
Abstract
Background
Chiari malformation type II (CM-II) is mainly characterized by elongation and descent of the cerebellum through the foramen magnum into the spinal canal. Moreover, CM-II is uniquely associated with myelomeningocele. Sprengel’s deformity refers to the malposition of the scapula, i.e. scapular elevation which is sometimes accompanied with scapula dysplasia. Although few familial cases of CM-II and Sprengel’s deformity have been previously reported, both of these defects are considered to be sporadic, thus the exact etiology and causative genes have largely remained unknown.
Case presentation
The patient was diagnosed with CM-II accompanied with Sprengel’s deformity. Further genetic investigation revealed a novel 666 kb microdeletion located in 3q29 (chr3:194,532,035–195,198,585; Hg19). Subsequently, genes within the affected region were summarized, and XXYLT1 and ACAP2 were identified as the candidate genes.
Conclusion
We reported a case of a patient with CM-II and Sprengel’s deformity harboring a microdeletion in 3q29. This case highlights the importance of 3q29 in early neural and skeletal development, as well as expands the phenotype spectrum of this rare disorder.
Similar content being viewed by others


Background
Chiari malformation type II (CM-II; OMIM #207950) is a rare congenital defect, characterized by elongation and descent of the cerebellum through the foramen magnum into the spinal canal. Other cerebral structures including pons, medulla, and fourth ventricle can also be affected, leading to the brain stem compression, or obliteration of the fourth ventricle and cisterna magna [1, 2]. Moreover, CM-II is uniquely associated with myelomeningocele (OMIM #182940), another congenital malformation identified as unfused neural tube occurring between postovulatory days 21 and 27 [3]. CM-II associated myelomeningocele is mainly observed in thoracic segments, while the nonsyndromic myelomeningocele often affects lumbosacral portions [2, 4, 5]. Sprengel’s deformity (OMIM #184400) is the most common congenital abnormality of the shoulder girdle [6]. It refers to the congenital elevation of the scapula, sometimes accompanied with scapula dysplasia. What’s more, complications like regional muscle hypoplasia or atrophy can occur, leading to limited shoulder movement. Although several familial cases of CM-II or Sprengel’s deformity have been reported, both defects are widely considered to be sporadic, due to the unknown etiology and undetermined causal genes [7,8,9,10].
Matsuoka et al. have studied the origin of neck and shoulder in mice model [11]. Their results indicated that the parts of the skeleton affected in CM-I/II, Sprengel’s deformity and the KlippelFeil syndrome all derive from a specific population of neural crest cells, suggesting these diseases might have a similar developmental origin. However, research on the association between spinal dysraphism and Sprengel’s deformity is scarce [12].
Herein, we reported a case of a patient diagnosed with CM-II accompanied with Sprengel’s deformity. One-copy deletion of 3q29 region (chr3: 194,532,035–195,198,585; Hg19) was identified. The affected region contains two genes, XXYLT1 (xyloside xylosyltransferase 1, also known as C3orf21; OMIM #614552) and ACAP2 (Arf-gap with coiled-coil, ankyrin repeat, and pleckstrin homology domains-2, also known as CENTB2; OMIM #607766). XXYLT1 encodes enzyme xyloside xylosyltransferase, which is responsible for the addition of xylose to O-glucosylated Notch epidermal growth factor (EGF)-like repeats, and mainly functions in Notch signaling transduction [13]. ACAP2 is the GTPase-activating proteins of Arf6 (ADP-ribosylation factor 6). ACAP2 mediates cellular events including endocytic recycling, phagocytosis, cytokinesis and neurite outgrowth through regulating Arf6, which is involved in the endosomal membrane trafficking and the actin cytoskeleton in the cell periphery [14].
Case presentation
The Review Board of the Xiangya Hospital of the Central South University approved this study. The informed consent was obtained from each subject.
