
Abstract
Background
Hand-foot-genital syndrome (HFGS) is a rare condition characterized by congenital malformations in the limbs and genitourinary tract. Generally, this syndrome occurs due to point mutations that cause loss of function of the HOXA13 gene, which is located on 7p15; however, there are some patients with HFGS caused by interstitial deletions in this region.
Case presentation
We describe a pediatric Mexican patient who came to the Medical Genetics Department at the National Institute of Pediatrics because he presented with genital, hand and feet anomalies, facial dysmorphisms, and learning difficulties. Array CGH reported a 12.7 Mb deletion that includes HOXA13.
Conclusions
We compared our patient with cases of HFGS reported in the literature caused by a microdeletion; we found a minimum shared region in 7p15.2. By analyzing the phenotype in these patients, we suggest that microdeletions in this region should be investigated in all patients with clinical characteristics of HFGS who also present with dysplastic ears, mainly low-set implantation with a prominent antihelix, as well as a low nasal bridge and long philtrum.
Similar content being viewed by others

Background
Hand-foot-genital syndrome (HFGS; OMIM #140000) is an autosomal dominant condition that has been reported in 23 families [1,2,3,4,5,6,7,8] and 5 sporadic cases [9,10,11,12]. It is characterized by congenital limb malformations, mainly affecting the hands, such as short thumbs, and feet, as well as the genitourinary tract, such as hypospadias and cryptorchidism in males or urogenital septum and bicorne uterus in females [13]. HFGS is produced by mutations that cause loss of function of the HOXA13 gene, which is located on 7p15 and is important for the development of fetal limbs and the urinary tract. The mutations responsible for HFGS include missense mutations, nonsense mutations, polyalanine expansions, and in very few cases, interstitial deletions at 7p15. We describe a Mexican patient with HFGS caused by a 12.7 Mb deletion on chromosome 7, ranging from p15.3 to p14.3, including the HOXA13 gene. We compared previously reported patients with the purpose of determining a minimum shared region, as well as clinical characteristics that could guide the diagnosis of HFGS caused by a microdeletion.
Case presentation
The patient is a 14-year-old male, the third child of a 21-year-old mother and a 30-year-old father, both of whom are healthy and non-consanguineous. He has two healthy brothers aged 20 and 18 years old. He was born after an uneventful pregnancy; his birth weight was 2300 g (<3th percentile), and his height was 49 cm (>15th percentile).
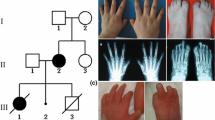
At 7 years old, the patient was referred to our hospital due to genital anomalies. Upon admission, a global developmental delay was detected, which was most profound in the area of language. He also presented with short stature, microcephaly, facial dysmorphisms, such as facial asymmetry, midface hypoplasia, a broad and prominent forehead, mild hypertelorism, strabismus, a broad nasal bridge, a long and smooth philtrum, a thin upper lip, a high palate (Fig. 1a), and low-set ears with a prominent antihelix (Fig. 1b). Additionally, we observed subglandular hypospadias and cryptorchidism, hands with fifth finger clinodactyly, abnormal palmar creases, and severe bilateral hallux valgus (Fig. 2a, b and 3a).

