
Abstract
Background
Ring chromosome 18 [r(18)] syndrome represents a relatively rare condition with a complex clinical picture including multiple congenital dysmorphia and varying degrees of mental retardation. The condition is cytogenetically characterized by a complete or mosaic form of ring chromosome 18, with ring formation being usually accompanied by the partial loss of both chromosomal arms. Here we observed a 20-year-old male patient who along with the features typical for r(18) carriers additionally manifested a severe congenital subaortic stenosis. To define the genetic basis of such a compound phenotype, standard cytogenetic and high-resolution molecular-cytogenetic analysis of the patient was performed.
Case presentation
Standard chromosome analysis of cultured lymphocytes confirmed 46, XY, r(18) karyotype. Array-based comparative genomic hybridization (array-CGH) allowed to define precisely the breakpoints of 18p and 18q terminal deletions, thus identifying the hemizygosity extent, and to reveal an additional duplication adjoining the breakpoint of the 18p deletion. Apart from the terminal imbalances, we found an interstitial microdeletion of 442 kb in size (18q12.1) that encompassed DTNA gene encoding α-dystrobrevin, a member of dystrophin-associated glycoprotein complex. While limited data on the role of DTNA missense mutations in pathogenesis of human cardiac abnormalities exist, a microdeletion corresponding to whole DTNA sequence and not involving other genes has not been earlier described.
Conclusions
A detailed molecular-cytogenetic characterization of the patient with multiple congenital abnormalities enabled to unravel a combination of genetic defects, namely, a ring chromosome 18 with terminal imbalances and DTNA whole-gene deletion. We suggest that such combination could contribute to the complex phenotype. The findings obtained allow to extend the knowledge of the role of DTNA haploinsufficiency in congenital heart malformation, though further comprehensive functional studies are required.
Similar content being viewed by others


Background
Ring chromosome 18 [r(18)] is a relatively rare structural chromosomal abnormality characterized by the replacement of a normal chromosome 18 by the ring chromosome. A ring formation is usually accompanied by the partial loss of both chromosomal arms that in turn leads to hemizygous state of genes from the deleted regions [1–3]. Patients bearing a r(18) have complex clinical presentation that combines phenotypic features of 18p (OMIM #146390) and 18q (OMIM #601808) deletion syndromes and includes multiple congenital defects with varying degrees of mental retardation [4, 5]. The most typical features include short stature, multiple facial dysmorphia such as a carp-shaped mouth, cleft lip/palate, broad flat nose and epicanthic folds, hypertelorism, hearing loss, different eye abnormalities, microcephaly, abnormal white matter, hypotonia, and, rarer, congenital heart defect most commonly in a form of pulmonary stenosis [2–6].
There is considerable phenotypic variability among patients with ring 18 that is mainly due to differences in extent of hemizygosity, ring chromosome instability and somatic mosaicism [3, 4, 7, 8]. Besides, the presence of additional chromosomal abnormalities also can be considered. In this regard, whole-genome high-resolution cytogenetic analysis proves to be indispensable for comprehensive diagnosis, accurate genetic counseling and clinical management of the families.
In this report, we characterize a patient who apart from r(18) clinical manifestations exhibited a severe congenital subaortic stenosis, a heart malformation usually not described in ring 18 cases. Array-based comparative genomic hybridization (aCGH) analysis allowed us to refine the boundaries of r(18) terminal deletions and thus to define a hemizygous region responsible for the main phenotypic features. We also revealed an interstitial heterozygous microdeletion encompassing a cardiac gene, which might have an additional impact on the heart malformation of the patient.
Case presentation
Clinical report
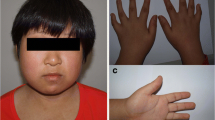
The proband was born at term as the first child of healthy non-consanguineous parents of Russian origin. At birth the patient presented multiple malformations typical for r(18) syndrome, namely microcephaly, low-set deformed ears, broad flat nose, a carp-shaped mouth, gothic palate, hypertelorism, broad and short great toe and syndactyly. At 3 years old, a subaortic stenosis and mental retardation were diagnosed. Two years later, at 5 years, subaortic stenosis was surgically corrected followed by a repeated operation and Ross-Konno procedure at the age of 13 and balloon angioplasty at the age of 18 years.
