
Abstract
Background
Copy-number variants (CNVs) drive many neurodevelopmental-related disorders. Although many neurodevelopmental-related CNVs can give rise to widespread phenotypes, it is necessary to identify the major genes contributing to phenotypic presentation. Copy-number variations in chromosome 6, such as independent 6p deletion and 6p duplication, have been reported in several live-born infants and present widespread abnormalities such as intellectual disability, growth deficiency, developmental delay, and multiple dysmorphic facial features. However, a contiguous deletion and duplication in chromosome 6p regions have been reported in only a few cases.
Case presentation
In this study, we reported the first duplication of chromosome band 6p25.3–p22.3 with deletion of 6p25.3 in a pedigree. This is the first case reported involving CNVs in these chromosomal regions. In this pedigree, we reported a 1-year-old boy with maternal 6p25-pter duplication characterized by chromosome karyotype. Further analysis using CNV-seq revealed a 20.88-Mb duplication at 6p25.3-p22.3 associated with a contiguous 0.66-Mb 6p25.3 deletion. Whole exome sequencing confirmed the deletion/duplication and identified no pathogenic or likely pathogenic variants related with the patient´s phenotype. The proband presented abnormal growth, developmental delay, skeletal dysplasia, hearing loss, and dysmorphic facial features. Additionally, he presented recurrent infection after birth. CNV-seq using the proband´s parental samples showed that the deletion/duplication was inherited from the proband´s mother, who exhibited a similar phenotype to the proband. When compared with other cases, this proband and his mother presented a new clinical finding: forearm bone dysplasia. The major candidate genes contributing to recurrent infection, eye development, hearing loss features, neurodevelopmental development, and congenital bone dysplasia were further discussed.
Conclusions
Our results showed a new clinical finding of a contiguous deletion and duplication in chromosome 6p regions and suggested candidate genes associated with phenotypic features, such as FOXC1, SERPINB6, NRN1, TUBB2A, IRF4, and RIPK1.
Similar content being viewed by others

Background
Neurodevelopmental disorders are the most prevalent and common chronic medical events in paediatrics, with a prevalence of 2–3% in the population around the world [1,2,3]. In a broader conceptualization, neurodevelopmental disorders include developmental delay (DD), intellectual disability (ID), autism or autism spectrum disorder (ASD), epilepsy, and behavioural abnormalities along with developmental disabilities [4]. Among them, developmental delay (DD) and intellectual disability (ID) are some of the most common conditions in neurodevelopmental disorders, affecting up to approximately 3% of the paediatric population [5,6,7,8]. DD and ID are complementary entities separated chronologically because IQ testing is not quite valid and reliable for younger children (younger than 5 years). Therefore, DD is reserved for younger children, and ID is applied for older children [9]. Generally, children’s developmental domains include gross/fine motor development, speech/language development, cognitive development, social/personal development, and activities of daily living. DD/ID is defined as a significant delay in two or more developmental domains. Patients express serious neurodevelopmental disorders during childhood [10].
Neurodevelopmental disorders, especially DD/ID, represent a heterogeneous group of disorders. The reasons for these diseases are quite complicated, including environmental factors and genetic factors. It has been reported that genetic factors contribute at least a quarter to half of the aetiological reasons for DD/ID, including copy-number variants, structural variants, and single-nucleotide variations. To date, copy-number variants comprising chromosomal deletions and duplications have been proven to be a major reason for neurodevelopmental disorders, especially DD and ID [3, 11]. Pathogenic CNVs can alter the structure and function of the genes, resulting in human genetic diseases. The majority of pathogenic CNVs that lose or gain genetic material are based on replication error or DNA repair mechanisms. Approximately 25.7% of children with developmental delay were due to deleterious CNVs larger than 400 kb [12].
Differences in size, the number of affected genes, and even the precise breakpoints of each CNV may cause different neurodevelopmental phenotypes. It has been reported that CNV variations on the short arm of chromosome 6 result in different phenotypes, especially different breakpoints, either interstitial (breakpoints within the 6p22p24 region) or terminal (breakpoints within the 6p24-pter region) [13]. In addition, Martinet et al. reported that the CNV variations in 6p are often more complex, they described an 8.1 Mb 6pter-6p24.3 deletion associated with a contiguous 5.8 Mb 6p24.36p24.1 inverted duplication [14]. Nakane et al. described a patient with a 2.1-Mb 6p25.3 deletion and a 4.14-Mb 6p25.3p25.2 duplication [15]. Other researchers also reported the deletion and duplication on 6p [16,17,18,19]. However, the phenotypes of patients are highly heterogeneous. Nevertheless, the 6p deletion associated with a contiguous duplication is rare, and fewer than 10 individuals have been reported [13,14,15, 17]. To evaluate the variability of clinical features in patients with 6p CNVs, it is necessary to report additional cases to better review genotype–phenotype correlations.
In this study, we reported a patient with neurodevelopmental delay with facial anomalies. His mother exhibited a clinical phenotype that was almost identical to that of the proband. The genomic variations of these pedigree were assessed by chromosome karyotype, whole exome sequencing and copy number variation analysis. The results showed a 6p25.3 terminal deletion associated with a 6p25.3-p22.3 duplication in the proband and his mother. We discuss the phenotype diversity with those of previously reported patients and compared the putative genes in related CNV regions, aiming to establish genotype–phenotype correlations.
Case presentation
The patient, who is currently a 1.9-year-old Chinese boy, was the first child of nonconsanguineous parents. He is the only child of his parents. He was delivered by caesarean section at 35 weeks and 2 days of gestation due to high blood pressure and hyperglycaemia of his mother. The pregnancy was unremarkable except for unclear nasal bone and imperforate anus noted on ultrasound scan at 28 weeks of gestation. His birth weight was 1400 g (Z score, − 2.85), length was 42 cm (Z score, − 1.83), and occipitofrontal circumference (OFC) was 26 cm (Z score, − 4.19). The neonate's crying manifested a degree of weakness, and the percutaneous oxygenation was inadequate to maintain pace with the infant's spontaneous breathing subsequent to birth. Further examinations showed that he had anal atresia, patent ductus arteriosus, atrial septal defect, and pulmonary hypertension.
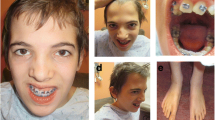
The patient revealed severe feeding difficulties, significant physical growth and neurobehavioural development delay after birth. At 1.4 years old, his weight was 7.73 kg (Z score, − 3.39), length was 65.2 cm (Z score, − 5.04), and occipitofrontal circumference (OFC) was 41.8 cm (Z score, − 4.29). As shown in Fig. 1A, he had multiple dysmorphic facial features, including a higharched palate, long philtrum, thin upper lip, abnormal nasal bridge morphology, upper eyelid entropion, alternating strabismus, nystagmus, and decreased width of the eyelids. His skull shape was abnormal, presenting as bilateral symmetrical defects of the parietal bone. In addition, he had a congenital choroidal defect in the left eye (Fig. 1B). He had congenital heart defects, such as patent foramen ovale, coronary sinus dilation, and persistent left superior chamber. Marked growth impairment was noted. He shows severe psychomotor developmental delay, including motor and intellectual development: at 1.4 years old, he could not sit unsupported, stand unassisted and walk independently. He was uncapable of speaking meaningful words. The Gesell developmental scale test revealed severe global development delay: gross motor skills DQ: 32; fine motor skills DQ: 34; adaptation ability DQ: 25; speaking skills DQ: 28; social capacity DQ: 10.