a. Facial phenotype of patient with midface hypoplasia, broad and prominent front, mild hypertelorism, strabism, broad nasal bridge, long and smooth philtrum, thin upper lip. b. Posteriorly rotated ears and prominent antihelix

a. Small hands with pointed fingers, brachydactyly, bilateral fifth finger clinodactyly. b. Abnormal creases are observed. c. Small distal phalanges, metacarpal shortening

a. Feet with brachydactyly and severe bilateral hallux valgus. b. Metatarsal shortening and hypoplastic phalanges of all toes. Most notable is the thumb, which presents with valgus deviation of the last phalange
The patient’s brain MRI was normal, while auditory evoked potentials revealed normal left hearing and right conductive superficial hearing loss (secondary to serous otitis media). An X-ray of the hands showed a three-year delayed bone age, feet with an absence of mid-foot bone structures, such as cuneiform bones, only cuboid and navicular bones were observed, as well as bilateral hallux valgus, which was surgically repaired at 13 years old (Figs. 2c and 3b). Karyotype and Array CGH Agilent 60 K reported a 12.7 Mb deletion at 7p14.2p15.3 (Fig. 4a and b; ISCN 2016), which was confirmed by FISH with a specific 54.95Kb probe targeted to 7p15.2 inside the deleted region (Fig. 4c). The specific nomenclature is as follows: 46,XY.ish del(7)(p15.2)(D7Z1+,HOXA6−,HOXA13−).arr[GRCh37/hg19] 7p15.3p14.3(22574164_35288260)x1dn (Fig. 5; ISCN 2016). Both parents were normal. Analysis of the genomic alterations and the genotype-phenotype correlation enabled a diagnosis of hand-foot-genital syndrome.

a. Karyotype with G-banding. b. Normal and deleted chromosome 7 (arrows) from different metaphases; c. FISH for the 7p15 region on metaphase chromosomes; alpha satellite probe for chromosome 7 centromere (D7Z1) marked with green fluorochrome Q-BIOgene (one thin arrow), and locus specific probe for 7p15.2 (where the HOXA6-HOXA13 genes are normally located) marked with red fluorochrome-SureFiSH, Agilent (two thick arrows). Note that deleted chromosomes only have the green fluorescent probe and are missing the red fluorescent probe

Array CGH of the patient. Array Agilent SurePrint G3 Human CGH microarray kit, 8x60K 46,XY.array [GRCh37/hg19] 7p15.3p14.3(22574164_35288260)x1dn (12,714,097 bp)
Discussion and conclusions
Clinical aspects of hand-foot-genital syndrome
Some of the most frequent limb anomalies in HFGS are short thumbs, medial phalanges, small feet and clinodactyly of the fifth finger [13]. Hallux valgus is an important diagnostic sign, when present [11]. These anomalies have complete penetrance, but they can be so mild that imaging studies may be necessary to observe them. Short phalanges and pointed thumbs, hypoplasia of the first metacarpus and metatarsus, a small calcaneus, trapezium-scaphoid fusion, and cuneiform-navicular fusion can be observed radiologically [10]. Occasional findings include fusion of the medial and distal phalanges to the second to fifth toes [14], as well as delayed ossification [8].
It is estimated that half of the affected patients show urogenital malformations with variable expressivity. Male patients show hypospadias and cryptorchidism, while women present with a double uterus and partial or complete longitudinal urogenital septum. In the presence of any of these findings, HFGS must be investigated due to a 0.5–4% prevalence of the fusion of müllerian structures in the general population [8]. Fertility is conserved in HFGS, though there is an increased risk of spontaneous abortion, preterm birth, and stillbirth [2, 10, 11, 13].
In both genders, common findings include chronic urinary tract infections, ectopic ureteric orifices, vesicoureteral reflux, and pelviureteric junction. Additionally, the disease is associated with complications, such as chronic pyelonephritis and renal failure, which may require transplant. Psychomotor development and cognitive abilities are within normal range [11].
Differential diagnosis to be considered are Fanconi anemia syndrome, Rothmund Thomson syndrome, Holt-Oram syndrome, SALL4-related disorders, Nager syndrome, Townes-Brocks syndrome, and Lacrimo-auriculo-dento-digital (LADD) syndrome, due to the thumb hypoplasia. Incomplete Müllerian fusion and/or longitudinal vaginal septum should be cause for consideration of acro-renal-mandibular, Bardet-Biedl, Beckwith-Wiedemann, Fraser, Fryns, Halal, and Meckel syndromes. Heterozygous mutations in the HOXA13 gene cause HFGS, however the specific c.1112A > T (p.Gln371Leu) mutation causes Guttmacher syndrome which shares some, but not all, features of HFGS and includes postaxial polydactyly of the hands, preaxial hand and foot deficiencies, short second toes, and glandular hypospadias [14]. Moreover, when patients present with short or absent nails and absent or hypoplastic distal phalanges without genital anomalies, brachydactyly type B1 and Cooks syndrome, must be considered [14].