Results
The standard cytogenetic analysis showed the presence of r(18) in proband’s karyotype (46, XY, r(18)) (Fig. 1, Additional file 1) with the ring chromosome being revealed in all analyzed metaphase plates. The deletion of both 18p and 18q terminal regions of r(18) was unambiguously confirmed by FISH with probes marking subtelomeric regions of chromosome 18 (Fig. 2c, d). That is, hybridization signals were specifically detected on a normal chromosome 18 homolog but were not revealed on r(18).

Standard cytogenetic analysis of the patient. GTG-banded karyotype showing the presence of ring chromosome 18 (karyotype 46, XY, 18(r)). Red arrow points to the ring chromosome (mar: marker)

High-resolution molecular cytogenetic analysis of the patient. a, b Comparative genomic hybridization using Agilent 60 K microarray. a The chromosome 18 view; oligonucleotides with log2 ratio ~ −1 (red dots, red rectangles) indicate deleted regions; oligonucleotides with log2 ratio ~ +0.6 (blue dots, blue rectangle) indicate a duplicated region. The boundaries of terminal deletions, a duplication and an interstitial deletion (18q12.1) were defined with high resolution. b The enlarged 18q12.1 region with imported DGV and OMIM databases tracks and genes annotations. The data illustrate the presence of DTNA deletion. c, d Fluorescent in situ hybridization (FISH) on metaphases from cultured peripheral blood lymphocytes. FISH with probes to centromeric (aqua) and subtelomic (yellow) regions of chromosome 18 (ToTelVysion Probe Kit, Abbott/Vysis; the probes to subtelomeric regions of chromosomes 11 and 12 (p – green, q – red) are added in the hybridization mix by the manufacturer). Inserts show the enlarged view of the normal and the ring chromosome 18. The analysis confirmed the deletion of both terminal regions on r(18)
Array-CGH analysis showed that 18p terminal deletion corresponded to the 18p11.32 cytoband (Fig. 2a) and spanned 305 kb (chr18: 132096–437282 bp, hg18 build). The 18q terminal deletion was assigned to the 18q22.3–q23 chromosomal region (Fig. 2a) with length about 7.5 Mb (chr18: 68533620–76083117 bp, hg18 build) that involves many annotated genes including OMIM morbid genes TSHZ1 and CTDP1. Additionally, a duplication adjoining the breakpoint of the 18p deletion and spanning approximately 2 Mb (chr18: 467510–2480365 bp, hg18 build) was detected. Apart from the terminal imbalances, an interstitial heterozygous deletion of 442 kb in size (chr18: 30327060–30769230 bp, hg18 build) was found on chromosome 18 (Fig. 2a, b). The deletion involved only the DTNA gene (http://www.ncbi.nlm.nih.gov/gene, ID: 1837; OMIM 601239) and encompassed the whole gene sequence. The deletion was verified by qPCR (Additional file 2). As parental biomaterial was not available, we could not determine the origin of the deletion.
Discussion
Here we report on one more case of ring chromosome 18. Whole-genomic microarray-CGH analysis allowed to refine the r(18) deletion breakpoints with high resolution and thus to define the content of hemizygous genes. The 18p deletion involved the very terminal chromosomal region and affected the entire USP14 and THOC1 genes and the most part of COLEC12 gene. These genes are not assigned to morbid ones, and, according to currently available data, they do not belong to critical regions and have not been discussed as putative causative genes for r(18) severe phenotypic features.