Clinical features of the proband and his mother. A Facial appearance of the patient at age 1 year and 5 months; B Congenital choroidal defect of the patient (Blue dotted box indicated the choroidal defect); C Picture of proband’s forearm. D Facial appearance of the mother at age 26 year; E Hand and arm picture of the mother
The results of routine serum biochemical and haematological tests were normal. Brain MRI showed abnormalities in the brain: bilateral ventricles were enlarged asymmetrically. Basic metabolic screening of haematuria, X-ray examination of the hip joint, and posterotemporal CT were also normal. He was found to have profound bilateral sensorineural deafness, with 50 dB in the right ear and 70 dB in the left ear. The patient underwent anal atresia plasty. In addition, although not obvious, the proband showed a slightly bowed radius and ulna. As shown in Fig. 1C, the picture indicated a widened radioulnar joint space and abnormal physiological curvature of the radius (excessive curvature). At present, the patient has been treated by family anal dilatation, and the shape and frequency of stool are basically normal. It should be noted that from birth to now, he had a history of at least five hospitalizations for common pneumonia. All detailed information is presented in Table 1.
The patient’s mother is a 26-year-old Chinese woman. She was married at 22 years old and was pregnant spontaneously at 25 years old. She is short in stature and 147 cm in height (< P3), and her weight is 41.5 kg (< P10). As shown in Fig. 1D, her facial dysmorphic features are similar to the proband´s features: long philtrum, thin upper lip, and abnormal nasal bridge morphology. She presented developmental delay after 6 months. Additionally, she had congenital moderate intellectual disability. She was uncapable of communicating with others normally and could hardly understand conversations now. Ultrasonic testing found that there was a hamartoma in her left kidney. In addition, her arms began to bend after birth, and now her arm and fingers could not be fully extended due to joint contractures (Fig. 1E), which was quite similar to the proband. The patient’s father was normal and developed well after birth. After a history of Japanese encephalitis at 3 years old, he had difficulty speaking, and neurocognitive development was impaired mildly. The pedigree of this family is shown in Fig. 2.

Pedigree of this family
To determine the cytogenetic and molecular variants of the proband, whether there were any chromosome abnormalities in the proband was evaluated, and chromosome analysis was utilized first. Two millilitres of peripheral blood from the patient was exsanguinated in a heparin anticoagulant tube. The blood sample was cultured with RPMI 1640 cell culture media, and 3–5 drops of colchicines (50 mg/mL) were added to the cells and further incubated for 1 h. Then, the cells were collected and incubated with 8 mL KCl buffer (0.075 mol/L) for 25 min. The cells were fixed and stained with Giemsa solution. Next, the cells were observed under a microscope, and the chromosome karyotype was analysed. Genomic DNA and metaphase chromosomes were obtained from the peripheral blood leukocytes of the proband. Chromosome analysis showed 46, XY, add(6)(p25), indicating a suspicious addition in chromosome 6 for unknown reason (Fig. 3A). No apparent pathogenic copy number variations were found in other chromosomal regions.

Genetic analysis of the proband. A Chromosome karyotype; B CNV-seq result showed a 20.88 Mb heterozygous duplication at 6p25.3-p22.3 and a 0.66 Mb heterozygous deletion at 6p25.3; C WES-CNV revealeda 0.3-Mb heterozygous deletion at 6p25.3 and a 20.49-Mb heterozygous duplication at 6p25.3-p22.3
To further detect the suspicious addition in chromosome 6, CNV-seq was applied for the proband. DNA was extracted from trio samples by MagMAX DNA Ultra 2.0 (Thermo Fisher, CA, USA). DNA samples were sequenced on the NGS platform (Berry Genomics, Beijing, China). A PCR-free-frag library was constructed for CNV-seq. Approximately 5 million 37-bp plus 8-bp (index) raw reads were generated for each sample after library sequencing on the NextSeq CN500 platform (Berry Genomics). Reads were processed, and CNVs were evaluated by an in-house pipeline using read counts based on a smoothness model (Berry Genomics, Beijing, China) according to the previous description. Surprisingly, the CNV-seq results not only showed a 20.88-Mb heterozygous duplication at 6p25.3-p22.3 (chr6:820,000–21,699,769, hg38) but also a 0.66-Mb heterozygous deletion at 6p25.3 (chr6:g.160000–820,000, hg38) (Fig. 3B).
The 20.88-Mb heterozygous duplication at 6p25.3p22.3 (chr6: 820,000–21,699,769, hg38) has not been recorded in the Database of Genomic Variants. This duplication region contains 89 RefSeq protein-coding genes, including FOXC1, TUBB2B, and NRN1. The upstream duplication breakpoint and downstream duplication breakpoint are in an intergenic region. The Decipher database and literature have reported several cases whose duplication regions overlapped or contained the proband’s duplication region. Almost all of these cases presented underdevelopment and multiple dysmorphic features. The Clingen CNV comprehensive score was greater than 0.99 (1.5). Therefore, this duplication region was classified as pathogenic CNV according to the ACMG rating guidelines for CNV [20].
The 0.66-Mb heterozygous deletion at 6p25.3 (chr6:g.160000–820,000, hg38) has not been recorded in the Database of Genomic Variants either. This deletion region contains 4 RefSeq protein-coding genes, including DUSP22, EXOC2, HUS1B and IRF4. Cases with deletion regions overlapping this deletion have rarely been reported, and only one male patient whose 6p25.2 presented a 0.72-Mb paternal deletion region was recorded in the Decipher database (Decipher#280,500). This patient presented with global developmental delay. The Clingen CNV comprehensive score was between − 0.89 and 0.89 (0). Therefore, this deletion region was classified as an uncertain significance CNV according to the ACMG rating guidelines for CNV.
To further exclude the possibility of other single-gene diseases, whole exome sequencing of the proband was performed. Whole exome sequencing results were compared by Sprinkl for CNV calling, and copy number calculation and CNV identification were performed in exons and long segment areas. The genomic coordinates of the WES results are indicated according to NCBI build 38 (hg38). After bioinformatics analysis, no pathogenic or likely pathogenic mutations related to phenotype were found. However, WES-CNV results revealed a 0.3-Mb heterozygous deletion at 6p25.3 (chr6:391,751–693,084, hg38) and a 20.49-Mb heterozygous duplication at 6p25.3-p22.3 (chr6:1,101,132–21,595,959, hg38) (Fig. 3C). Taking the CNV region and WES probe location together, the upstream breakpoint should be between chr6:1,053,627 and chr6:1,101,132, and the downstream breakpoint should be between chr6:21,595,959 and chr6:22,136,495.