Genetics of HFGS
HOX genes are evolutionarily conserved; they participate in developmental processes and maintain their expression until adulthood [14]. They are organized into four clusters, HOXA, HOXB, HOXC, and HOXD, localized in 7p15.2, 17q21.32, 12q13.2, and 2q31.1, respectively. They contain between 9 and 11 genes each and codify transcription factors essential for morphogenesis [2]. In particular, HOXA13 has 2 exons: exon 1 contains five polyalanine regions, and exon 2 has a homeobox domain that is involved in the development of distal limbs and the fetal urogenital tract [12]. HOXA13 was proposed as candidate gene for HFGS and its involvement in limb development was corroborated in murine models [15].
The first identified pathogenic variant that causes HFGS in humans was the nonsense mutation c.1107G > A (p.Trp369Ter) [15]. Since then, there have been reports of pathogenic variants in both familial and de novo cases, including missense mutations (p.Arg326Gly, p.Asn372His, p.Gln371 Leu), nonsense mutations (p.Ser1360Ter, p.Gln1960Ter, p.Gln3650Ter and p.Trp3690Ter), and triplet expansions that code for a polyalanine region (6, 8–12, and 14 additional polyalanines) [7, 12]. Approximately 35% of these variants are localized within the coding region and, of these, 50 to 60% are found in the polyalanine regions [13, 15]. The missense pathogenic variants produce a gain of function mechanism associated with a more severe phenotype, while the nonsense mutations and polyalanine expansion variants are responsible for loss of protein function, presenting a milder phenotype [15].
Microdeletions in HFGS
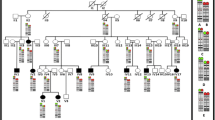
Interstitial deletions including the 7p15 region are a rare cause of hand-foot-genital syndrome. To date, there are few reported patients with de novo deletions, which mostly occur between 7p15 and 7p21, with a variable size ranging from 2.5 to 12.7 Mb [9, 16,17,18,19,20,21]. We compared previously reported patients in the literature according to the genetic cause of the syndrome, polyalanine expansions, point mutations, and microdeletions; our patient was included in the latter group (Table 1). We found clinical features in patients with microdeletions that are not shared with patients carrying point mutations or those with polyalanine expansions (data not shown). Likewise, we found a minimum deleted region of 584,758 bp located at 7p15.2 that is shared by patients with HFGS. This region contains the HOXA cluster, the SKAP2 and EVX1 genes, and miRNA196b (Fig. 6).

Genome Browser image comparing HFGS cases by microdeletion, included in Table 1. The red rectangle demarcates the minimum region (including the HOXA cluster) proposed as responsible for the phenotype of these patients. The most important genes that correlate with the proposed phenotype are: the HOXA13 gene (gray circle), the HOXA1 and HOXA2 genes (blue circle), the HOXA5 and HOXA11 genes (orange circle), and miRNA196b (green circle)
From this analysis, we derived important phenotypic data that might suggest the presence of a microdeletion in the 7p15 region, which include the following: a) Neurodevelopmental delay or intellectual disability present in all patients with the microdeletion. HOXA1 gene is localized in the minimum shared region and is associated with central nervous system development [22]. Also, in this region is miRNA196b (Fig. 6; green circle) [22], in which the gene target NRC2 is implicated in neuronal development [23]. b) Short stature is shared by more than half of the patients (5 of 9) with the microdeletion. Both HOXA5 and HOXA11 are located within the HOXA cluster (Fig. 6; orange circle), dysregulation of which might affect growth and consequently generate short stature [22, 24]. c) Facial dysmorphias that are not present in patients with point mutations are found in 9 out of 9 patients with the microdeletion; the most common finding is dysplastic ears, particularly low-set ears with a prominent antihelix, observed in 8 out of 9 reported patients (in the ninth patient, the ears were not phenotypically described). Other facial features shared by most patients with microdeletion are a depressed nasal bridge and a long philtrum. The HOXA cluster is located within the minimum shared region and includes the HOXA1 and HOXA2 genes (Fig. 6; blue circle), both of which are related to external ear development. This has been verified in mouse models, where high Hoxa1 and Hoxa2 messenger expression levels and low mmu-miR-10a (a modulator of Hoxa1 expression during ear development) levels have been reported [25].