An additional relatively large duplication at the site of the 18p deletion breakpoint was revealed, which agrees with the available data on high-resolution molecular-cytogenetic analysis of patients with ring 18. That is, about 20 % of ring chromosome cases, including r(18), have been shown to be accompanied by duplication of the regions bordering the terminal deletion breakpoints [3, 9]. Together with hemizygosity extent, ring chromosome instability and somatic mosaicism, such duplications are regarded as a cause of r(18) phenotypic heterogeneity. In the case presented here, the duplication spanned about 2 Mb and involved eight protein-coding genes, including OMIM genes COLEC12, CETN1, TYMS, ENOSF1, YES1 and ADCYAP1. In DECIPHER database, several CNV (copy number variant) gains similar in position, length and gene content have been described (Decipher IDs 289491, 252149, 253424) with their pathogenicity and clinical significance being largely uncertain. The phenotypes annotated for these cases have included ataxia, oculomotor apraxia, and proportionate short stature. The association between increased dosage of the above-mentioned genes and congenital craniofacial and/or cardiac defects has not been described. A patient exhibiting ADCYAP1 (or, PACAP) overexpression, as a consequence of a partial trisomy 18p, has suffered from severe mental retardation [10]. This is a reasonable link, since PACAP is known to encode a neuropeptide that plays a role in regulation of neuronal development, differentiation and survival [11–13]. Taking all of this into account, the revealed 18p duplication does not seem to be responsible for the severe congenital defects (facial dysmorphia and the heart defect) presented by the patient but could contribute to the individual phenotype, in particular, to mental delay.
The 18q terminal deleted region was quite extended and encompassed 27 RefSeq protein coding genes including several OMIM morbid genes known to be associated with a number of particular congenital dysmorphia and intellectual disability, which correlates with the patient’s clinical manifestations. That is, heterozygous disruption of TSHZ1 (teashirt zinc finger homeobox 1) gene was found to be responsible for congenital aural atresia phenotype in mice [14] and humans [15, 16]. Additionally, deletion of this gene has been implicated in congenital foot malformations/deformations [15, 16] as well as in cleft lip and palate formation [14, 16]. Finally, together with ZADH2 gene, TSHZ1 belongs to a critical region associated with mood disorder in people with 18q distal deletion [17]. Mutations in CTDP1 gene were identified in Roma/Gypsy population as a cause of Congenital Cataracts Facial Dysmorphism Neuropathy (CCFDN) syndrome (OMIM 604168) characterized by anomalies of the eye, impaired physical growth, mild facial dysmorphism and a hypo/demyelinating, symmetric, distal peripheral neuropathy [18]. The loss of SALL3, another gene located in the 18q-deleted region, has been shown to underlie craniofacial development in mice [19]. Some other genes from the region are also known to be involved in regulation of crucial biological functions, but their implications in r(18) clinical features still need to be clarified.
On the whole, we conclude that the patient’s phenotypic picture distinctive for ring chromosome 18 cases is largely determined by haploinsufficiency as a consequence of hemizygosity of the 18q22.3–q23 region. However, it is worth noting that the patient also presented a severe congenital heart defect, namely, subaortic stenosis. At present, there is a lack of data on genotype-phenotype correlations for heart malformations in ring 18 individuals. Recently, van Trier and coauthors reported on two patients with 18q terminal deletion and complex cardiac abnormalities and compared the data with earlier published del(18q) cases [20]. As a result, the authors have suggested the most distal part of 18q as a critical overlapping region for heart malformation.
According to the published data, the cardiac anomalies that are predominantly described for r(18) and 18q deletion syndrome include pulmonary stenosis and atrial septal defects [3, 16, 20, 21]. Congenital subaortic stenosis is a heart malformation not typical for ring chromosome 18 condition. The only example of association between hypertrophic subaortic stenosis and a ring chromosome from E group, presumably r(18), was described by Wald and coauthors [22]. In that study, karyotyping was carried out on the basis of chromosome morphology and tritiated tymedin incorporation pattern, and unambiguous identification of the chromosome formed a ring was not possible. Besides, the boundaries of ring chromosome terminal deletions, as well as any additional subtle chromosomal rearrangements contributing to the clinical picture could not be examined without comprehensive cytogenetic and molecular-cytogenetic analysis.
