi protein Galphai2 is an essential mediator of thrombo-inflammatory organ damage in mice. Proc Natl Acad Sci U S A. 2015;112:6491–6.
Boknik P, Grote-Wessels S, Barteska G, Jiang M, Muller FU, Schmitz W, et al. Genetic disruption of G proteins, G(i2) alpha or G(o) alpha, does not abolish inotropic and chronotropic effects of stimulating muscarinic cholinoceptors in atrium. Br J Pharmacol. 2009;158:1557–64.
Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, et al. Cell type- and brain region-resolved mouse brain proteome. Nat Neurosci. 2015;18:1819–31.
Jiang M, Gold MS, Boulay G, Spicher K, Peyton M, Brabet P, et al. Multiple neurological abnormalities in mice deficient in the G protein go. Proc Natl Acad Sci U S A. 1998;95:3269–74.
Choi JM, Kim SS, Choi CI, Cha HL, Oh HH, Ghil S, et al. Development of the main olfactory system and main olfactory epithelium-dependent male mating behavior are altered in go-deficient mice. Proc Natl Acad Sci U S A. 2016;113:10974–9.
Nakamura K, Kodera H, Akita T, Shiina M, Kato M, Hoshino H, et al. De novo mutations in GNAO1, encoding a Galphao subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am J Hum Genet. 2013;93:496–505.
Talvik I, Moller RS, Vaher M, Vaher U, Larsen LH, Dahl HA, et al. Clinical phenotype of De novo GNAO1 mutation: case report and review of literature. Child neurology open. 2015;2:2329048X15583717.
Strittmatter SM, Valenzuela D, Vartanian T, Sudo Y, Zuber MX, Fishman MC. Growth cone transduction: go and GAP-43. J Cell Sci Suppl. 1991;15:27–33.
Ghil SH, Kim BJ, Lee YD, Suh-Kim H. Neurite outgrowth induced by cyclic AMP can be modulated by the alpha subunit of go. J Neurochem. 2000;74:151–8.
Worley PF, Baraban JM, Van Dop C, Neer EJ, Snyder SH. Go, a guanine nucleotide-binding protein: immunohistochemical localization in rat brain resembles distribution of second messenger systems. Proc Natl Acad Sci U S A. 1986;83:4561–5.
Luo Y, Denker BM. Interaction of heterotrimeric G protein Galphao with Purkinje cell protein-2. Evidence for a novel nucleotide exchange factor. J Biol Chem. 1999;274:10685–8.
Prather PL, Martin NA, Breivogel CS, Childers SR. Activation of cannabinoid receptors in rat brain by WIN 55212-2 produces coupling to multiple G protein alpha-subunits with different potencies. Mol Pharmacol. 2000;57:1000–10.
Iscru E, Serinagaoglu Y, Schilling K, Tian J, Bowers-Kidder SL, Zhang R, et al. Sensorimotor enhancement in mouse mutants lacking the Purkinje cell-specific Gi/o modulator, Pcp2(L7). Mol Cell Neurosci. 2009;40:62–75.
Cerminara NL, Lang EJ, Sillitoe RV, Apps R. Redefining the cerebellar cortex as an assembly of non-uniform Purkinje cell microcircuits. Nat Rev Neurosci. 2015;16:79–93.
Ichikawa R, Hashimoto K, Miyazaki T, Uchigashima M, Yamasaki M, Aiba A, et al. Territories of heterologous inputs onto Purkinje cell dendrites are segregated by mGluR1-dependent parallel fiber synapse elimination. Proc Natl Acad Sci U S A. 2016;113:2282–7.
Watanabe M, Kano M. Climbing fiber synapse elimination in cerebellar Purkinje cells. Eur J Neurosci. 2011;34:1697–710.
Kaneko M, Yamaguchi K, Eiraku M, Sato M, Takata N, Kiyohara Y, et al. Remodeling of monoplanar Purkinje cell dendrites during cerebellar circuit formation. PLoS One. 2011;6:e20108.
Sotelo C, Dusart I. Intrinsic versus extrinsic determinants during the development of Purkinje cell dendrites. Neuroscience. 2009;162:589–600.
Schuller U, Lamp EC, Schilling K. Developmental expression of heterotrimeric G-proteins in the murine cerebellar cortex. Histochem Cell Biol. 2001;116:149–59.
Kano M, Hashimoto K, Kurihara H, Watanabe M, Inoue Y, Aiba A, et al. Persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking mGluR1. Neuron. 1997;18:71–9.