These genes, as members of the HOX gene family, play an important role in embryonic patterning and organogenesis, specifically in the developing region of the rhombencephalus and branchial arch [26,27,28]. Mouse Hoxa2 knockout models, as well as pig models with nonsense mutations in Hoxa1, have been associated with congenital microtia [29, 30].
Deleted region sizes are variable, so we looked for a minimum shared region in patients with HFGS caused by a 7p15 microdeletion to compare phenotypes and to determine if larger genomic losses cause more dysmorphisms or more severe phenotypes. We found that the patient that had the smallest deletion (2.5 Mb; including the 584,758 bp minimum shared region; Fig. 6) [17], as well as our patient, who has the largest deletion reported so far (12.7 Mb), the classical clinical characteristics of HFGS were sufficient evidence to suggest haploinsufficiency of HOXA13, despite the absence of several other genes, responsible of a more severe phenotype, making diagnosis more difficult.
According to the information reviewed in every previously reported patient and the characteristics of our patient, we can suggest that a microdeletion in the 7p15 region must be investigated in every HFGS patient that presents with dysplastic ears, mainly low-set ears with a prominent antihelix, as well as a depressed nasal bridge and a long philtrum. Finally, the patient’s follow-up and management was based on the literature reports [16], and independent of the etiology, the recurrence risk for each pregnancy is 50% for carrier parents and less than 1% for healthy parents.
References
Stern AM, Gall JC Jr, Perry BL, Stimson CW, Weitkamp LR, Poznanski AK. The hand-foot-uterus syndrome: a new hereditary disorder characterized by hand and foot dysplasia, dermatoglyphic abnormalities, and partial duplication of the female genital tract. J Pediatr. 1970;77:109–16.
Mortlock DP, Innis JW. Mutation of HOXA13 in hand-foot-genital syndrome. Nat Genet. 1997;15:179–80. 10.1038/ng0297-179.
Goodman FR, Bacchelli C, Brady AF, Brueton LA, Fryns JP, Mortlock DP, et al. Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am J Hum Genet. 2000;67:197–202.
Debeer P, Bacchelli C, Scambler PJ, De Smet L, Fryns JP, Goodman FR. Severe digital abnormalities in a patient heterozygous for both a novel missense mutation in HOXD13 and a polyalanine tract expansion in HOXA13. J Med Genet. 2002;39:852–6.
Utsch B, Becker K, Brock D, Lentze MJ, Bidlingmaier F, Ludwig M. A novel stable polyalanine [poly(a)] expansion in the HOXA13 gene associated with hand-foot-genital syndrome: proper function of poly(a)-harbouring transcription factors depends on a critical repeat length? Hum Genet. 2002;110:488–94.
Frisén L, Lagerstedt K, Tapper-Persson M, Kockum I, Nordenskjöld A. A novel duplication in the HOXA13 gene in a family with atypical hand-foot-genital syndrome. J Med Genet. 2003;40:e49. 10.1136/jmg.40.4.e49.
Innis JW, Mortlock D, Chen Z, Ludwig M, Williams ME, Williams TM, et al. Polyalanine expansion in HOXA13: three new affected families and the molecular consequences in a mouse model. Hum Mol Genet. 2004;13:2841–51.