We showed that the patient was a carrier of an interstitial deletion corresponded to whole DTNA gene. DTNA encodes α-dystrobrevin, a member of dystrophin-associated glycoprotein (DAG) complex, which is thought to provide the integrity and maintenance of sarcolemma and to be involved in muscle contraction and relaxation signaling [23–25]. The previous studies gave evidence that α-dystrobrevin includes four main structural-functional domains and directly interacts with some members of DAG complex, in particular, with dystrophin, syntrophin and sarcoglycan complex [23, 24, 26]. The current notion of the DAG complex network in muscle and its schematic illustration was provided by Nakamori and Takahashi [26]. It is now well known that abnormalities in different members of this complex cause various muscular dystrophies and cardiomyopathies. Specifically, mutations in the dystrophin gene are responsible for Duchenne (OMIM #310200) and Backer (OMIM #300376) muscular dystrophies as well as for X-linked dilated cardiomyopathy (OMIM #302045). Sequence variations in other DAG members including α-, β-, γ-, and δ- sarcoglycans, integrin α7 and laminin α2 are also found to underlie muscular dystrophies and cardiomyopathies [27–32].
Much less is known about α-dystrobrevin. According to several studies, α-dystrobrevin deficiency is also associated with congenital muscular dystrophy [26, 33–35]. At present, the data on α-dystrobrevin functioning in heart and its role in pathogenesis of cardiovascular diseases are especially limited. Mice deficient in α-dystrobrevin exhibited both skeletal and cardiac myopathies [36]. Besides, the hearts of α-DB null (adbn−/−) mice were shown to be highly susceptible to injury during cardiac stress [25]. In human, it has been shown that DTNA missense mutations were associated with left ventricular noncompaction 1 cases (OMIM #604169) - both isolated cases and/or conditions with associated congenital heart defects including coronary artery anomalies, conotruncal anomalies, ventricular and atrial septal defects, pulmonic stenosis, anomalous venous pulmonary veins, hypoplastic left heart syndrome, Ebstein’s anomaly [37–39].
With regard to DTNA, it proves to be especially important to focus on genotype/phenotype correlations not only in cases of single nucleotide changes but whole-gene deletion as well. There are five known α-dystrobrevin isoforms resulting from alternative splicing [23, 40, 41], therefore in case of complete gene deletion the depleted protein function can not be compensated by remaining isoforms. By now, the condition of complete α-dystrobrevin gene deletion has been modeled in mutant mice [42]. To our knowledge, just a few cases of 18q deletion involving DTNA have been described in human. In particular, the data on two deletions from PubMed literature [43, 44] and five deletions from the DECIPHER database [45] are available (Decipher IDs 250878, 260121, 276030, 286198, 288657). Along with severe intellectual disability and motor delay, in some of these cases the heart and great vessels’ malformation as well as hypotonia were observed. At the same time, these deletions spanned as much as 3.2–14.5 Mb in size, encompassed several morbid genes and, in some cases, were associated with additional chromosomal micro-rearrangements. For this reason, the interpretation of genotype-phenotype correlation as well as the assessment of DTNA deletion impact on the phenotypes is ambiguous. Here, for the first time we presented a case of the 18q12.1 deletion restricted only to DTNA gene in a patient with subaortic stenosis that allows to suggest that DTNA belongs to dosage sensitive genes and to extend our knowledge of the role of DTNA haploinsufficiency in congenital heart malformation.
