
Abstract
Background
Neurodevelopmental disorders are genetically heterogeneous pediatric conditions. The first tier diagnostic method for uncovering copy number variations (CNVs), one of the most common genetic etiologies in affected individuals, is chromosomal microarray (CMA). However, this methodology is not yet a routine molecular cytogenetic test in many parts of the world, including Hungary. Here we report clinical and genetic data of the first, relatively large Hungarian cohort of patients whose genetic testing included CMA.
Methods
Clinical data were retrospectively collected for 78 children who were analyzed using various CMA platforms. Phenotypes of patients with disease-causing variants were compared to patients with negative results using the chi squared/Fisher exact tests.
Results
A total of 30 pathogenic CNVs were identified in 29 patients (37.2%). Postnatal growth delay (p = 0.05564), pectus excavatum (p = 0.07484), brain imaging abnormalities (p = 0.07848), global developmental delay (p = 0.08070) and macrocephaly (p = 0.08919) were more likely to be associated with disease-causing CNVs.
Conclusion
Our results allow phenotypic expansion of 14q11.2 microdeletions encompassing SUPT16H and CHD8 genes. Variants of unknown significance (n = 24) were found in 17 patients. We provide detailed phenotypic and genetic data of these individuals to facilitate future classification efforts, and spotlight two patients with potentially pathogenic alterations. Our results contribute to unraveling the diagnostic value of rare CNVs.
Similar content being viewed by others


Background
Neurodevelopmental disorders (NDDs), including intellectual disability (ID), global developmental delay (GDD), autism spectrum disorder (ASD), attention deficit/hyperactivity disorder (ADHD), specific learning disorders, motor disorders (developmental coordination disorder, stereotypic movement disorder, etc.), and communication disorders (language disorder, speech sound disorder, etc.) pose a considerable challenge in everyday pediatric practice. Children with these conditions face difficulties in adaptive functions, social interactions and self-care, which may translate to entire families under serious emotional (and often financial) stress. Early and accurate diagnosis is therefore not only important for optimal care of the affected child, but for alleviating the burden and guilt on the parents.
The etiology underlying NDDs is exceedingly complex, and genetic underpinnings show further heterogeneity. One of the largest groups of genetic etiologic factors are copy number variations [CNVs, a few dozen kilobase pair (Kb) to several megabase pair (Mb) large structural variations in the genome]. The recommended gold standard techniques for diagnosing such alterations (excluding those that cause clinically recognizable syndromes) are chromosomal microarray analyses (CMA) - array comparative genomic hybridization (array CGH) and single nucleotide polymorphism (SNP) arrays. These methodologies have elevated the diagnostic yield of traditional cytogenetics from approximately 5% to 15–20% [1,2,3,4]. CMA is however not yet a part of routine genetic testing in Hungary. Delineating clinical features that could help with indicating this laborious test would be beneficial to both families and clinicians. Here we report our experiences with the first relatively large Hungarian pediatric cohort who underwent CMA testing.
Results
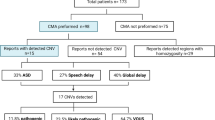
During the period between 2011 and 2020, 88 patients of Semmelweis University’s II. Department of Pediatrics underwent CMA testing. Children were selected for investigation if they had idiopathic DD/ID or a major congenital anomaly, and at least one additional suggestive feature (other NDDs, characteristic facies, multiple congenital anomalies, etc.), and the family consented to further genetic testing. At our Department, the typical DiGeorge syndrome (proximal A-D 22q11.11 deletion, including TBX1 gene), Williams syndrome and the deletion form of Prader-Willi syndrome are diagnosed by fluorescent in situ hybridization (FISH), therefore these cases are not included in this study. 78 patients underwent CMA after negative routine cytogenetic evaluations, while 10 patients were tested to refine the findings of previous tests. These latter patients with microscopically visible rearrangements (genetic and phenotypic data listed in Additional File 1) have been excluded from further statistical analysis.
The final investigation cohort consisted of 78 individuals, of which 47 were males (60.2%; male:female ratio 1.52:1). The average age at first clinical genetics consultation was 4.17 years (median 2.00 years, range from 4 days old to 20 years 7 months old). Disease-causing variants (n = 30) were identified in 29 patients (15 males and 14 females), which translates to an overall diagnostic yield of 37.18%. The disease-causing CNVs were on average 3.481 Mb large (median 1.124 Mb); 19/30 were deletions and 11/30 were duplications (Additional file 2). Chromosome 16 was most frequently affected (9/30; 30.0%), followed by chromosome 22 (4/30; 13.3%), and chromosomes 14, 17 and X (3/30; 10.0% each) (Additional file 3).