Jorgensen EM, Ruman JI, Doherty L, Taylor HS. A novel mutation of HOXA13 in a family with hand-foot-genital syndrome and the role of polyalanine expansions in the spectrum of Müllerian fusion anomalies. Fertil Steril. 2010;94:1235–8. 10.1016/j.fertnstert.2009.05.057.
Devriendt K, Jaeken J, Matthijs G, Van Esch H, Debeer P, Gewillig M, Fryns JP. Haploinsufficiency of the HOXA gene cluster, in a patient with hand-foot-genital syndrome, velopharyngeal insufficiency, and persistent patent Ductus botalli. Am J Hum Genet. 1999;65:249–51.
Parker L, Mangwani J, Wakeling E, Singh D. Hallux valgus interphalangeus and a novel mutation in HOXA13. Part of the broadening spectrum of hand-foot-genital syndrome. Foot Ankle Surg. 2011;17:e28–30. 10.1016/j.fas.2010.12.003.
Wallis M, Tsurusaki Y, Burgess T, Borzi P, Matsumoto N, Miyake N2, True D, Patel C. Dual genetic diagnoses: atypical hand-foot-genital syndrome and developmental delay due to de novo mutations in HOXA13 and NRXN1. Am J Med Genet A. 2016;170:717–24. 10.1002/ajmg.a.37478.
Imagawa E, Kayserili H, Nishimura G, Nakashima M, Tsurusaki Y, Saitsu H, Ikegawa S, Matsumoto N, Miyake N. Severe manifestations of hand-foot-genital syndrome associated with a novel HOXA13 mutation. Am J Med Genet A. 2014;164A:2398–402. 10.1002/ajmg.a.36648.
Tas E, Sebastian J, Madan-Khetarpal S, Sweet P, Yatsenko AN, Pollock N, et al. Familial deletion of the HOXA gene cluster associated with hand–foot–genital syndrome and phenotypic variability. Am J Med Genet A. 2017;173:221–4. 10.1002/ajmg.a.37981.
Quinonez SC, Innis JW. Human HOX gene disorders. Mol Genet Metab. 2014;111:4–15. 10.1016/j.ymgme.2013.10.012.
Watson CM, Crinnion LA, Harrison SM, Lascelles C, Antanaviciute A, Carr IM, Bonthron DT, Sheridan E. Chromosome 7 Pericentric inversion defined at single-nucleotide resolution using diagnostic whole genome sequencing in a patient with hand-foot- genital syndrome. PLoS One. 2016;11:e0157075. 10.1371/journal.pone.0157075.
Pezzani L, Milani D, Manzoni F, Baccarin M, Silipigni R, Guerneri S, Esposito S. HOXA genes cluster: clinical implications of the smallest deletion. Ital J Pediatr. 2015;41:31. 10.1186/s13052-015-0137-3.
Hosoki K, Ohta T, Fujita K, Nishigaki S, Shiomi M, Niikawa N, Saitoh S. Hand-foot-genital syndrome with a 7p15 deletion: clinically recognizable syndrome. Pediatr Int. 2012;54:e22–5. 10.1111/j.1442-200X.2011.03550.x.
Dunø M, Hove H, Kirchhoff M, Devriendt K, Schwartz M. Mapping genomic deletions down to the base: a quantitative copy number scanning approach used to characterise and clone the breakpoints of a recurrent 7p14.2p15.3 deletion. Hum Genet. 2004;115:459–67.
Kosaki R, Higuchi M, Mitsui N, Matsushima K, Ohashi H, Kosaki K. Deletion involving the TWIST locus and the HOXA cluster: a contiguous gene syndrome on 7p? Congenit Anom. 2005;45:35–8.
Jun KR, Seo EJ, Lee JO, Yoo HW, Park IS, Yoon HK. Molecular cytogenetic and clinical characterization of a patient with a 5.6-Mb deletion in 7p15 including HOXA cluster. Am J Med Genet A. 2011;155:642–7.