Surgical Repairation
The patient, a 7-year-old girl from the Central-south region of China (Xiangtan, Hunan Province), was born to a non-consanguineous family. No family history of congenital defects was noted. The gestation process was uncomplicated by maternal illness or teratogen exposure. Our patient was born at 41 weeks by spontaneous vaginal delivery. The birth weight and length of the proband were normal, i.e. 2900 g (25th centile) and 43 cm (25th centile) respectively. Moreover, her head circumference was 34 cm (25th centile). Nonetheless, she was observed having spinal meningocele in the cervicothoracic region (Fig. 1a). No facial phenotypes were noted.

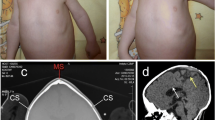
Phenotypic appearance of the affected individual in our study. a X-ray shows spinal meningocele in the cervicothoracic region in our patient at birth. b This X-ray was taken in 2011 when the proband came to our institution for spina bifida repair. It shows that besides myelomeningocele, the proband also had elongated and descended cerebellum. These results led to her diagnosis of Chiari malformation. c Patient’s X-ray from 2016, revealing an elevated right scapula. d After partial scapulectomy and collum costatectomy, the shoulder function was partially restored. e Human 660w–Quad SNP-array results. A 666 Kb microdeletion (chr3:194,532,035–195,198,585; Hg19) located in the 3q29 region. XXYLT1 (C3orf21) is affected by this deletion
Her spinal defect was repaired at the age of two, i.e. in 2011 when she came to our institution. MRI and X-ray were performed as part of a comprehensive preoperative physical examination. MRI results showed an extended cerebellum, and brain stem extending into the foramen magnum (Fig. 1b). Based on her medical history of spina bifida, she was diagnosed with CM-II [15]. What’s more, X-ray showed soft tissue mass shadows in the cervicothoracic region, while the cardiac and pulmonary features were normal. In 2016, the patient came again to our institution due to left acromioclavicular dislocation and significantly elevated right scapula (Fig. 1c). Indentations were observed in the axilla and shoulder region, and were accompanied with muscular hypertonia. The physiological functions of her articulatio humeri were assessed, and the obtained results showed impaired pronation and supination. Subsequently, partial scapulectomy and collum costatectomy were performed. The procedure was smooth, and the patient partially regained shoulder function (Fig. 1d).
Cytogenetic analysis
Five milliliters of peripheral blood were collected from the patient and her parents. The blood samples were subjected to lymphocyte culture as previously described [16]. Chromosome analysis was performed by conventional G-Banded techniques at the level of 550 bands. Karyotype analysis of the patient was normal. We hypothesized that CNVs might contribute to the multiple birth defects. Then, the genomic DNA was prepared using a DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) on the QIAcube automated DNA extraction robot (Qiagen, Hiden, Germany) [17]. Single nucleotide polymorphism (SNP)-array analysis was employed to test it (Human660W–Quad Chip, Bead station Scanner, and Genome Studio V2011 software) [18]. The call rates of the samples were greater than 99.5%. We performed a family-based copy number variations (CNVs) validation with the PennCNV algorithm to obtain authentic CNVs [19]. We identified a 666 kb microdeletion located in 3q29 (chr3:194,532,035–195,198,585; Hg19). This deletion might lead to haploinsufficiency of XXYLT1 and ACAP2, due to function loss of the other allele (Fig. 1e). The function of these two genes is briefly summarized in the Table-1.
In conclusion, our patient was diagnosed with CM-II accompanied with Sprengel’s deformity. Surgical intervention was effective, with good postoperative recovery.
Discussion
Herein, we described the clinical features and genotype of a patient with CM-II and Sprengel’s deformity. She was born with open spina bifida, and she received repair surgery at the age of two. The diagnosis of CM-II malformation was made with the use of magnetic resonance imaging (MRI). Five years later, she returned to our institution because of Sprengel’s deformity diorthosis. To determine the underlying genetic cause for our patient’s conditions, we performed SNP array and identified a novel 666 Kb microdeletion in the terminal 3q29 region (chr3: 194,532,035–195,198,585; Hg19).