Conclusions
In conclusion, we report on a patient with ring chromosome 18 phenotype combined with a congenital subaortic stenosis. The results of array-CGH analysis imply that a combination of several genetic aberrations such as terminal imbalances of r(18) and 18q12.1 interstitial microdeletion might contribute to the complex phenotype. Our study draws attention to DTNA gene whose deficiency was possibly implicated in the described congenital heart malformation, but further careful functional studies are needed. Here we support the utility of array-CGH analysis for accurate individual diagnosis, disease prognosis and genetic counseling of patients with multiple congenital defects and non-typical clinical picture.
Methods
Routine analysis of G-banded metaphase chromosomes at a resolution of 400 bands was performed using phytohaemagglutinin (PHA)-stimulated peripheral blood lymphocytes, with fifteen metaphase plates being analyzed. Fluorescent in situ hybridization (FISH) with molecular probes specific to centromeric and subtelomeric regions of chromosome 18 (ToTelVysion Probe Kit, Abbott/Vysis) was carried out on metaphase spreads of the same lymphocyte suspension.
Whole-genome DNA analysis was performed using oligonucleotide array-based CGH. As a platform, Agilent 60 K array with median probe spacing 41 kb was used (SurePrint G3 Human CGH Microarray, Agilent Technologies, Santa Clara, CA, USA). The study was performed according to Helsinki Declaration and study approval was obtained from Institutional Ethical Review Board at the Almazov Federal Medical Research Centre in St. Petersburg. Written informed consent was obtained from the patient and his parents prior to investigation. Genomic DNA was extracted from peripheral blood cells using a Puregene DNA Extraction Kit (Gentra, Qiagen, USA). The sample preparation and the hybridization procedure were carried out according to manufacturer’s recommendations. The data obtained was processed and analyzed using CytoGenomics Software (v3.0.1.1, Agilent Technologies) with imported tracks of publically available databases of normal and pathogenic human genome variants, such as Database of Genomic Variants (DGV) [46] and Online Mendelian Inheritance in Man database (OMIM) [47]. Additionally, the revealed copy number variants were compared with the data from DECIPHER [45] and pubmed [48] databases. CNVs (gains and losses) were called using an aberration detection statistical algorithm ADM-2, with a sensitivity threshold of 6.0. All genomic coordinates refer to the human reference assembly NCBI36 (hg18).
The loss of 18q12.1 region was confirmed by real-time quantitative PCR (qPCR) using SYBR Green Master Mix according to manufacturer’s recommendations (http://www.evrogen.com/). The assay was performed for DTNA gene sequence (exon 6; NG_009201.1) with the following primers designed using NCBI Primer Blast tool [49]: F 5′-TTGCGGGAAAATGCTCTGAAC-3′; R 5′- TAAGGAGGAGGCTGATGGACT-3′. The quantity assessment of the target sequence was carried out relative to a normal control DNA. The relative copy number was evaluated using the comparative ΔΔCt method with the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) being used for normalization.
The structural variants were submitted to the DGV archive [50] with accession number estd226.
Ethics, consent and permissions
Written informed consent was obtained from the patient and his parent prior to investigation for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- aCGH:
-
array-based comparative genomic hybridization
- CNV:
-
copy number variant
- DAG:
-
dystrophin-associated glycoprotein complex
- DECIPHER:
-
database of chromosomal imbalance and phenotype in humans using ensembl resources
- DGV:
-
database of genomic variants
- FISH:
-
fluorescent in situ hybridization
- OMIM:
-
online mendelian inheritance in man database
- PHA:
-
phytohaemagglutinin
References
Chen CP, Kuo YT, Lin SP, Su YN, Chen YJ, Hsueh RY, et al. Mosaic ring chromosome 18, ring chromosome 18 duplication/deletion and disomy 18: perinatal findings and molecular cytogenetic characterization by fluorescence in situ hybridization and array comparative genomic hybridization. Taiwan J Obstet Gynecol. 2010;49(3):327–32.
Spreiz A, Guilherme RS, Castellan C, Green A, Rittinger O, Wellek B, et al. Single-nucleotide polymorphism array-based characterization of ring chromosome 18. J Pediatr. 2013;163(4):1174–8.