To further verify the CNV parental origin of the proband, CNV-seq analysis was utilized for the proband’s mother and father. The CNV-seq result of the proband’s father revealed no pathogenic deletion or duplication variations (Additional file 1: Fig. S1A). The CNV-seq results of the mother indicated the presence of the same deletion/duplication that was identified in the proband. (Additional file 1: Fig. S1B). In addition, the chromosome karyotype of the mother showed 46, XY, add(6)(p25) (Additional file 1: Fig. S1C).
Discussion and conclusions
In this study, we report a genetic analysis-based thorough investigation of a patient with contiguous deletion and duplication in chromosome 6, manifesting with global developmental delay. G-banded chromosomal karyotype and CNV-seq findings, as well as WES-CNV, would be compatible with a duplication of 6p22.3-p25.3 complicated by a distal 6p25.3 deletion. The patient’s mother presented a similar phenotype, and chromosome abnormalities were consistent with her son, indicating maternal inheritance.
We noticed that there were slight differences between the phenotypes of the proband and his mother. First, the eye abnormalities of the proband were more severe than those of his mother. The proband presented upper eyelid entropion, alternating strabismus, and nystagmus. His mother showed upper eyelid entropion but without alternating strabismus and nystagmus. We speculated that this may be related to environmental factors, other unknown pathogenic genes or some regulatory factors. Second, we found some differences in the clinical symptom severity of the arm and hands. At present, we have only observed slight bend growth in the proband, but his mother presented obvious bent long bone and even joint contractures in the hands’ fingers. Her arm and fingers could not be fully extended. After investigating her medical history, it should be noted that her radius and ulna began to bend grow at a very young age and gradually developed more seriously. Therefore, we should indicate that these differences may be due to age of onset. Finally, the proband presented with recurrent upper and lower respiratory tract infections. However, his mother did not show similar recurrent infections at present. Due to the unavailability of adequate information from his mother's parents and her current inability to recall and recount her medical history from a younger age, we were unable to ascertain whether she had experienced recurrent infections during her childhood. Therefore, it is difficult to compare the symptoms of the proband and his mother at the same age. With regard to the proband’s repeated infection, but his mother is not as an adult, we speculated that the proband’s recurrent infection may be associated with the following reason: when compared with adults, children’s immune cells have not developed memory or antigen specificity for most of the antigens, which may result in infections. Therefore, infections are always more severe at younger ages and may be greatly eased with increasing age.
Duplication region difference between the proband and his mother
Our proband and his mother presented quite similar phenotypes. Utilizing CNV-seq, the proband showed a 20.88-Mb heterozygous duplication at 6p25.3-p22.3 (chr6:820,000–21,699,769, hg38) and a 0.66-Mb heterozygous deletion at 6p25.3 (chr6:g.160000–820,000, hg38). The results of the mother showed a 0.66-Mb heterozygous deletion at 6p25.3 (chr6:g.160000–820,000, hg38) and a 20.86-Mb heterozygous duplication at 6p25.3-p22.3 (chr6:820,000–21,699,769, hg38). Therefore, it could be assumed that the deletion and duplication in chr6 of the proband is inherited from the mother.
However, it should be noted that the duplication between the proband and his mother exhibited subtle differences, and the variation region of the mother was 20 kb smaller than that of the proband. We assumed that this difference was due to the technical restriction of CNV-seq. CNV-seq is a low-depth whole genome sequencing method with an average sequencing depth of 0.1× . The processed reads were divided into contiguous 20 kb bins. In other words, CNV variation differences under 20 kb were credible.
In addition, we noticed that the CNVs detected via WES-CNV and CNV-seq had some discrepancies: CNVs detected via CNV-seq were larger than those detected via WES. This may be due to the method differences between WES-CNV and CNV-seq. WES-CNV is better capable of assessing the breakpoints in exonic regions, even deletions or duplications in a single exon. However, WES-CNV could not detect regions beyond the exons. When compared, although the limited resolution of CNV-seq is not allowed to detect deletion or duplication regions smaller than 100 kb, CNV-seq can not only be evaluated at the genome level for contiguous deletions or duplications in exonic regions but also in intron regions [21,22,23]. Therefore, the different advantages and limitations of WES-CNV and CNV-seq are the major reason why the CNVs detected via WES-CNV and CNV-seq have discrepancies.
Genotype and phenotype comparison
To date, as shown in Fig. 4 and Table 1, at least 4 patients with contiguous duplication and deletion in chromosome 6 have been reported to have multiple clinical phenotypes. When compared with these cases, the duplication region of our proband was larger, and the deletion region was smaller.

A contiguous deletion and duplication in chromosome 6p regions of our patient and previously reported patients. Red indicated deletion and blue indicated duplication
We noticed that all these patients, including our patients, had variable deletions in the 6p25-pter [24]. Submicroscopic deletions of 6p, especially at 6p25 to 6pter, have been reported in several studies and could lead to hearing impairment, ocular dysgenesis, and brain abnormalities. In addition, most patients show dysmorphic features, such as a low nasal bridge and downslanting palpebral fissures. Other features, such as skeletal, renal, and cardiac malformations, are variable [25, 26]. We have noticed that the clinical phenotype and severity of all previously reported 6p25 cytogenetic abnormalities are dependent on altered doses of a gene or genes in the duplicated or deleted regions (Tables 2 and 3). Our proband and his mother have main physical findings: eye abnormalities, developmental delay, language impairment, intellectual disability, growth retardation, and dysmorphic features [27]. The Database of Genomic Variants reported several duplications in the 6p25-p22 region, but they were much smaller. We assumed that when no triple-dosage sensitive gene exists, chromosomal deletions may cause more severe and more serious clinical effects than duplications.
Linhares et al. reported a girl who exhibited 6p25.3p25.1 terminal deletion associated with a 6p25.1 duplication (Patient 1 in Table 1) [13]. Our patient (proband and his mother) showed similar clinical features to this patient, including dysmorphic features (frontal bossing, depressed nasal bridge), eye problems (strabismus), hearing loss, developmental delay, and intellectual disability. In addition, this patient also presented some overlapping phenotypes with our patients, such as dysmorphic features (severe hypertelorism, midface hypoplasia, high palate) and skeletal abnormalities (total absence of carpal bones and epiphysis ossification, slender long bones, tall vertebral bodies). It should be noted that this patient showed extra phenotypes, such as brain abnormalities (dilated brain ventricles, cerebellar hypoplasia, rotation of the vermis away from the brainstem, highlighted leukopathy), eye abnormalities (strabismus and anterior eye), and seizures. We assumed that these phenotypes may be due to multiple genes in a larger deletion region in 6p25.3. Her deletion region was 4.778 Mb ranging from chr6:335,924 to chr6:5,113,947. This deletion contains eye development-associated genes (FOXC1) [28,29,30] and neurodevelopmental disorder- and seizure-associated genes (GMDS, TUBB2A, TUBB2B) [31,32,33,34,35,36,37]. Therefore, we assumed that these factors mainly contributed to these extra phenotypes.