Information regarding the disease-causing CNVs is detailed in Table 1, the accompanying phenotypes of the patients are elaborated in Table 2. One child (patient SEG2_82) carried multiple disease-causing CNVs, specifically a terminal duplication of chromosome 19 (57243585_58445449; GRCh38) and a terminal deletion of chromosome 22 (49368551_50759338; GRCh38). These rearrangements originated from a paternal balanced translocation, and subsequent targeted FISH analyses revealed the CNVs in the patient’s younger sister as well. Of further note are patients SEG2_62 and SEG2_68. The former carried a deletion of chromosome X (6537108_8167604 Mb; GRCh37) encompassing the STS gene (OMIM*300,747), and was subsequently diagnosed with a mild form of X-linked ichthyosis (OMIM#308,100). An additional finding on the SNP array was a large loss of heterozygosity (LOH) on chromosome 14 (20052038_106871264 Mb; GRCh37). This finding was followed up by methylation analysis and maternal uniparental disomy 14 (Temple syndrome, OMIM#616,222) was confirmed, which was ultimately the underlying cause of his primary phenotype (see Table 2). SEG2_68’s parents are first cousins, which resulted in the identification of LOH on 21 regions of 12 chromosomes (overall 196.8 Mb of the genome).
Furthermore, we identified 24 variants of unknown significance (VUS) in 17 children (21.79% of the cohort); five individuals carried two, while one individual carried three VUS simultaneously (Additional file 4 lists detailed genetic and phenotypic information). Four children carried VUS in addition to disease-causing variants. The average size of the VUS was 585.9 Kb (median 228.2 Kb), 12 were losses and 12 were copy number gains (Additional file 2).
We compared the main phenotypic features of the disease-causing CNV carrier patients and the negative CMA group (visualized on Fig. 1). Due to the patient selection criteria, NDDs were common in both groups, as were congenital anomalies of the internal organs. Postnatal growth delay was the only symptom to approach significance (p = 0.05564). Pectus excavatum (p = 0.07484), brain imaging abnormalities (p = 0.07848), global DD (0.08070), the sub-phenotype of speech and language delay (p = 0.08070) and macrocephaly (p = 0.08919) were more commonly, but none-significantly associated with disease-causing CNVs. Conversely, errors of refraction were more common in the negative group, the difference reached significance (p = 0.02880).

Phenotypic comparison of the patients with disease-causing variants and those with negative CMA results
We were able to investigate inheritance in only 12.8% of patients; including three families where only the mother agreed to testing (neither mother carried the CNV of the respective child). Samples were available from both parents for seven patients. These seven children carried in total 9 CNVs, three proved to be de novo alterations, three were maternally, and three were paternally inherited. Six of the carrier parents were healthy, while the other four showed mild symptoms (history of learning difficulties and/or pediatric obesity, behavioral disorders, etc.; Additional file 5).
Discussion
In our study, CMA enabled an etiologic diagnosis in 29 children in a cohort of 78 patients with NDDs and/or congenital anomalies. A relatively high proportion of VUS were identified as well. Our diagnostic yield (37.18%) is quite high compared to the estimated 15–20% yield of CMA methods, albeit these estimates are often based on studies where selection criteria were rather broad and CMA resolution was relatively low [2]. Our diagnostic yield is comparable to several studies with smaller patient groups and/or similarly strict selection criteria [5,6,7,8,9,10,11,12,13,14,15].
Our rigorous selection also impacted our statistical observations (keeping in mind the limitations of the small sample size as well). In contrast to our results, congenital heart defects and other major congenital anomalies are often observed in significantly higher frequencies in patients with definitive CMA results [3, 10, 16]. The association of postnatal growth delay, global DD, speech and language delay and brain imaging abnormalities is in line with literature data [3, 16, 17]. Macrocephaly and pectus excavatum are well-known suggestive features/minor anomalies, and are therefore not unexpected results. Only one child had an error of refraction in the disease-causing CNV group, in contrast to 7 patients in the negative group.