Carter E, Heard P, Hasi M, Soileau B, Sebold C, Hale DE, et al. Ring 18 molecular assessment and clinical consequences. Am J Med Genet A. 2015;167A(1):54–63.
de Grouchy J. Chromosome 18: A topologic approach. J Pediatr. 1965;66:414–31.
Schinzel A. Catalogue of unbalanced chromosome aberrations in man. 2nd ed. Berlin, New York: Walter de Gruyter; 2001.
Stankiewicz P, Brozek I, Hélias-Rodzewicz Z, Wierzba J, Pilch J, Bocian E, et al. Clinical and molecular-cytogenetic studies in seven patients with ring chromosome 18. Am J Med Genet. 2001;101(3):226–39.
Sodre´ CP, Guilherme RS, Meloni VFA, Brunoni D, Juliano Y, Andrade JAD, et al. Ring chromosome instability evaluation in six patients with autosomal rings. Genet Mol Res. 2010;9:134–43.
Guilherme RS, Meloni VFA, Kim CA, Pellegrino R, Takeno SS, Spinner NB, et al. Mechanism of ring chromosome formation, ring instability and clinical consequences. BMC Med Genet. 2011;12:171.
Rossi E, Riegel M, Messa J, Gimelli S, Maraschio P, Ciccone R, et al. Duplications in additional to terminal deletions are present in a proportion of ring chromosomes: Clues to the mechanisms of formation. J Med Genet. 2008;45:147–54.
Freson K, Hashimoto H, Thys C, Wittevrongel C, Danloy S, Morita Y, et al. The pituitary adenylate cyclase-activating polypeptide is a physiological inhibitor of platelet activation. J Clin Invest. 2004;113(6):905–12.
Gonzalez BJ, Basille M, Vaudry D, Fournier A, Vaudry H. Pituitary adenylate cyclase-activating polypeptide promotes cell survival and neurite outgrowth in rat cerebellar neuroblasts. Neuroscience. 1997;78(2):419–30.
Lu N, DiCicco-Bloom E. Pituitary adenylate cyclase-activating polypeptide is an autocrine inhibitor of mitosis in cultured cortical precursor cells. Proc Natl Acad Sci U S A. 1997;94(7):3357–62.
Fahrenkrug J. PACAP: a multifacetted neuropeptide. Chronobiol Int. 2006;23(1–2):53–61.
Coré N, Caubit X, Metchat A, Boned A, Djabali M, Fasano L. Tshz1 is required for axial skeleton, soft palate and middle ear development in mice. Dev Biol. 2007;308(2):407–20.
Feenstra I, Vissers LELM, Pennings RJE, Nillessen W, Pfundt R, Kunst HP, et al. Disruption of teashirt zinc finger homeobox 1 is associated with congenital aural atresia in humans. Am J Hum Genet. 2011;89(6):813–9.
Cody JD, Sebold C, Heard P, Carter E, Soileau B, Hasi-Zogaj M, et al. Consequences of chromosome 18q deletions. Am J Med Genet C Semin Med Genet. 2015;169(3):265–80.
Daviss WB, O’Donnell L, Soileau BT, Heard P, Carter E, Pliszka S, et al. Mood disorders in individuals with distal 18q deletions. Am J Med Genet Part B. 2013;162B(8):879–88.
Kalaydjieva L. Congenital cataracts-facial dysmorphism-neuropathy. Orphanet J Rare Dis. 2006;1:32.
Parrish M, Ott T, Lance-Jones C, Schuetz G, Schwaeger-Nickolenko A, Monaghan AP. Loss of the Sall3 gene leads to palate deficiency, abnormalities in cranial nerves, and perinatal lethality. Mol Cell Biol. 2004;24(16):7102–12.
van Trier DC, Feenstra I, Bot P, de Leeuw N, Draaisma JM. Cardiac anomalies in individuals with the 18q deletion syndrome; report of a child with Ebstein anomaly and review of the literature. Eur J Med Genet. 2013;56(8):426–31.