Thus far, more than 40 cases of 3q29 microdeletion have been reported [20, 21]. Most patients share a common 2 Mb deletion from 196 Mb to 198 Mb, which are mainly characterized by psychiatric manifestations, including autism and schizophrenia. Three genes, FBXO45 (OMIM #609112), DLG1 (OMIM #601014), and PAK2 (OMIM #605022), have been proposed as a cause of these mental disorders due to their roles in synaptic transmission [21, 22]. Other genes located in the affected region may also contribute to the pathogenic process.
However, in the present study, an atypical 3q29 deletion with the affected region range from 194 Mb to 195 Mb comprising two genes, ACAP2 and XXYLT1, was detected (Table 1). XXYLT1 encodes the protein Xyloside Xylosyltransferase, which is localized in the endoplasmic reticulum. This enzyme mediates xylosylation, a conserved post-translational modification characterized as the addition of the second xylose residue to EGF-like repeats with C-X-S-X-(P/A)-C motif of the Notch protein. The O-glucose residues and the first xylose residues are added by Protein O-glucosyltransferase 1 (POGLUT-1) and Glucoside Xylosyltransferase-1 or − 2 (GXYLT1/2) respectively (Fig. 2) [13]. Both glucosylation and xylosylation function as modulators of Notch signaling pathway. Previous studies in Drosophila and Mus musculus revealed that xylose residues on EGF16–20 can negatively regulate the surface expression of the Notch receptor, while overexpressed Notch1 can inhibit osteoblastogenesis by suppressing Wnt/β-Catenin signaling [13, 23, 24]. It is very likely that this 3q29-deletion induced XXYLT1 dysfunction may impair EGF xylosylation, lead to up-regulated Notch signaling, and eventually result in impaired osteoblastogenesis, including spinal defects and Sprengel’s deformity (Fig. 3).

The O-glucose residues and the first xylose residues are added to EGF repeats with C-X-S-X-(P/A)-C motif of the Notch by POGLUT-1 and GXYLT1/2, respectively. The second xylosylation is mediated by XXYLT1

In the absence of β-catenin, TCF/LEF-1 and Groucho can form a complex and suppress transcriptional events. When Wnt signaling is activated, and β-catenin is stabilized, Groucho is displaced, and it binds TCF/LEF-1 causing a transcriptional activation. In our case, XXYLT1 deletion leads to impaired Notch xylosylation, causing abnormally up-regulated Notch function. This process decreases β-catenin and increases HES-1, which interacts with Groucho and prevents the following activation of gene transcriptions. As a result, osteoblastogenesis mediated by Wnt/β-catenin is inhibited by over-functional Notch signaling, which may contribute to the pathogenic process and finally lead to the congenital skeletal defects observed in our patient
While XXYLT1 mainly functions in Notch signaling during development, the other affected gene, ACAP2, is thought to participate in various cellular events including endocytic recycling, phagocytosis, cytokinesis and neurite outgrowth [14, 25]. ACAP2 is one of the Arf6-GAPs, and mainly functions as the inhibitor of Arf6 by catalyzing Arf6 into the inactive GDP-bound state. Previous study has revealed that ACAP2 regulates neurite outgrowth by inactivating Arf6 at the pericentrosomal endosomes: when ACAP2 is down-regulated by shRNA, dramatic reduction occurs in the neurite length [14]. In this scenario, truncated ACAP2 may disrupt primary neurulation, lead to nonclosure of neural tube, and finally cause myelomeningocele. Further investigations are required to clarify the exact function of XXYTL1 and ACAP2 during development, as well as their pathogenic roles.
Additionally, we compared the clinical features in our patient with previous cases diagnosed with CM-II and Sprengel’s deformity (Table 2) [12]. Although the genotypes were not available for all these cases, we found multiple segmentation defects of the spine, ribs, and shoulder among these patients, which suggests that these symptoms might result from a common genetic defect affecting somitogenesis.