Chromosomes 16, 22, 14, 17 and X were most frequently affected by disease-causing imbalances, which aligns with literature data. Six of the nine chromosome 16 CNVs, two of the four imbalances of chromosome 22, two of three on chromosome 17 and one on chromosome X corresponded to recurrent microdeletion/microduplication regions, and thus are the consequence of non-allelic homologous recombination mediated by segmental duplications [18]. Amongst the 16p alterations, a noteworthy four were gains or losses of the proximal 16p11.2 region, which corresponds to 5.1% of the entire cohort, and 13.8% of children diagnosed. Recurrent rearrangements are known to present with highly variable phenotypes, and are incompletely penetrant [19] The presented patients’ phenotypes are in accordance with their respective recurrent rearrangements.
Surprisingly, microdeletions of a non-recurrent region were also slightly enriched in our study: 3/78 patients (3.8% of the studied cohort, 10.3% of diagnosed patients) carried approximately 500 Kb large deletions of 14q11.2. Microdeletions of chromosome region 14q11.2 are associated with DD/ID, ASD, other neuropsychiatric disorders, macrocephaly and characteristic facial features (hypertelorism, down-slanting palpebral fissures, broad nose, long philtrum, prominent Cupid’s bow and abnormalities of the auricular pinnae) [20, 21]. The authors of the original report [20] defined a minimal critical region (MCR) of 35 Kb encompassing two genes: SUPT16H (OMIM*605,012) and CHD8 (OMIM*610,528). CHD8 is currently thought to be one of the most common genetic drivers of ASD, and is often additionally associated with macrocephaly, overgrowth, facial minor anomalies and gastrointestinal problems [21,22,23,24]. SUPT16H was recently linked to a NDD consisting of DD/ID, ASD, seizures, precocious puberty, craniofacial minor anomalies, and corpus callosum abnormalities, but not macrocephaly [25]. Notably, neither of our three patients had increased head circumference; on the contrary, SEG2_49 had microcephaly (with generalized somatic underdevelopment). We speculate that this is due to other, as of yet unknown genetic factors counteracting the effect of the CHD8 pathogenic alteration. Taken together with one other reported child with normal head circumference [26], our results highlight the fact that macrocephaly is not an obligatory symptom in SUPT16H-CHD8 microdeletions. Our patients enable further phenotypic expansion of the microdeletion: somatic DD/short stature, muscular hypertonia/spasticity, ventriculomegaly and hirsutism/hypertrichosis, deep-set eyes and STPC are all novel associated features noted in 2/3 patients in the current study (Table 2).
To this day, classification of CNVs is often confounded by lack of sufficient evidence, and is thus dynamic as the accumulation of data, case reports, functional analyses, etc. can lead to reclassification of VUS. Improved genetic counseling and optimal patient care necessitates further studies to resolve the ambiguity of the not insignificant number of uncertain variants. Two of the VUS identified in the presented cohort merit further discussion. Patient SEG2_57 is a young boy with severe DD/ID, behavioral stereotypies and multiplex congenital anomalies (bilateral talipes equinovarus, bilateral complete syndactyly of the fingers and the toes, nail dystrophy, macrocephaly and postnatal overgrowth, craniofacial minor anomalies, spasticity, mild congenital heart disease, strabismus; see Fig. 2 and Additional file 4). The etiology of the child’s phenotype is complicated by two factors: there is a high likelihood of perinatal hypoxia; and four days after a syndactyly correcting operation he presented sudden loss of vision, multiple symptomatic focal epileptic attacks and diffuse hypoxic-ischaemic encephalopathy with cytotoxic cerebral oedema. This happened unexpectedly, and no cause has been identified to date (genetic variants causing malignant hyperthermia were ruled out from a blood sample, but were not tested from muscle biopsy). There were no further seizures from this point onward, and treatment (carbamazepine) was discontinued after one year. Pediatric neurologists retrospectively identified very mild cerebral atrophy when reevaluating the patient’s pre-surgery brain MRI. At the 6 month follow-up, MRI revealed severe cortical necrosis as sequelae to the encephalopathy, as well as diffuse mild cerebral atrophy. The boy has consistently slightly elevated lactate levels. His karyotype was determined to be 46,XY; pathogenic GLI3 (OMIM*1,654,240) variants causing Greig cephalopolysyndactyly (OMIM#175,700) were ruled out at an early stage of genetic investigations; WES was performed later and identified no pathogenic variants or VUS that might explain the