Cody JD, Ghidoni PD, DuPont BR, Hale DE, Hilsenbeck SG, Stratton RF, et al. Congenital anomalies and anthropometry of 42 individuals with deletions of chromosome 18q. Am J Med Genet. 1999;85(5):455–62.
Wald S, Engel E, Nance WE, Devies J, Puyau FA, Sinclair-Smith BC. E ring chromosome with persistent left superior vena cava and hypertrophic subaortic stenosis. J Med Genet. 1969;6(3):328–33.
Sadoulet-Puccio HM, Rajala M, Kunkel LM. Dystrobrevin and dystrophin: an interaction through coiled-coil motifs. Proc Natl Acad Sci U S A. 1997;94(23):12413–8.
Yoshida M, Hama H, Ishikawa-Sakurai M, Imamura M, Mizuno J, Araishi K, et al. Biochemical evidence for association of dystrobrevin with the sarcoglycan-sarcospan complex as a basis for understanding sarcoglycanopathy. Hum Mol Genet. 2000;9(7):1033–40.
Strakova J, Dean JD, Sharpe KM, Meyers TA, Odom GL, Townsend D. Dystrobrevin increases dystrophin’s binding to the dystrophin-glycoprotein complex and provides protection during cardiac stress. J Mol Cell Cardiol. 2014;76:106–15.
Nakamori M, Takahashi MP. The role of α-dystrobrevin in striated muscle. Int J Mol Sci. 2011;12(3):1660–71.
Roberds SL, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD, et al. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78(4):625–33.
Bönnemann CG, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, et al. Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat Genet. 1995;11(3):266–73.
Noguchi S, McNally EM, Ben Othmane K, Hagiwara Y, Mizuno Y, Yoshida M, et al. Mutations in the dystrophin-associated protein gamma-sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270(5237):819–22.
Nigro V, de Sa ME, Piluso G, Vainzof M, Belsito A, Politano L, et al. Autosomal recessive limbgirdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta sarcoglycan gene. Nat Genet. 1996;14(2):195–8.
Tomé FM, Evangelista T, Leclerc A, Sunada Y, Manole E, Estournet B, et al. Congenital muscular dystrophy with merosin deficiency. CR Acad Sci III. 1994;317(4):351–7.
Helbling-Leclerc A, Zhang X, Topaloglu H, Cruaud C, Tesson F, Weissenbach J, et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. 1995;11(2):216–8.
Metzinger L, Blake DJ, Squier MV, Anderson LV, Deconinck AE, Nawrotzki R, et al. Dystrobrevin deficiency at the sarcolemma of patients with muscular dystrophy. Hum Mol Genet. 1997;6(7):1185–91.
Jones KJ, Compton AG, Yang N, Mills MA, Peters MF, Mowat D, et al. Deficiency of the syntrophins and alpha-dystrobrevin in patients with inherited myopathy. Neuromuscul Disord. 2003;13(6):456–67.
Nakamori M, Kimura T, Kubota T, Matsumura T, Sumi H, Fujimura H, et al. Aberrantly spliced alpha-dystrobrevin alters alpha-syntrophin binding in myotonic dystrophy type 1. Neurology. 2008;70(9):677–85.
Grady RM, Grange RW, Lau KS, Maimone MM, Nichol MC, Stull JT, et al. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol. 1999;1(4):215–20.
Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium: a study of eight cases. Circulation. 1990;82:507–13.
Burke A, Mont E, Kutys R, Virmani R. Left ventricular noncompaction: A pathological study of 14 cases. Hum Pathol. 2005;36:403–11.
Ichida F. Left ventricular noncompaction. Circ J. 2009;73(1):19–26.
Nawrotzki R, Loh NY, Ruegg MA, Davies KE, Blake DJ. Characterisation of alpha-dystrobrevin in muscle. J Cell Sci. 1998;111(Pt 17):2595–605.