Offermanns S, Hashimoto K, Watanabe M, Sun W, Kurihara H, Thompson RF, et al. Impaired motor coordination and persistent multiple climbing fiber innervation of cerebellar Purkinje cells in mice lacking Galphaq. Proc Natl Acad Sci U S A. 1997;94:14089–94.
Hashimoto K, Watanabe M, Kurihara H, Offermanns S, Jiang H, Wu Y, et al. Climbing fiber synapse elimination during postnatal cerebellar development requires signal transduction involving G alpha q and phospholipase C beta 4. Prog Brain Res. 2000;124:31–48.
Furuya S, Makino A, Hirabayashi Y. An improved method for culturing cerebellar Purkinje cells with differentiated dendrites under a mixed monolayer setting. Brain Res Brain Res Protoc. 1998;3:192–8.
Fujishima K, Horie R, Mochizuki A, Kengaku M. Principles of branch dynamics governing shape characteristics of cerebellar Purkinje cell dendrites. Development. 2012;139:3442–55.
Rieff HI, Raetzman LT, Sapp DW, Yeh HH, Siegel RE, Corfas G. Neuregulin induces GABA(a) receptor subunit expression and neurite outgrowth in cerebellar granule cells. J Neurosci. 1999;19:10757–66.
Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by sonic hedgehog. Neuron. 1999;22:103–14.
Leto K, Arancillo M, Becker EB, Buffo A, Chiang C, Ding B, et al. Consensus Paper: Cerebellar Development. Cerebellum. 2016;15:789–828.
Choi CI, Yoon SP, Choi JM, Kim SS, Lee YD, Birnbaumer L, et al. Simultaneous deletion of floxed genes mediated by CaMKIIalpha-Cre in the brain and in male germ cells: application to conditional and conventional disruption of Goalpha. Exp Mol Med. 2014;46:e93.
Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5.
Donald S, Humby T, Fyfe I, Segonds-Pichon A, Walker SA, Andrews SR, et al. P-Rex2 regulates Purkinje cell dendrite morphology and motor coordination. Proc Natl Acad Sci U S A. 2008;105:4483–8.
von Bohlen Und Halbach O. Structure and function of dendritic spines within the hippocampus. Ann Anat. 2009;191:518–31.
Espinosa JS, Luo L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. J Neurosci. 2008;28:2301–12.
Legue E, Gottshall JL, Jaumouille E, Rosello-Diez A, Shi W, Barraza LH, et al. Differential timing of granule cell production during cerebellum development underlies generation of the foliation pattern. Neural Dev. 2016;11:17.
Largent BL, Jones DT, Reed RR, Pearson RC, Snyder SH. G protein mRNA mapped in rat brain by in situ hybridization. Proc Natl Acad Sci U S A. 1988;85:2864–8.
Brann MR, Collins RM, Spiegel A. Localization of mRNAs encoding the alpha-subunits of signal-transducing G-proteins within rat brain and among peripheral tissues. FEBS Lett. 1987;222:191–8.
Korbo L, Andersen BB, Ladefoged O, Moller A. Total numbers of various cell types in rat cerebellar cortex estimated using an unbiased stereological method. Brain Res. 1993;609:262–8.
Weisheit G, Gliem M, Endl E, Pfeffer PL, Busslinger M, Schilling K. Postnatal development of the murine cerebellar cortex: formation and early dispersal of basket, stellate and Golgi neurons. Eur J Neurosci. 2006;24:466–78.
Qin L, Wine-Lee L, Ahn KJ, Crenshaw EB 3rd. Genetic analyses demonstrate that bone morphogenetic protein signaling is required for embryonic cerebellar development. J Neurosci. 2006;26:1896–905.
Hashimoto K, Kano M. Synapse elimination in the developing cerebellum. Cell Mol Life Sci. 2013;70:4667–80.
Hashimoto K, Ichikawa R, Takechi H, Inoue Y, Aiba A, Sakimura K, et al. Roles of glutamate receptor delta 2 subunit (GluRdelta 2) and metabotropic glutamate receptor subtype 1 (mGluR1) in climbing fiber synapse elimination during postnatal cerebellar development. J Neurosci. 2001;21:9701–12.
Lyon AM, Tesmer JJ. Structural insights into phospholipase C-beta function. Mol Pharmacol. 2013;84:488–500.
Brown DA, Sihra TS. Presynaptic signaling by heterotrimeric G-proteins. Handb Exp Pharmacol. 2008:207–60.