phenotype. Mitochondrial genome analysis performed on peripheral blood was negative. CMA analysis identified three VUS (Additional file 4), one of which encompasses an NDD-associated disease-causing gene. His 384 Kb large chromosome 4 duplication (arr[GRCh37]4q24(102058416_102443207)x3) contains PPP3CA (OMIM*114,105). The encoded calcineurin A protein (the catalytic subunit of calcineurin, which is responsible for calmodulin-calcineurin interactions) has an important role in synaptic vesicle recycling through regulation of response to calcium levels [27]; and is associated with epilepsy and other NDDs. Loss-of-function pathogenic variants cause autosomal dominant developmental and epileptic encephalopathy 91 (OMIM#617,711). This disorder is characterized by early-onset epilepsy (severity is variable and may be correlated with the specific variant), moderate/severe DD/ID, developmental regression, autistic behavior, generalized muscular hypotonia or spasticity, talipes equinovarus, feeding difficulties, cortical vision loss, cerebral atrophy and delayed myelinization [28,29,30]. Gain-of-function pathogenic variants lead to a syndrome characterized by arthrogryposis, cleft palate, craniosynostosis and ID (OMIM#618,265). Postnatal growth delay, plagio- or trigonocephaly, vesicoureteral reflux, gracile bones, brachydactyly, generalized seizures, behavioral stereotypies may also be present [31]. The other two VUS of this patient are a 504 Kb size 20p11.23 duplication (arr[GRCh37]20(19240620_19745197)x3), encompassing SLC24A3 gene (OMIM*609,839), important for calcium homeostasis [32]; and an 81 Kb size Xq25 deletion (arr[GRCh37]X(124088718_124169834)x0), containing part of TENM1 gene (OMIM*300,588), relevant in synaptic organization [33]. The phenotype of the presented SEG2_57 patient greatly overlaps with the disorders associated with PPP3CA (severe DD/ID, talipes equinovarus, cerebral atrophy, vision loss, and arthrogryposis, abnormal cranial morphology, behavioral stereotypies). The duplication of this gene plausibly contributed to the patient’s complex disorder, genotype-phenotype correlation is however confounded by the presence of multiple uncertain environmental factors. The syndromic origins of the child’s epilepsy and vision loss are questionable, but they might be attributable to decompensation of an existing genetic disorder. The complete syndactyly affecting all four extremities remains unexplained, which seems counter-intuitive at first glance, however, approximately 60% of syndactyly cases are sporadic, and hereditary cases are often isolated and multifactorial [34].

Phenotype of patient SEG2_57
Left: close-up of one of the child’s feet (syndactyly and pes equinovarus). Right: age 3 years. At this age he was unable to sit without assistance. He learned to sit and hold himself up while leaning on something at age 5 years, and to pull himself to a standing position at age 7 years
One further potentially relevant VUS is the 154 Kb large chromosome 20 duplication (arr[GRCh37]20p11.21(24554628_24708699)x3) encompassing SYNDIG1 (OMIM*614,311) gene. This CNV was identified in a 5-year-old boy (patient SEG2_81, Additional file 4) who presented with moderate global developmental delay. His speech delay is particularly severe as he has no spoken words; receptive speech is also impaired, albeit to a lesser degree than expressive. He was initially diagnosed with ASD, but this diagnosis was later revised due to the complete failure of autism-specific developmental therapies and the overall longitudinal clinical picture. He is currently under the care of an expert neuropsychologist; extensive medical evaluations have unfortunately been repeatedly delayed. Preliminary neuropsychiatric opinion states that the patient has a complex NDD affecting the quality of many neurodevelopmental domains, including social interactions, behavior, and processing functions. A previous brain MRI (performed in 2017) was normal except for mild atrophy of the posterior third of the truncus corporis callosi. He has no other major or minor congenital anomalies. Patient SEG2_81’s 20p11.21 duplication was inherited from the father. The encompassed gene, SYNDIG1, is a brain-specific transmembrane protein proven to be a regulator of excitatory synapse development in rat brain [35]. Further study has shown that SYNDIG1-deficient excitatory synapses have impaired structure and function, thus suggesting an important role in normal synapse development [36]. No comparable duplications of SYNDIG1 have been reported in the literature or online databases. In the presented case, paternal inheritance further complicates classification. Nevertheless, the currently suspected pathogenesis of patient SEG2_81’s phenotype implicates faulty synaptic development.