Patient 2 was a Japanese girl reported with a 6.4-Mb duplication at 6p25.3-p25.1 and a 220-kb deletion at 6p25.3 [17]. When compared, we found that the clinical features between patient 2 and our proband were more similar than those of patient 1 [13]. We surmise that the reason is that the duplication and deletion regions of patient 2 are more similar than those of patient 1. Patient 2 and our proband showed similar facial features, such as a short nose with a flat nasal root, hypoplastic alaenasi, a long and flat philtrum, and a thin upper lip vermilion. In addition, motor, intellectual and language development were all delayed. However, except for these similar symptoms, patient 2 showed obvious cardiovascular system abnormalities (ventricular septal defects, patent ductus arteriosus, and tetralogy of Fallot), seizures, and renal problems. She was diagnosed with nephrotic syndrome at 2 years old. Despite extensive treatment, her renal situation worsened. She underwent renal transplantation at nine years old. It should be noted that patient 2, with both smaller deletion and duplication regions, presented severe symptoms. We found that the duplication in our proband is tandem, and patient 2 would be compatible with an inverted duplication of 6p25.3–p25.1 complicated by a distal 6p25.3 deletion. This structural variation may contribute to the differences between patient 2 and the proband. To confirm this, the breakpoint of patient 2 should be analysed to check whether there exists a fusion gene or whether gene transcription regulation is affected. Another reason may be that the age of our proband is younger than the age at onset of renal symptoms; thus, long-term follow-up is needed to observe whether the proband develops similar symptoms.
Patient 3 and patient 7 (Table 1) both had a deletion and duplication in 6p, which may be due to a typical “inv dup del” pattern derived from U-type exchange [14, 15]. They showed similar phenotypes to our patients, such as facial abnormalities, eye abnormalities (iris hypoplasia and congenital glaucoma), and epilepsy. The deletion regions of patients 3 and 7 included the FOXC1 gene, which has been indicated that its haploinsufficiency can be responsible for glaucoma.
Patients with 6p duplications reported in the literature or database often show intrauterine growth retardation, short stature, microcephaly, prominent forehead, and others, as shown in patient 4 (Decipher#402,407), patient 5 (Decipher#282,057) and patient 6 (Decipher#345,282), which could explain the prominent forehead and short stature of these family patients. In addition, we found that some clinical features could not be explained by 6p deletion or 6p duplication, including recurrent respiratory system infection and imperforate anus. Therefore, the genotype–phenotype correlation in our proband should be discussed further.
Genotype–Phenotype correlation
Gene associated with eye phenotype
Our patient presented with a congenital choroidal defect and an eye phenotype. The eye phenotype is commonly observed in 6p deletion patients, including glaucoma, refractive error, strabismus, corectopia, far-sightedness, and corneal opacity. Several reports have suggested that the FOXC1 gene may be the major contributor to the eye phenotype of patients with 6p25 deletion syndromes. The FOXC1 gene is a member of the forkhead family of transcription factors [38].
It has been validated that the FOXC1 gene is haploinsufficient and that deletion or pathogenic variations of the FOXC1 gene could cause a variety of anterior eye chamber abnormalities associated with glaucoma, including Axenfeld-Rieger syndrome (OMIM*602,482) and anterior segment dysgenesis 3, multiple subtypes (OMIM*601,631) [39,40,41,42,43]. Patients 1 and 3 in Fig. 4 and Table 1 with deletion of the FOXC1 gene presented refractive error, corectopia, far-sightedness, and corneal opacity, which were consistent with previous reports.
Our proband with FOXC1 gene duplication also showed an eye phenotype. Unlike Patients 1 and 3, the proband had mainly congenital choroidal defects. Interestingly, patient 2, presenting far-sightedness, also had a FOXC1 gene duplication. Nishimura et al. analysed the FOXC1 gene in 70 probands with congenital anterior chamber defects [39]. Among them, 2 patients with iris hypoplasia or Peters anomaly had duplications of 6p25, which encompasses the FOXC1 gene. The authors suggested that both deletion and duplication of FOXC1 may cause anterior chamber defects of the eye. Therefore, we have reasons to assume that FOXC1 gene duplication contributes majorly to eye symptoms.
It should be noted that although we emphasized the function of the FOXC1 gene in eye symptoms here, FOXC1 is currently the most recognized crucial causative gene for 6pter-p24 deletion syndrome, which is involved in a wide range of biological functions and may be associated with abnormalities of multiple systems in patients. Researchers have found that the FOXC1 gene also plays an important role in cardiac, craniofacial, auditory and cerebellar development. These will be discussed in the following discussion section.
Gene associated with hearing loss
Hearing loss could be observed in both 6p deletion syndrome and 6p duplication syndrome. The SERPINB6 gene (OMIM*173,321) is a kind of serpin peptidase inhibitor. The SERPINB6 gene was first defined as a hearing loss-associated gene in a consanguineous Turkish family in 2010[44, 45]. All affected members presented nonsyndromic sensorineural hearing loss. Sirmaci et al. found that SERPINB6 protein was expressed in hair cells [45]. Tan et al. found that after homozygous replacement of SERPINB6A (the orthologue of human SERPINB6) in mice, they showed progressive hearing loss concomitant with cochlear degeneration after 2 weeks of age [46]. The effect appeared developmentally from outer hair cells, inner hair cells, primary auditory neurons, and fibrocytes. The SERPINB6 gene is located at 6p25.3 and was duplicated in our proband [47]. Current cases with hearing loss reported were mostly SERPINB6 gene deletions. Remarkably, the SERPINB6 gene of patient 3 in Fig. 4 and Table 1 was also duplicated, and he also showed hearing loss. Therefore, we have reasons to assume that alterations in another gene in CNV regions may contribute to hearing loss and that the SERPINB6 gene may cause hearing loss.
The FOXC1 gene, as mentioned above, is essential for development. The FOXC1 gene can affect the maintenance of many kinds of cells, such as haematopoietic stem cells, progenitor cell maintenance, and hair follicle stem cells [48, 49]. Scientific reports found that the FOXC1 gene could lead to hearing loss. Mears et al. reported a patient who carried a heterozygous mutation in the FOXC1 gene and showed deafness [50]. In addition, several papers have described patients with hearing loss and developmental delay, and further analysis confirmed the deletion of FOXC1 at 6p25 [51,52,53,54]. Therefore, we could not rule out the effect of FOXC1 on hearing loss in our patients.