Fryssira H, Makrythanasis P, Kattamis A, Stokidis K, Menten B, Kosaki K, Willems P, Kanavakis E. Severe developmental delay in a patient with 7p21.1-p14.3 microdeletion spanning the TWIST gene and the HOXA gene cluster. Mol Syndromol. 2011;2:45–9.
UCSC Genome Browser Database. https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&lastVirtModeType=default&lastVirtModeExtraState=&virtModeType=default&virtMode=0&nonVirtPosition=&position=chr7%3A26706135-27290892&hgsid=598513081_4nTQV0mYW5UIO52GaWMNBnraE8RI. Accessed 5 jun 2017.
NCBI Gene Database. https://www.ncbi.nlm.nih.gov/gene?cmd=Retrieve&dopt=full_report&list_uids=7182. Accessed 5 jun 2017.
Foucher I, Volovitch M, Frain M, Kim JJ, Souberbielle JC, Gan L, Unterman TG, Prochiantz A, Trembleau A. Hoxa5 overexpression correlates with IGFBP1 upregulation and postnatal dwarfism: evidence for an interaction between Hoxa5 and Forkhead box transcription factors. Development. 2002;129:4065–74.
Torres L, Juárez U, García L, Miranda-Ríos J, Frias S. External ear microRNA expression profiles during mouse development. Int J Dev Biol. 2015;59:497–503. https://doi.org/10.1387/ijdb.150124sf.
Trainor PA, Krumlauf R. Hox genes, neural crest cells and branchial arch patterning. Curr Opin Cell Biol. 2001;13:698–705.
Barrow JR, Stadler HS, Capecchi MR. Roles of Hoxa1 and Hoxa2 in patterning the early hindbrain of the mouse. Development. 2000;127:933–44.
O’Gorman S. Second branchial arch lineages of the middle ear of wild-type and Hoxa2 mutant mice. Dev Dyn. 2005;234:124–31.
Qiao R, He Y, Pan B, Xiao S, Zhang X, Li J, et al. Understanding the molecular mechanisms of human microtia via a pig model of HOXA1 syndrome. Dis Model Mech. 2015;8:611–22. 10.1242/dmm.018291.
Minoux M, Kratochwil CF, Ducret S, Amin S, Kitazawa T, Kurihara H, et al. Mouse Hoxa2 mutations provide a model for microtia and auricle duplication. Development. 2013;140:4386–97. 10.1242/dev.098046.
Acknowledgements
The aCGH was carried out at Genetadi Laboratory (Bilbao, Spain).
Funding
This study was partially supported by CONACYT FOSSIS-S0008–2008-C01–87792 and Fondos Federales 2013, Instituto Nacional de Pediatría (México). The funding body did not participate in the design of the study, in the collection, analysis, or interpretation of the data, or in the writing of the manuscript.
Availability of data and materials
Authors declare that all relevant data are included in the article.
Author information
Authors and Affiliations
Contributions
EY and SF contributed to the design and development of the protocol, analyzed the results and wrote the manuscript. DLSP wrote the early manuscript and interpreted results. SS and AR performed FISH and designed the figs. BM and RJ performed the G-banded karyotype. EL contributed to the clinical description and revised the English language usage in the manuscript. SA and JLC performed the array-CGH. VC contributed to the design of the protocol, clinical description and analysis of results. All authors read and approved the final version and are responsible for all aspects of the manuscript. SF and VC share the responsibility of corresponding authors for this paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethics and research committees of the National Institute of Pediatrics (Mexico) (Project No. 06/2009).
Consent for publication
The parents read, accepted and signed the informed consent for the protocol and for the publication of photos and images of their son.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yokoyama, E., Smith-Pellegrin, D.L., Sánchez, S. et al. 7p15 deletion as the cause of hand-foot-genital syndrome: a case report, literature review and proposal of a minimum region for this phenotype. Mol Cytogenet 10, 42 (2017). https://doi.org/10.1186/s13039-017-0345-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-017-0345-1