The main limitations of this study are the small sample size, the different CMA platforms utilized (not all patients were tested using SNP arrays, therefore LOHs suggestive of syndromes caused by UPDs could have been missed), and the lack of information regarding inheritance in the majority of patients.
Conclusion
The diagnostic yield of CMA in our Hungarian cohort of pediatric patients with NDDs and/or congenital anomalies was 37.2%, promoting the continued usefulness of molecular cytogenetics. Our high diagnostic rate is partly due to rigorous patient selection criteria necessitated by availability and funding considerations. We identified several phenotypic features that increased the likelihood of finding a diagnosis in the patient group: postnatal growth delay, brain imaging abnormalities, global DD, macrocephaly and pectus excavatum. We highlight new clinical features associated with the rare, non-recurrent 14q11.2 microdeletion. Classification, and therefore genetic counseling of VUS is still confounded by the lack of scientific knowledge, highlighting the benefits of continued data sharing. We provide detailed phenotypic information corresponding to all VUS identified in our cohort. Amongst these, we identified two novel rare variants containing genes (PPP3CA and SYNDIG1, respectively) possibly relevant for the associated clinical phenotypes, bolstering said genes’ conceivable role in human disease. Further studies and case reports are necessary to elucidate the pathogenicity of the presented CNVs.
Methods
Clinical data up to the point of genetic diagnosis/negative CMA result were retrospectively collected and organized. Karyotypes (at standard band resolution of 450–550) were determined by analysis of 20 Giemsa-stained metaphases each from standard 72-hour peripheral blood lymphocyte cultures. The platforms and analysis software used for CMA were NimbleGen Array (CGX 1.4 M) with NimbleGen MS 200 Microarray Scanner (30 patients; Roche NimbleGen Inc., Madison, WI, USA), Agilent qChip Post (60 K; 5 patients) and Agilent 180 K oligo-array (18 patients) with Agilent Genomic Workbench 7.0 (Agilent Technologies, Santa Clara, CA, USA), Affymetrix CytoScan Optima (300 K; 5 patients), Affymetrix CytoScan 750 K (17 patients), and Affymetrix CytoScan HD (3 patients) with Affymetrix Genechip Scanner or Chromosome Suite Ananlysis (ChAS) 4.0 (Thermo Fisher Scientific, Inc.; Waltham, MA, USA). CNV interpretation was based on guidelines published by the American College of Medical Genetics and Genomics (ACMG) [37, 38]. Relevant CNVs were validated using FISH or quantitative multiplex PCR of short fluorescent fragments (QMPSF) analysis. Parental studies were possible in 10 families: parents of three families and a mother of a fourth child underwent CMA, two sets of parents and two additional mothers were tested using QMPSF, and targeted FISH testing was performed for two parent pairs (Additional file 5). The main phenotypic features of the patients with disease-causing CNVs were compared to the patients with negative CMA results using the chi squared test. If any cell of the contingency table had an expected value less than five, the Fisher exact test was applied.
Data availability
Not applicable.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- ACMG:
-
autism spectrum disorder
- CHG:
-
comparative genomic hybridization
- CMA:
-
chromosomal microarray
- CNV:
-
copy number variation
- DSD:
-
disorders of sexual development
- FISH:
-
fluorescent in situ hybridization
- Kb:
-
kilobase pairs
- LOH:
-
loss of heterozygosity
- Mb:
-
megabase pairs
- NDD:
-
neurodevelopmental disorder
- OMIM:
-
Online Inheritance in Man
- PCR:
-
polymerase chain reaction
- QMPSF:
-
quantitative multiplex PCR of short fluorescent fragments
- SNP:
-
single nucleotide polymorphism
- VUS:
-
variant of unknown significance.
- WES:
-
whole exome sequencing
References
van Karnebeek CDM, Jansweijer MCE, Leenders AGE, Offringa M, Hennekam RCM. Diagnostic investigations in individuals with mental retardation: a systematic literature review of their usefulness. Eur J Hum Genet. 2005 Jan;13(1):6–25.
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010 May;86(5):749–64.