Gene associated with neurodevelopmental phenotype
NRN1 gene (OMIM*607,409), which encodes neuritin-1, is a glutamate and neurotrophin receptor target gene. Neuritin is a GPI-anchored protein and can promote neurite outgrowth, which is crucial for the branching of neuritic processes in primary hippocampal and cortical cells [55]. Many reports associated with brain NMR abnormalities have been performed in 6p25 deletion syndrome, which contains the NRN1 gene. Brain NMR abnormalities, including Dandy-Walker and white matter abnormalities, include brain leukopathy, dilated brain ventricles, cerebellar hypoplasia, short/thin corpus callosum, small cerebellar vermis and dilated fourth ventricle [56]. Several researchers have stressed that the NRN1 gene should be a major gene for neurodevelopment of the patient features. The NRN1 gene may be critically associated with neurobiology and influence cognitive dysfunction [57,58,59]. In our pedigree, both the proband and his mother presented intellectual disabilities, which belong to the neurodevelopmental phenotype. Linhares ND et al. reported that a patient with NRN1 gene duplication also showed neurodevelopmental delay [13]. Although there are situations in which haploinsufficiency and increased gene dosage may cause the same phenotype, we still suggest that the NRN1 gene may be associated with the neurocognitive phenotype.
In addition, as we mentioned before, in addition to NRN1, we still suspect the function of the FOXC1 gene in neurodevelopment. Maclean et al. reported several patients with FOXC1 variations, these patients had mild to moderate developmental delay, and magnetic resonance imaging showed that they had CNS anomalies such as hydrocephalus and hypoplasia of the cerebellum, brainstem, and corpus callosum [51]. Although the majority of probands detected had FOXC1 de novo mutations or deletions, Nishimura et al. reported 70 probands with congenital anterior chamber defects and other phenotypes, such as neurodevelopmental delay, and members from 2 families encompassed the duplication of the FOXC1 gene [39]. These results indicated that not only FOXC1 haploinsufficiency but also increased FOXC1 gene dosage may cause phenotypes. Therefore, we speculated that the FOXC1 gene may contribute to the neurodevelopmental phenotype.
The TUBB2A gene [OMIM*615101], encoding tubulins, is significant for microtubules and functions in mitosis, intracellular transport, neuron morphology, and ciliary and flagellar motility. Currently, it has been validated that TUBB2A gene variations could lead to cortical dysplasia, complex, and other brain malformations 5 [OMIM*615763], and patients may present severe neurodevelopmental disorders, such as diffuse simplified gyral patterns in the brain, enlarged ventricles, and mildly enlarged posterior fossa. In addition, seizures and global developmental delay are also common [34, 60]. Although reported cases carried TUBB2A deletion, indicating that the specific developmental brain malformations and neurodevelopmental phenotype were due to TUBB2A haploinsufficiency, cases in our pedigree and patient 2 in Table 1 with TUBB2A duplication also presented neurodevelopmental delay. Therefore, we believe that the TUBB2A gene may also contribute to the neurodevelopmental phenotype.
Gene associated with skeletal phenotype
In this pedigree, we found a new clinical finding in the proband and his mother when compared with other cases: forearm bone dysplasia. Both the proband and his mother showed a bowed radius and ulna, which have not been reported in the literature. After analysing the genes involved, we indicated that the TUBB2A gene may be essential for skeletal development.
The TUBB2A gene [OMIM*615101], as we mentioned above, is related to neurodevelopment. Additionally, we indicated that the TUBB2A gene may also be associated with the skeletal phenotype. Although there have been no reports indicating the relationship between duplication of the TUBB2A gene and skeletal phenotype, after enquiring the MINT database, Linhares et al. found that the TUBB2A protein could interact with CUL7 [OMIM*609577], which could lead to 3-M syndrome [OMIM*273750] [13]. 3-M syndrome is an autosomal recessive disorder characterized by main skeletal anomalies. The patients showed long and slender tubular bones, delayed bone age, and other skeletal manifestations [61]. Although there has been no direct evidence demonstrating the function of the TUBB2A gene in skeletal development, the interaction between TUBB2A and CUL7 suggests that TUBB2A may be involved in the skeletal phenotype after there are no pathogenic mutations in the known genes related to skeletal features.
Gene associated with recurrent infection
The proband in this pedigree showed recurrent infections, including upper respiratory tract infection and lower respiratory tract infection (pneumonia). He has been hospitalized at least eight times for recurrent respiratory infections since birth. After analysing the genes involved in deletion and duplication regions, although several genes may contribute, we assumed that the IRF4 gene (OMIM*601,900) may be the major gene for recurrent infection [62]. The IRF4 gene was first reported to be associated with skin/hair/eye pigmentation (OMIM*611,724) in the OMIM database [63, 64]. The single-nucleotide polymorphisms in the IRF4 gene have the strongest association with hair colour, skin colour, eye colour, and skin tanning response to sunlight [65]. In addition, researchers have found that the IRF4 gene is essential for the development of T helper-2 (Th2) cells, Th17 cells, and Th9 cells, and it is interferon regulatory factor 4 [66].
Although the IRF4 gene has not been found to be associated with some kind of immunodeficiency and the inheritance is unclear, it has been confirmed that the IRF4 gene plays a role in immunity [62, 67]. The fusion gene with IRF4 or mutation could contribute to an aberrant IRF4 regulatory network and further fuse the gene expression programs of normal plasma cells and activated B cells [66, 68]. Yu et al. reported six case reports of large B-cell lymphoma with IRF4 rearrangement [69]. Benatti S reported that the IRF4 mutation (L116R) could promote the proliferation of chronic lymphocytic leukaemia B cells [70]. It should be noted that Bravo García-Morato M reported a 5-month-old Spanish girl to nonconsanguineous parents. The girl presented primary immune deficiencies (PIDs), including bronchopneumonia and long periods of fever without focus. After genetic analysis, they found that the combined PID was caused by a homozygous splicing mutation in IRF4 (NM_001195286:c.1213-2A > G,p.V405GfsTer127) [71].
Mittrucker et al. created IRF4 protein-deficient mice by knocking out exons 2 and 3 [72]. They found that mutant mice had poor T- and B-lymphocyte proliferative responses and lacked production of all serum Ig subclasses. Therefore, IRF4 is essential for T- and B-lymphocyte function. Ochiai et al. generated mixed bone marrow chimaeras with mouse IRF4+/+ and IRF4−/− progenitors and proposed a model of kinetic control in which signalling-induced dynamics of IRF4 in activated B cells controls their cell-fate outcomes: IRF4 could bind with interferon sequence response elements and further enrich for genes involved in plasma cell differentiation [73]. Staudenraus D et al. mentioned that the point mutation L116R in IRF4 differentially impacts key cytokine production in Th2, Th9, and Th17 cells [74]. Cook SL et al. reported that IRF4 haploinsufficiency may impair the affinity maturation of B cells [75].
In summary, although there are few case reports of recurrent infection due to IRF4 deletion, IRF4 is essential for immune cells, and IRF4 deletion may contribute to immune system problems such as recurrent infections.