Conclusion
The spinal defects in combination with Sprengel’s deformity occur very rarely, and thus far only few cases have been reported. What’s more, relevant developmental etiology and genetic causes still remain poorly understood. Although pathogenesis of both spinal dysraphism and Sprengel’s deformity remain unclear, there is evidence suggesting that both conditions might be linked to abnormal somitogenesis. In this case, deleted XXYTL1 and ACAP2 were identified in a patient with CM-II and Sprengel’s deformity. These two genes could have a critical role during somitogenesis, and their dysfunction may disrupt embryonic development, and lead to spinal dysraphism and shoulder defects. Further investigations are required to clarify their exact function during development, as well as their role in spinal dysraphism and Sprengel’s deformity.
Abbreviations
- CM-II:
-
Chiari Malformation type II
- CNV:
-
Copy Number Variation
- EGF repeats:
-
Epidermal Growth Factor-like Repeats
- GXYLT1/2:
-
Glucoside Xylosyltransferase-1 or − 2
- POGLUT-1:
-
Protein O-glucosyltransferase 1
- XXYTL1:
-
Xyloside Xylosyltransferase 1
References
Messing-Junger M, Rohrig A. Primary and secondary management of the Chiari II malformation in children with myelomeningocele. Childs Nerv Syst. 2013;29(9):1553–62.
Bell WO, Charney EB, Bruce DA, Sutton LN, Schut L. Symptomatic Arnold-Chiari malformation: review of experience with 22 cases. J Neurosurg. 1987;66(6):812–6.
Boltshauser E, Schneider J, Kollias S, Waibel P, Weissert M. Vanishing cerebellum in myelomeningocoele. Eur J Paediatr Neurol. 2002;6(2):109–13.
Song RB, Glass EN, Kent M. Spina bifida, Meningomyelocele, and Meningocele. Vet Clin North Am Small Anim Pract. 2016;46(2):327–45.
Wang GQ, Kang YJ, Li YW, Wang B. A rare presentation in a girl with occult spinal bifida. Spine J. 2016;16(4):e247.
Harvey EJ, Bernstein M, Desy NM, Saran N, Ouellet JA. Sprengel deformity: pathogenesis and management. J Am Acad Orthop Surg. 2012;20(3):177–86.
Bindoudi A, Kariki EP, Vasiliadis K, Tsitouridis I. The rare sprengel deformity: our experience with three cases. J Clin Imaging Sci. 2014;4:55.
Isidor B, David A. Two girls with short stature, short neck, vertebral anomalies, Sprengel deformity and intellectual disability. Eur J Med Genet. 2015;58(1):47–50.
Soares de Araujo JS, Regis CT, Gomes RG, Tavares TR, Rocha Dos Santos C, Assuncao PM, et al. Microcephaly in north-east Brazil: a retrospective study on neonates born between 2012 and 2015. Bull World Health Organ. 2016;94(11):835–40.
Mizuguchi K, Morota N, Kubota M. Respiratory complications in children with Chiari malformation type II associated with myelomeningocele. No To Hattatsu. 2016;48(1):25–8.
Matsuoka T, Ahlberg PE, Kessaris N, Iannarelli P, Dennehy U, Richardson WD, et al. Neural crest origins of the neck and shoulder. Nature. 2005;436(7049):347–55.
van Aalst J, Vles JS, Cuppen I, Sival DA, Niks EH, Van Rhijn LW, et al. Sprengel's deformity and spinal dysraphism: connecting the shoulder and the spine. Childs Nerv Syst. 2013;29(7):1051–8.
Lee TV, Sethi MK, Leonardi J, Rana NA, Buettner FF, Haltiwanger RS, et al. Negative regulation of notch signaling by xylose. PLoS Genet. 2013;9(6):e1003547.
Kobayashi H, Fukuda M. Rab35 regulates Arf6 activity through centaurin-beta2 (ACAP2) during neurite outgrowth. J Cell Sci. 2012;125(Pt 9):2235–43.