Chaves TF, Baretto N, Oliveira LF de, Ocampos M, Barbato IT, Anselmi M, et al. Copy Number Variations in a Cohort of 420 Individuals with Neurodevelopmental Disorders From the South of Brazil. Sci Rep [Internet]. 2019;9(1):17776. Available from: https://doi.org/10.1038/s41598-019-54347-z.
Wayhelova M, Smetana J, Vallova V, Hladilkova E, Filkova H, Hanakova M, et al. The clinical benefit of array-based comparative genomic hybridization for detection of copy number variants in Czech children with intellectual disability and developmental delay. BMC Med Genomics. 2019 Jul;12(1):111.
Iourov IY, Vorsanova SG, Kurinnaia OS, Zelenova MA, Silvanovich AP, Yurov YB. Molecular karyotyping by array CGH in a Russian cohort of children with intellectual disability, autism, epilepsy and congenital anomalies. Mol Cytogenet. 2012 Dec;5(1):46.
Kashevarova AA, Nazarenko LP, Skryabin NA, Salyukova OA, Chechetkina NN, Tolmacheva EN, et al. Array CGH analysis of a cohort of Russian patients with intellectual disability. Gene. 2014 Feb;536(1):145–50.
Perovic D, Damnjanovic T, Jekic B, Dusanovic-Pjevic M, Grk M, Djuranovic A, et al. Chromosomal microarray in postnatal diagnosis of congenital anomalies and neurodevelopmental disorders in Serbian patients. J Clin Lab Anal. 2022 Jun;36(6):e24441.
Lay-Son G, Espinoza K, Vial C, Rivera JC, Guzmán ML, Repetto GM. Chromosomal microarrays testing in children with developmental disabilities and congenital anomalies. J Pediatr (Rio J). 2015;91(2):189–95.
Ho KS, Wassman ER, Baxter AL, Hensel CH, Martin MM, Prasad A, et al. Chromosomal Microarray Analysis of Consecutive Individuals with Autism Spectrum Disorders Using an Ultra-High Resolution Chromosomal Microarray Optimized for Neurodevelopmental Disorders. Int J Mol Sci. 2016 Dec;17(12).
Wu X-L, Li R, Fu F, Pan M, Han J, Yang X, et al. Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr. 2017 May;17(1):117.
Kessi M, Xiong J, Wu L, Yang L, He F, Chen C, et al. Rare Copy Number Variations and Predictors in Children With Intellectual Disability and Epilepsy. Front Neurol. 2018;9:947.
Mihaylova M, Staneva R, Toncheva D, Pancheva M, Hadjidekova S. Benign. Pathogenic and Copy Number Variations of Unknown Clinical Significance in Patients with Congenital Malformations and Developmental Delay. Balkan J Med Genet. 2017 Jun;20(1):5–12.
Wang R, Lei T, Fu F, Li R, Jing X, Yang X, et al. Application of chromosome microarray analysis in patients with unexplained developmental delay/intellectual disability in South China. Pediatr Neonatol. 2019 Feb;60(1):35–42.
Werling AM, Grünblatt E, Oneda B, Bobrowski E, Gundelfinger R, Taurines R, et al. High-resolution chromosomal microarray analysis for copy-number variations in high-functioning autism reveals large aberration typical for intellectual disability. J Neural Transm. 2020 Jan;127(1):81–94.
Liu Y, Lv Y, Zarrei M, Dong R, Yang X, Higginbotham EJ, et al. Chromosomal microarray analysis of 410 Han Chinese patients with autism spectrum disorder or unexplained intellectual disability and developmental delay. NPJ genomic Med. 2022 Jan;7(1):1.
Shoukier M, Klein N, Auber B, Wickert J, Schröder J, Zoll B, et al. Array CGH in patients with developmental delay or intellectual disability: are there phenotypic clues to pathogenic copy number variants? Clin Genet. 2013 Jan;83(1):53–65.
Heide S, Keren B, Billette de Villemeur T, Chantot-Bastaraud S, Depienne C, Nava C, et al. Copy Number Variations Found in Patients with a Corpus Callosum Abnormality and Intellectual Disability. J Pediatr. 2017 Jun;185:160–6.e1.
Golzio C, Katsanis N. Genetic architecture of reciprocal CNVs. Curr Opin Genet Dev. 2013 Jun;23(3):240–8.
Deshpande A, Weiss LA. Recurrent reciprocal copy number variants: Roles and rules in neurodevelopmental disorders. Dev Neurobiol. 2018 May;78(5):519–30.