Beside IRF4 gene, RIPK1 gene [OMIM: 603453] was also suspected to be responsible for recurrent infection. RIPK1 gene encodes a cytosolic kinase and could control multiple signaling pathways leading to inflammation and apoptotic or necroptotic cell death. It has been reported that RIPK1 is an essential molecule in programmed necrosis pathway, which is crucial for immunity, development, and tissue response [76]. At present, RIPK1 gene has been reported to be associated with two disease: autoinflammation with episodic fever and lymphadenopathy (AIEFL, OMIM: 618,852, AD) and immunodeficiency 57 with autoinflammation (OMIM: 618,108, AR). Although there has no report about RIPK1 gene duplication leading to disease, Tao et al., have reported two AIEFL patients with heterozygous RIPK1 gene mutation D324H and D324V via exome sequencing and further confirmed by Sanger sequencing. When compared with control group, the patients serum presented higher level of inflammatory cytokines and chemokines like IL-6, TNF, and gamma-IFN. In addition, the patients’ immune cells showed excessive inflammatory response and activated MAPK signalling pathway, and increased sensitivity could be reversed by inhibiting RIPK1. Meanwhile, in vitro studies utilizing patient fibroblasts indicated the mutation could block the RIPK1 cleavage by caspase-8. Taken together, it should be suspected that these mutations could result in a gain-of-function effect, and further influence inflammatory response [77]. In our patient, RIPK1 gene was duplicated. although RIPK1 gene has not been valiadated as triplosensitive gene, it is still considered as a candidate gene contributing to the recurrent infection of our proband.
Gene associated with renal abnormalities
Patients with 6p duplications mostly presented renal complications such as protein uria, renal failure, hypoplastic/aplastic kidney, hydronephrosis, ectopic kidney, horseshoe kidney, et al. The variation of the renal complications in 6p duplication syndrome would be compatible with congenital anomalies of the kidney and urinary tract (CAKUT) [17, 78,79,80], but no genes was localized within CNV variations of patient 2. Finally, FOXC1 gene was suspected to have some etiological contribution in renal abnormalities: (i) several literatures reported that FOXC1 gene mutation could cause renal dysplasia, duplex kidney and double ureters in mice [81, 82]; (ii) Patients with renal abnormalities carried a 3-bp (GGC) insertion variation in the six GGC repeats in the coding region of FOXC1 were reported [83]. However, it is not clear this in-frame insertion variation is causal for these patients.
Patient 2 presented renal abnormalities and FOXC1 gene was duplicated. When compared, as shown in Fig. 4, FOXC1 gene duplication was also seen in our proband (III3) and his mother (II4). However, our patients have not presented any renal abnormalities at present. Patient 1 and 3 whose FOXC1 genes were deleted did not show renal abnormalities either. Therefore, we suspected FOXC1 gene is suspected to have some etiological contribution in renal abnormalities but not be a single genetic factor, which is consistent with the conclusion with previous studies [17].
In this study, we reported a patient with developmental delay, recurrent infections, congenital choroidal defects, and craniofacial abnormalities, such as frontal bossing, higharched palate, long philtrum, thin upper lip, abnormal nasal bridge morphology, narrow eyelids, and upper eyelid entropion, suggestive of 6p deletion and 6p duplication syndrome. Since the resolution of the chromosome karyotype is larger than 5 Mb, only a duplication in 6p of our proband was detected. Further molecular analysis helped to more accurately assess a 0.66-Mb telomeric deletion and disclosed a contiguous duplication. In addition, it should be emphasized that the patient’s mother showed a similar phenotype and genotype as her son, which further indicated the pathogenicity of the CNVs. Our observations contribute to the clinical features of contiguous deletion and duplication in 6p. In addition, although the interpretation of genotype–phenotype correlations is not a simple task even in the era of molecular techniques, we still tried to connect the genotype with phenotype. After taking all the evidence into account, we suggested that FOXC1, NRN1, SERPINB6, TUBB2A, IRF4, and RIPK1 gene may contribute to the phenotype of our patient.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CNVs:
-
Copy number variations
- CNV-seq:
-
Copy number variation sequencing
- WES:
-
Whole exome sequencing
References
Morris-Rosendahl DJ, Crocq MA. Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialog Clin Neurosci. 2020;22(1):65–72.
D’Souza H, Karmiloff-Smith A. Neurodevelopmental disorders. Wiley Interdiscip Rev Cogn Sci. 2017;8(1–2):e1398.
Commission on Novel Technologies for Neurodevelopmental Copy Number, V., Neurodevelopmental copy-number variants: A roadmap to improving outcomes by uniting patient advocates, researchers, and clinicians for collective impact. Am J Hum Genet, 2022. 109(8): p. 1353–1365
Moeschler JB, Shevell M, Committee G. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014;134(3):e903–9018.
Bowling KM, Thompson ML, Amaral MD, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9(1):43.
Oguz S, Arslan UE, Kiper PÖŞ, et al. Diagnostic yield of microarrays in individuals with non-syndromic developmental delay and intellectual disability. J Intellect Disabil Res. 2021;65(12):1033–48.
Flore LA, Milunsky JM. Updates in the genetic evaluation of the child with global developmental delay or intellectual disability. Semin Pediatr Neurol. 2012;19(4):173–80.
Lee JS, Hwang H, Kim SY, et al. Chromosomal microarray with clinical diagnostic utility in children with developmental delay or intellectual disability. Ann Lab Med. 2018;38(5):473–80.
Vasudevan P, Suri M. A clinical approach to developmental delay and intellectual disability. Clin Med (Lond). 2017;17(6):558–61.
Belanger SA, Caron J. Evaluation of the child with global developmental delay and intellectual disability. Paediatr Child Health. 2018;23(6):403–19.
Scionti F, Di Martino MT, Pensabene L, et al. The Cytoscan HD array in the diagnosis of neurodevelopmental disorders. High Throughput. 2018;7(3):28.
Grayton HM, Fernandes C, Rujescu D, et al. Copy number variations in neurodevelopmental disorders. Prog Neurobiol. 2012;99(1):81–91.
Linhares ND, Svartman M, Rodrigues TC, et al. Subtelomeric 6p25 deletion/duplication: Report of a patient with new clinical findings and genotype-phenotype correlations. Eur J Med Genet. 2015;58(5):310–8.
Martinet D, Filges I, Schmutz NB, et al. Subtelomeric 6p deletion: clinical and array-CGH characterization in two patients. Am J Med Genet A. 2008;146A(16):2094–102.
Nakane T, Kousuke N, Sonoko H, et al. 6p subtelomere deletion with congenital glaucoma, severe mental retardation, and growth impairment. Pediatr Int. 2013;55(3):376–81.
Pace NP, Maggouta F, Twigden M, et al. Molecular cytogenetic characterisation of a novel de novo ring chromosome 6 involving a terminal 6p deletion and terminal 6q duplication in the different arms of the same chromosome. Mol Cytogenet. 2017;10:9.