Zhang HQ, Deng A, Liu SH, Chen LQ, Guo CF, Tang MX, et al. Adult thoracolumbar or lumbar scoliosis with Chiari malformation and syringomyelia: a retrospective study of correction and fusion strategies. Arch Orthop Trauma Surg. 2011;131(4):475–80.
Sun G, Tan Z, Fan L, Wang J, Yang Y, Zhang W. 1q21.1 microduplication in a patient with mental impairment and congenital heart defect. Mol Med Rep. 2015;12(4):5655–8.
Wu PF, Guo S, Fan XF, Fan LL, Jin JY, Tang JY, et al. A novel ZRS mutation in a Chinese patient with Preaxial Polydactyly and Triphalangeal thumb. Cytogenet Genome Res. 2016;149(3):171–5.
Pinto D, Darvishi K, Shi X, Rajan D, Rigler D, Fitzgerald T, et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol. 2011;29(6):512–20.
Chen JL, Yang YF, Huang C, Wang J, Yang JF, Tan ZP. Clinical and molecular delineation of 16p13.3 duplication in a patient with congenital heart defect and multiple congenital anomalies. Am J Med Genet A. 2012;158A(3):685–8.
Quintero-Rivera F, Sharifi-Hannauer P, Martinez-Agosto JA. Autistic and psychiatric findings associated with the 3q29 microdeletion syndrome: case report and review. Am J Med Genet A. 2010;152A(10):2459–67.
Cox DM, Butler MG. A clinical case report and literature review of the 3q29 microdeletion syndrome. Clin Dysmorphol. 2015;24(3):89–94.
Biamino E, Di Gregorio E, Belligni EF, Keller R, Riberi E, Gandione M, et al. A novel 3q29 deletion associated with autism, intellectual disability, psychiatric disorders, and obesity. Am J Med Genet B Neuropsychiatr Genet. 2016;171B(2):290–9.
Deregowski V, Gazzerro E, Priest L, Rydziel S, Canalis E. Notch 1 overexpression inhibits osteoblastogenesis by suppressing Wnt/beta-catenin but not bone morphogenetic protein signaling. J Biol Chem. 2006;281(10):6203–10.
Sciaudone M, Gazzerro E, Priest L, Delany AM, Canalis E. Notch 1 impairs osteoblastic cell differentiation. Endocrinology. 2003;144(12):5631–9.
Jackson TR, Brown FD, Nie Z, Miura K, Foroni L, Sun J, et al. ACAPs are arf6 GTPase-activating proteins that function in the cell periphery. J Cell Biol. 2000;151(3):627–38.
Acknowledgments
We wish to thank the patient and her parents for participating in this study. We wish to thank the Laboratory of Medical Genetics of Central South University for technical assistance. We wish to thank all subjects for participating in this study.
Funding
This study was supported by the National Natural Science Foundation of China (81370394) and the Fundamental Research Funds for Central Universities of Central South University (2017zzts360).
Availability of data and materials
All study data are available via email shirlesmile@csu.edu.cn.
Author information
Authors and Affiliations
Contributions
SG performed SNP-array and was a major contributor in writing the manuscript. X-FF described clinical features in our patient. J-YJ made the Karyotype analysis and DNA extraction. L-LF analyzed the SNP-array results. Z-BZ and LZ reviewed the clinical features in previous cases and the affected genes in 3q29, respectively. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The Review Board of the Xiangya Hospital of the Central South University approved this study. A written consent form was signed by the subject.
Consent for publication
Consent for publication of this study was signed by patient’s father.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Guo, S., Fan, XF., Jin, JY. et al. A novel proximal 3q29 chromosome microdeletion in a Chinese patient with Chiari malformation type II and Sprengel’s deformity. Mol Cytogenet 11, 8 (2018). https://doi.org/10.1186/s13039-018-0358-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-018-0358-4