Zahir F, Firth HV, Baross A, Delaney AD, Eydoux P, Gibson WT, et al. Novel deletions of 14q11.2 associated with developmental delay, cognitive impairment and similar minor anomalies in three children. J Med Genet. 2007 Sep;44(9):556–61.
Yasin H, Gibson WT, Langlois S, Stowe RM, Tsang ES, Lee L, et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J Hum Genet [Internet]. 2019;64(4):271–80.
Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014 Jul;158(2):263–76.
Thygesen JH, Wolfe K, McQuillin A, Viñas-Jornet M, Baena N, Brison N, et al. Neurodevelopmental risk copy number variants in adults with intellectual disabilities and comorbid psychiatric disorders. Br J Psychiatry [Internet]. 2018/04/25. 2018;212(5):287–94.
Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, et al. Mutations in Epigenetic Regulation Genes Are a Major Cause of Overgrowth with Intellectual Disability. Am J Hum Genet. 2017 May;100(5):725–36.
Bina R, Matalon D, Fregeau B, Tarsitano JJ, Aukrust I, Houge G, et al. De novo variants in SUPT16H cause neurodevelopmental disorders associated with corpus callosum abnormalities. J Med Genet. 2020 Jul;57(7):461–5.
Paderova J, Drabova J, Holubova A, Vlckova M, Havlovicova M, Gregorova A, et al. Under the mask of Kabuki syndrome: Elucidation of genetic-and phenotypic heterogeneity in patients with Kabuki-like phenotype. Eur J Med Genet. 2018 Jun;61(6):315–21.
Myers CT, Stong N, Mountier EI, Helbig KL, Freytag S, Sullivan JE, et al. De Novo Mutations in PPP3CA Cause Severe Neurodevelopmental Disease with Seizures. Am J Hum Genet. 2017 Oct;101(4):516–24.
Rydzanicz M, Wachowska M, Cook EC, Lisowski P, Kuźniewska B, Szymańska K, et al. Novel calcineurin A (PPP3CA) variant associated with epilepsy, constitutive enzyme activation and downregulation of protein expression. Eur J Hum Genet. 2019 Jan;27(1):61–9.
Yang S, Shen X, Kang Q, Kuang X, Ning Z, Liu S, et al. Clinical and Genetic Study on a Chinese Patient with Infantile Onset Epileptic Encephalopathy carrying a PPP3CA Null Variant: a case report. BMC Pediatr. 2020 Jun;20(1):315.
Panneerselvam S, Wang J, Zhu W, Dai H, Pappas JG, Rabin R, et al. PPP3CA truncating variants clustered in the regulatory domain cause early-onset refractory epilepsy. Clin Genet. 2021 Aug;100(2):227–33.
Mizuguchi T, Nakashima M, Kato M, Okamoto N, Kurahashi H, Ekhilevitch N, et al. Loss-of-function and gain-of-function mutations in PPP3CA cause two distinct disorders. Hum Mol Genet. 2018 Apr;27(8):1421–33.
Kraev A, Quednau BD, Leach S, Li XF, Dong H, Winkfein R, et al. Molecular cloning of a third member of the potassium-dependent sodium-calcium exchanger gene family, NCKX3. J Biol Chem. 2001 Jun;276(25):23161–72.
Mosca TJ, Hong W, Dani VS, Favaloro V, Luo L. Trans-synaptic Teneurin signalling in neuromuscular synapse organization and target choice. Nature. 2012 Mar;484(7393):237–41.
Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27.
Díaz E. SynDIG1 regulation of excitatory synapse maturation. J Physiol. 2012 Jan;590(1):33–8.
Chenaux G, Matt L, Hill TC, Kaur I, Liu X-B, Kirk LM, et al. Loss of SynDIG1 Reduces Excitatory Synapse Maturation But Not Formation In Vivo. eNeuro. 2016;3(5).
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011 Jul;13(7):680–5.
Riggs ER, Andersen EF, Cherry AM, Kantarci S, Kearney H, Patel A, et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet Med. 2020 Feb;22(2):245–57.
Kendall KM, Rees E, Escott-Price V, Einon M, Thomas R, Hewitt J, et al. Cognitive Performance Among Carriers of Pathogenic Copy Number Variants: Analysis of 152,000 UK Biobank Subjects. Biol Psychiatry. 2017 Jul;82(2):103–10.