Yoshimura-Furuhata M, Nishimura-Tadaki A, Ehara T, et al. Renal complications in 6p duplication syndrome: microarray-based investigation of the candidate gene(s) for the development of congenital anomalies of the kidney and urinary tract (CAKUT) and focal segmental glomerular sclerosis (FSGS). Am J Med Genet A. 2015;167A(3):592–601.
Atli EI, Gurkan H, Atli E, et al. De Novo subtelomeric 6p25.3 Deletion with duplication of 6q23.3-q27: genotype-phenotype correlation. J Pediatr Genet. 2020;9(1):32–9.
Qi Z, Jeng LJB, Slavotinek A, et al. Haploinsufficiency and triploinsensitivity of the same 6p25.1p24.3 region in a family. BMC Med Genomics. 2015;8:38.
Riggs ER, Andersen EF, Cherry AM, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020;22(2):245–57.
Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinform. 2009;10:80.
Sreenivasan R, Bell K, Bergen JVD, et al. Whole exome sequencing reveals copy number variants in individuals with disorders of sex development. Mol Cell Endocrinol. 2022;546:111570.
Testard Q, Vanhoye X, Yauy K, et al. Exome sequencing as a first-tier test for copy number variant detection: retrospective evaluation and prospective screening in 2418 cases. J Med Genet. 2022;13(4):421.
Lin RJ, Cherry AM, Chen KC, et al. Terminal deletion of 6p results in a recognizable phenotype. Am J Med Genet A. 2005;136(2):162–8.
DeScipio C. The 6p subtelomere deletion syndrome. Am J Med Genet C Semin Med Genet. 2007;145C(4):377–82.
Le-Caignec C, De Mas P, Vincent MC, et al. Subtelomeric 6p deletion: clinical, FISH, and array CGH characterization of two cases. Am J Med Genet A. 2005;132A(2):175–80.
Burd L, Martsolf JT, Kerbeshian J, et al. Partial 6p trisomy associated with infantile autism. Clin Genet. 1988;33(5):356–9.
Zhang HZ, Li P, Wang D, et al. FOXC1 gene deletion is associated with eye anomalies in ring chromosome 6. Am J Med Genet A. 2004;124A(3):280–7.
Akula M, Park JW, West-Mays JA. Relationship between neural crest cell specification and rare ocular diseases. J Neurosci Res. 2019;97(1):7–15.
Li M, Zhu LL, Liu JF, et al. Loss of FOXC1 contributes to the corneal epithelial fate switch and pathogenesis. Signal Transduct Target Ther. 2021;6(1):5.
Haliburton GD, McKinsey GL, Pollard KS. Disruptions in a cluster of computationally identified enhancers near FOXC1 and GMDS may influence brain development. Neurogenetics. 2016;17(1):1–9.
Aldinger KA, Lehmann OJ, Hudgins JL, et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p253 Dandy-Walker malformation. Nat Genet. 2009;41(9):1037–42.
Sullivan FX, Kumar R, Kriz R, et al. Molecular cloning of human GDP-mannose 4,6-dehydratase and reconstitution of GDP-fucose biosynthesis in vitro. J Biol Chem. 1998;273(14):8193–202.
Cushion TD, Paciorkowski AR, Pilz DT, et al. De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy. Am J Hum Genet. 2014;94(4):634–41.
Schmidt L, Wain KE, Hajek C, et al. Expanding the phenotype of TUBB2A-related tubulinopathy: three cases of a novel, heterozygous TUBB2A pathogenic variant p.Gly98Arg. Mol Syndromol. 2021;12(1):33–40.
Jaglin XH, Poirier K, Saillour KY, et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat Genet. 2009;41(6):746–52.
Guerrini R, Mei D, Cordelli DDM, et al. Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations. Eur J Hum Genet. 2012;20(9):995–8.
Pierrou S, Hellqvist M, Samuelsson L, et al. Cloning and characterization of seven human forkhead proteins: binding site specificity and DNA bending. EMBO J. 1994;13(20):5002–12.
Nishimura DY, Searby CC, Alward WL, et al. A spectrum of FOXC1 mutations suggests gene dosage as a mechanism for developmental defects of the anterior chamber of the eye. Am J Hum Genet. 2001;68(2):364–72.
Fuse N, Takahashi K, Yokokura S, et al. Novel mutations in the FOXC1 gene in Japanese patients with Axenfeld-Rieger syndrome. Mol Vis. 2007;13:1005–9.
Weisschuh N, Dressler P, Schuettauf F, et al. Novel mutations of FOXC1 and PITX2 in patients with Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci. 2006;47(9):3846–52.
Wu X, Xie HN, Wu T, et al. A novel mutation of FOXC1 in a Chinese family with Axenfeld-Rieger syndrome. Exp Ther Med. 2019;18(3):2255–61.
Komatireddy S, Chakrabarti S, Mandal AK, et al. Mutation spectrum of FOXC1 and clinical genetic heterogeneity of Axenfeld-Rieger anomaly in India. Mol Vis. 2003;9:43–8.
Sun J, Rose JB, Bird P. Gene structure, chromosomal localization, and expression of the murine homologue of human proteinase inhibitor 6 (PI-6) suggests divergence of PI-6 from the ovalbumin serpins. J Biol Chem. 1995;270(27):16089–96.
Sirmaci A, Erbek S, Price J, et al. A truncating mutation in SERPINB6 is associated with autosomal-recessive nonsyndromic sensorineural hearing loss. Am J Hum Genet. 2010;86(5):797–804.
Tan J, Prakash MD, Kaiserman D, et al. Absence of SERPINB6A causes sensorineural hearing loss with multiple histopathologies in the mouse inner ear. Am J Pathol. 2013;183(1):49–59.
Coughlin P, Nicholl J, Sun J, et al. Chromosomal mapping of the human proteinase inhibitor 6 (PI6) gene to 6p25 by fluorescence in situ hybridization. Genomics. 1995;26(2):431–3.
Omatsu Y, Seike M, Sugiyama T, et al. Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature. 2014;508(7497):536–40.
Wang L, Siegenthaler JA, Dowell RD, et al. Foxc1 reinforces quiescence in self-renewing hair follicle stem cells. Science. 2016;351(6273):613–7.
Mears AJ, Jordan T, Mirzayans F, et al. Mutations of the forkhead/winged-helix gene, FKHL7, in patients with Axenfeld-Rieger anomaly. Am J Hum Genet. 1998;63(5):1316–28.
Maclean K, Smith J, St Heaps L, et al. Axenfeld-Rieger malformation and distinctive facial features: Clues to a recognizable 6p25 microdeletion syndrome. Am J Med Genet A. 2005;132A(4):381–5.
Gauthier AC, Wiggs JL. Childhood glaucoma genes and phenotypes: focus on FOXC1 mutations causing anterior segment dysgenesis and hearing loss. Exp Eye Res. 2020;190: 107893.