Peters MF, Sadoulet-Puccio HM, Grady MR, Kramarcy NR, Kunkel LM, Sanes JR, et al. Differential membrane localization and intermolecular associations of alpha-dystrobrevin isoforms in skeletal muscle. J Cell Biol. 1998;142(5):1269–78.
Wang D, Kelly BB, Albrecht DE, Adams ME, Froehner SC, Feng G. Complete deletion of all alpha-dystrobrevin isoforms does not reveal new neuromuscular junction phenotype. Gene Expr. 2007;14(1):47–57.
Gilling M, Lauritsen MB, Møller M, Henriksen KF, Vicente A, Oliveira G, et al. A 3.2 Mb deletion on 18q12 in a patient with childhood autism and high-grade myopia. Eur J Hum Genet. 2008;16(3):312–9.
Chen CP, Huang MC, Chen YY, Chern SR, Wu PS, Chen YT, et al. Prenatal diagnosis of de novo interstitial deletions involving 5q23.1-q23.3 and 18q12.1-q12.3 by array CGH using uncultured amniocytes in a pregnancy with fetal interrupted aortic arch and atrial septal defect. Gene. 2013;531(2):496–501.
Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources (DECIPHER): https://decipher.sanger.ac.uk/. Accessed 7 Sept 2015.
Database of Genomic Variants (DGV): http://dgv.tcag.ca/dgv/app/. Accessed 24 Aug 2015.
Online Mendelian Inheritance in Man (OMIM) database: http://www.omim.org/. Accessed 14 Jan 2016.
Pubmed databases: http://www.ncbi.nlm.nih.gov/pubmed/. Accessed 19 Jan 2016.
NCBI Primer Blast tool: http://www.ncbi.nlm.nih.gov/tools/primer-blast/. Accessed 3 Aug 2015.
Database of Genomic Variants (DGV) archive: http://www.ebi.ac.uk/dgva/. Accessed 17 Sept 2015.
Acknowledgements
This work was partially financially supported by the Government of Russian Federation, Grant 074-U01 and “Russian scientific foundation”, grant agreement no. 14-15-00745.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None of the authors have any competing interests in the manuscript.
Authors’ contributions
AZ carried out all molecular cytogenetic experiments, participated in the design of the study and drafted the manuscript, TN and NY carried out the cytogenetic studies and provided genetic counseling, OM, TP and EG made the clinical evaluation of the patient and communicated with the family, AK conceived of the study, coordinated the project and made critical revisions of the manuscript. All the authors have read and approved the final manuscript.
Additional files
Additional file 1:
Standard cytogenetic analysis of the patient (additional material). Two GTG-banded metaphase plates showing the presence of ring chromosome 18 (karyotype 46, XY, 18(r)). Red arrows point to a normal chromosome homolog 18 and to a ring chromosome r(18). (PNG 437 kb)
Additional file 2:
Confirmation of DTNA deletion in the patient using quantitative real-time PCR analysis (qPCR). Description of data: qPCR data revealed one copy of the DTNA gene (18q12.1) in a patient DNA sample as compared to two copies of the gene in a normal control DNA sample. The data was normalized against GAPDH gene using the comparative ΔΔCt method. RQ (relative quantity) value is presented along the vertical axis. Each reaction was reproduced (repeated) in triplicate for both DNA samples (patient and control) and both genes (DTNA and GAPDH). The series of four ten-fold dilutions were included into analysis with the starting amount of DNA ~ 1 ng. The results obtained for one of the dilutions are depicted in the figure; for the rest dilutions, the ratio of quantity values between test and control samples was the same. (PNG 8 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Zlotina, A., Nikulina, T., Yany, N. et al. Ring chromosome 18 in combination with 18q12.1 (DTNA) interstitial microdeletion in a patient with multiple congenital defects. Mol Cytogenet 9, 18 (2016). https://doi.org/10.1186/s13039-016-0229-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-016-0229-9