Yamamoto T, Shimojima K, Yamazaki S, Ikeno K, Tohyama J. A 16q12.2q21 deletion identified in a patient with developmental delay, epilepsy, short stature, and distinctive features. Vol. 56, Congenital anomalies. Australia; 2016. pp. 253–5.
Lengyel A, Pinti É, Eggermann T, Fekete G, Haltrich I. Deletion of 16q22.2q23.3 in a Boy with a Phenotype Reminiscent of Silver-Russell Syndrome. Mol Syndromol [Internet]. 2021;12(5):300–4.
Nagamani SCS, Zhang F, Shchelochkov OA, Bi W, Ou Z, Scaglia F, et al. Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J Med Genet. 2009 Dec;46(12):825–33.
Bruno DL, Anderlid B-M, Lindstrand A, van Ravenswaaij-Arts C, Ganesamoorthy D, Lundin J, et al. Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J Med Genet. 2010 May;47(5):299–311.
Giannikou K, Fryssira H, Oikonomakis V, Syrmou A, Kosma K, Tzetis M, et al. Further delineation of novel 1p36 rearrangements by array-CGH analysis: narrowing the breakpoints and clarifying the “extended” phenotype. Gene. 2012 Sep;506(2):360–8.
Carvalheira G, Oliveira MM, Takeno S, Lima FT de, Meloni VA, Melaragno MI. 19q13.33→qter trisomy in a girl with intellectual impairment and seizures. Meta gene. 2014 Dec;2:799–806.
Wang J, Wang Y, Wang L, Chen WY, Sheng M. The diagnostic yield of intellectual disability: combined whole genome low-coverage sequencing and medical exome sequencing. BMC Med Genomics. 2020 May;13(1):70.
Burnside RD. 22q11.21 Deletion Syndromes: A Review of Proximal, Central, and Distal Deletions and Their Associated Features. Cytogenet Genome Res. 2015;146(2):89–99.
Wincent J, Bruno DL, van Bon BWM, Bremer A, Stewart H, Bongers EMHF, et al. Sixteen New Cases Contributing to the Characterization of Patients with Distal 22q11.2 Microduplications. Mol Syndromol. 2010;1(5):246–54.
Chung BHY, Mullegama S, Marshall CR, Lionel AC, Weksberg R, Dupuis L, et al. Severe intellectual disability and autistic features associated with microduplication 2q23.1. Eur J Hum Genet. 2012 Apr;20(4):398–403.
Hijazi H, Coelho FS, Gonzaga-Jauregui C, Bernardini L, Mar SS, Manning MA, et al. Xq22 deletions and correlation with distinct neurological disease traits in females: Further evidence for a contiguous gene syndrome. Hum Mutat. 2020 Jan;41(1):150–68.
Rossi MR, DiMaio MS, Xiang B, Lu K, Kaymakcalan H, Seashore M, et al. Clinical and genomic characterization of distal duplications and deletions of chromosome 4q: study of two cases and review of the literature. Am J Med Genet A. 2009 Dec;149A(12):2788–94.
Acknowledgements
We would like to express our gratitude towards the patients and their families, and the referring physicians.
Funding
Not applicable.
Open access funding provided by Semmelweis University.
Author information
Authors and Affiliations
Contributions
AL, GyF and IH conceived the study. AL, ÉP and GyF contributed to genetic counseling and data interpretation. HP, ÁK, TA, ZN and IH performed the genetic tests and contributed to data interpretation. AL performed data analysis and prepared the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This study was approved by the Semmelweis University Regional and Institutional Committee of Science and Research Ethics (accession number 44/2022).
Consent to participate
Informed consent to participate was obtained from legal guardians. According to national guidelines, verbal consent was sufficient; as this retrospective study publishes no identifiable information, and all tests were conducted for diagnostic purposes (the legal guardians have given written informed consent for the diagnostic genetic examinations at the time of testing.)
Consent for publication
Additional written informed consent was obtained for the publication of images for patient SEG2_57 (Fig. 2) from both the mother and the father, in English and in Hungarian.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lengyel, A., Pinti, É., Pikó, H. et al. Clinical evaluation of rare copy number variations identified by chromosomal microarray in a Hungarian neurodevelopmental disorder patient cohort. Mol Cytogenet 15, 47 (2022). https://doi.org/10.1186/s13039-022-00623-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-022-00623-z