Kreitzer AC, Regehr WG. Retrograde signaling by endocannabinoids. Curr Opin Neurobiol. 2002;12:324–30.
Zhang W, Linden DJ. Neuromodulation at single presynaptic boutons of cerebellar parallel fibers is determined by Bouton size and basal action potential-evoked ca transient amplitude. J Neurosci. 2009;29:15586–94.
Hoxha E, Tempia F, Lippiello P, Miniaci MC. Modulation, plasticity and pathophysiology of the parallel Fiber-Purkinje cell synapse. Front Synaptic Neurosci. 2016;8:35.
Furukawa T, Miura R, Mori Y, Strobeck M, Suzuki K, Ogihara Y, et al. Differential interactions of the C terminus and the cytoplasmic I-II loop of neuronal Ca2+ channels with G-protein alpha and beta gamma subunits. II. Evidence for direct binding. J Biol Chem. 1998;273:17595–603.
Kinoshita-Kawada M, Oberdick J, Xi Zhu M. A Purkinje cell specific GoLoco domain protein, L7/Pcp-2, modulates receptor-mediated inhibition of Cav2.1 Ca2+ channels in a dose-dependent manner. Brain Res Mol Brain Res. 2004;132:73–86.
Miyazaki T, Hashimoto K, Shin HS, Kano M, Watanabe M. P/Q-type Ca2+ channel alpha1A regulates synaptic competition on developing cerebellar Purkinje cells. J Neurosci. 2004;24:1734–43.
Hashimoto K, Tsujita M, Miyazaki T, Kitamura K, Yamazaki M, Shin HS, et al. Postsynaptic P/Q-type Ca2+ channel in Purkinje cell mediates synaptic competition and elimination in developing cerebellum. Proc Natl Acad Sci U S A. 2011;108:9987–92.
Chen CA, Manning DR. Regulation of G proteins by covalent modification. Oncogene. 2001;20:1643–52.
Fernandez E, Collins MO, Uren RT, Kopanitsa MV, Komiyama NH, Croning MD, et al. Targeted tandem affinity purification of PSD-95 recovers core postsynaptic complexes and schizophrenia susceptibility proteins. Mol Syst Biol. 2009;5:269.
Aller MI, Jones A, Merlo D, Paterlini M, Meyer AH, Amtmann U, et al. Cerebellar granule cell Cre recombinase expression. Genesis. 2003;36:97–103.
Slugocka A, Wiaderkiewicz J, Barski JJ. Genetic targeting in cerebellar Purkinje cells: an update. Cerebellum. 2017;16:191–202.
Zhang XM, Ng AH, Tanner JA, Wu WT, Copeland NG, Jenkins NA, et al. Highly restricted expression of Cre recombinase in cerebellar Purkinje cells. Genesis. 2004;40:45–51.
Ahn S, Joyner AL. Dynamic changes in the response of cells to positive hedgehog signaling during mouse limb patterning. Cell. 2004;118:505–16.
Acknowledgements
We thank Dr. Serena Dudek (National Institute of Environmental Health Sciences) for helpful comments.
Funding
This research was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (2018M3A9G1082594 to HS-K and 2017R1D1A1B03028120 to J-MC) and by the Intramural Research Program of the NIH (Project Z01-ES-101643 to LB).
Author information
Authors and Affiliations
Contributions
HLC, J-MC, H-HO and NB performed experiments; HLC, J-MC, Y-DL and S-SK conducted analyses of the data; LB and HS-K supervised the research; HLC, J-MC, LB and HS-K wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
All experimental procedures involving the use of animals were reviewed and approved by the Institutional Animal Research Ethics Committee at Ajou University Medical Center.
Consent for publication
Not applicable.
Competing interests
HS-K is a member of scientific board of Cell&Brain, a company studying the potential therapeutic use of mesenchymal stem cell. The all authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
The gross anatomy of the cerebellum is normal in Gnao−/− mice. (PDF 187 kb)
Additional file 2:
PFs in the ML visualized in vGluT1-stained cerebellum. (PDF 85 kb)
Additional file 3:
Expression of Pcp2 in the cerebellum of Gnao−/− mice. (PDF 380 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cha, H.L., Choi, JM., Oh, HH. et al. Deletion of the α subunit of the heterotrimeric Go protein impairs cerebellar cortical development in mice. Mol Brain 12, 57 (2019). https://doi.org/10.1186/s13041-019-0477-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13041-019-0477-9