Yamazaki H, Nakamura T, Hosono K, et al. Sensorineural hearing loss and hypoplastic cochlea in Axenfeld-Rieger syndrome with FOXC1 mutation. Auris Nasus Larynx. 2021;48(6):1204–8.
Reis LM, Maheshwari M, Capasso J, et al. Axenfeld-Rieger syndrome: more than meets the eye. J Med Genet. 2022. https://doi.org/10.1136/jmg-2022-108646.
Naeve GS, Ramakrishnan M, Kramer R, et al. Neuritin: a gene induced by neural activity and neurotrophins that promotes neuritogenesis. Proc Natl Acad Sci U S A. 1997;94(6):2648–53.
Shimada T, Yoshida T, Yamagata K. Neuritin mediates activity-dependent axonal branch formation in part via FGF signaling. J Neurosci. 2016;36(16):4534–48.
Sato H, Fukutani Y, Yamamoto Y, et al. Thalamus-derived molecules promote survival and dendritic growth of developing cortical neurons. J Neurosci. 2012;32(44):15388–402.
Yao JJ, Zhao QR, Lu JM, et al. Functions and the related signaling pathways of the neurotrophic factor neuritin. Acta Pharmacol Sin. 2018;39(9):1414–20.
Choi Y, Lee K, Ryu J, et al. Neuritin attenuates cognitive function impairments in tg2576 mouse model of Alzheimer’s disease. PLoS ONE. 2014;9(8): e104121.
Eid M, Eid O, Hegazy I, et al. Further insights into developmental brain malformations and leukoencephalopathy associated with 6p25.3 Deletion. Neuropediatrics. 2020;51(1):76–82.
Badina A, Pejin Z, Odent T, et al. Hip dislocation in 3-M syndrome: risk of misdiagnosis. Clin Dysmorphol. 2011;20(2):114–6.
Grossman A, Mittrücker HW, Nicholl J, et al. Cloning of human lymphocyte-specific interferon regulatory factor (hLSIRF/hIRF4) and mapping of the gene to 6p23-p25. Genomics. 1996;37(2):229–33.
Praetorius C, Grill C, Stacey SN, et al. A polymorphism in IRF4 affects human pigmentation through a tyrosinase-dependent MITF/TFAP2A pathway. Cell. 2013;155(5):1022–33.
Visser M, Palstra RJ, Kayser M. Allele-specific transcriptional regulation of IRF4 in melanocytes is mediated by chromatin looping of the intronic rs12203592 enhancer to the IRF4 promoter. Hum Mol Genet. 2015;24(9):2649–61.
Han J, Pope J, Sanson J, et al. A genome-wide association study identifies novel alleles associated with hair color and skin pigmentation. PLoS Genet. 2008;4(5): e1000074.
Staudt V, Bothur E, Klein M, et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity. 2010;33(2):192–202.
Negishi H, Ohba Y, Yanai H, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci U S A. 2005;102(44):15989–94.
Shaffer AL, Emre NC, Lamy L, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454(7201):226–31.
Yu Y, Sun Q, Li CW, et al. Large B-cell lymphoma with IRF4 rearrangement: six case reports and a literature review. Zhonghua Xue Ye Xue Za Zhi. 2022;43(6):475–80.
Benatti S, Atene CG, Fiorcari S, et al. IRF4 L116R mutation promotes proliferation of chronic lymphocytic leukemia B cells inducing MYC. Hematol Oncol. 2021;39(5):707–11.
Bravo Garcia-Morato M, Aracil Santos FJ, Briones AC, et al. New human combined immunodeficiency caused by interferon regulatory factor 4 (IRF4) deficiency inherited by uniparental isodisomy. J Allergy Clin Immunol. 2018;141(5):1924-1927e18.
Mittrucker HW, Matsuyama T, Grossman A, et al. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science. 1997;275(5299):540–3.
Ochiai K, Maienschein-Cline M, Simonetti G, et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity. 2013;38(5):918–29.
Staudenraus D, Porapu A, Leister H, et al. Point mutation L116R in interferon-regulatory factor 4 differentially impacts key cytokine production in Th2, Th9, and Th17 cells. Eur J Immunol. 2022;52(10):1680–3.
Cook SL, Sievert EP, Sciammas R. B Cell-Intrinsic IRF4 haploinsufficiency impairs affinity maturation. J Immunol. 2021;207(12):2992–3003.
Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–27.
Tao P, Sun J, Wu Z, et al. A dominant autoinflammatory disease caused by non-cleavable variants of RIPK1. Nature. 2020;577(7788):109–14.
Song R, Yosypiv IV. Genetics of congenital anomalies of the kidney and urinary tract. Pediatr Nephrol. 2011;26(3):353–64.
Saisawat P, Tasic V, Vega-Warner V, et al. Identification of two novel CAKUT-causing genes by massively parallel exon resequencing of candidate genes in patients with unilateral renal agenesis. Kidney Int. 2012;81(2):196–200.
Hwang DY, Dworschak GC, Kohl S, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int. 2014;85(6):1429–33.
Kume T, Deng K, Hogan BL. Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development. 2000;127(7):1387–95.
Komaki F, Miyazaki Y, Niimura F, et al. Foxc1 gene null mutation causes ectopic budding and kidney hypoplasia but not dysplasia. Cells Tissues Org. 2013;198(1):22–7.
Nakano T, Niimura F, Hohenfellner K, et al. Screening for mutations in BMP4 and FOXC1 genes in congenital anomalies of the kidney and urinary tract in humans. Tokai J Exp Clin Med. 2003;28(3):121–6.
Acknowledgements
The authors would like to thank the participation of the family members in this study. The authors would also like to thank the Berry Gnomics Co., for the technical support.
Funding
This research was funded by Shaanxi Provincial Science and Technology Project (No. 2019KJXX-055) and the project of Xi’an Children’s Hospital (2022C04).
Author information
Authors and Affiliations
Contributions
Conceptualization, YY; Methodology, YG; Validation, LZ, YY; Formal analysis, XX; Investigation, LZ, XT, FC, GW, BL; Resources, XT; Data curation, LZ, YY; Writing—original draft preparation, LZ, XT; Writing—review and editing, YY; Visualization, LZ, YY; Supervision, YY; Project administration, YY; Funding acquisition, YY. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted in accordance with the Declarationof Helsinki and approved by the Institutional Review Board (or Ethics Committee) of Xi’an Children’s Hospital, Xi’an (protocol code 20220090 and 24October 2022).
Consent for publication
Informed consent was obtained from the parents of the patient.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information

Additional file 1: Fig. S1
. The cytogenetic and molecular analysis of proband’s parents. A CNV-seq result of father; B CNV-seq result of mother; C Chromosome karyotype of mother
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Zhang, L., Tie, X., Che, F. et al. Novel maternal duplication of 6p22.3-p25.3 with subtelomeric 6p25.3 deletion: new clinical findings and genotype–phenotype correlations. Mol Cytogenet 16, 11 (2023). https://doi.org/10.1186/s13039-023-00640-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-023-00640-6