
Abstract
Background
Pallister-Killian syndrome (PKS) is a sporadic genetic disorder caused by the presence of a tissue-specific mosaicism for isochromosome 12p - i(12) (p10) and is characterized by facial dysmorphism including coarse facies, upslanting palpebral fissures, bitemporal alopecia, pigmentary skin anomalies, developmental delay, hypotonia and seizures. Although typical clinical features of PKS commonly exist, clinicians often do not raise the possibility of this diagnosis.
Results
We reviewed the medical records of 10 patients with confirmed PKS followed in our service (since 1990 to 2015). Age at diagnosis varied from prenatal to 3 years and clinical features were consistent with those described in the literature. In all patients, peripheral blood karyotypes were normal and cytogenomic study was performed in order to confirm the diagnosis. Three of these patients had PKS diagnosis confirmed by buccal smear MLPA.
Conclusion
An early conclusion from our results demonstrated that MLPA on buccal smears is a good and non-invasive method to detect extra copies of 12p and should be considered as the first exam, before a skin biopsy for a fibroblast karyotype is performed.
Similar content being viewed by others


Background
Pallister-Killian syndrome (PKS) is a sporadic and rare chromosomal disorder caused by the mosaicism for isochromosome 12p – i(12)(p10). There are just over 200 cases described in the literature [1]. The syndrome is known by several names, such as Pallister mosaic syndrome, Pallister-Killian syndrome, Pallister-Killian-Teschler-Nicola syndrome, tetrasomy 12p and isochromosome 12p [2, 3].
Clinical findings of PKS are distinctive, especially the combination of coarse facies, pigmentary skin anomalies, developmental delay, hypotonia and seizures. The facial appearance includes coarse facies (coarseness becomes more pronounced with age), prominent forehead, hypertelorism, upslanting palpebral fissures with epicanthal folds, small nose with upturned nares, high arched palate, macrostomia, long philtrum, micrognathia, bitemporal alopecia and an extension of the philtral skin into the vermilion border of the upper lip, which is termed “Pallister lip”. Other associated findings include the following: congenital heart defects, diaphragmatic hernia, cryptorchidism, renal malformations, imperforate or anteriorly placed anus [2–4]. Patients have hypotonia, mental and motor retardation from early infancy, which can vary from mild to severe and profound [2, 5–7].
PKS is caused by a tissue-limited mosaicism for supernumerary i(12)(p10) [4, 8]. Although different levels of mosaicism and genetic variation in isochromosome composition have already been described in the literature, there is currently no evidence for genotype-phenotype correlation [3].
A fibroblast karyotype from skin biopsy is commonly used to achieve PKS diagnosis, but other tissues such as lung, tissue from a buccal smear, bone marrow and even amniotic fluid yield higher percentages of mosaicism i(12)(p10), which can help diagnose PKS [9–11].
Results
All ten patients had PKS confirmed by a fibroblast or amniotic fluid karyotype or by buccal smear MLPA. The presence of i(12)(p10) in the fibroblast karyotype ranged from 50 % to 100 % and in amniotic fluid from 75 % to 100 % (Table 1).
Peripheral blood karyotypes were normal in nine patients. In one who died at 5 days of age it was not performed but diagnosis was confirmed by amniotic fluid karyotype.
Three patients, with clinical features of PKS, had buccal smear MLPA confirming the diagnosis. In two patients fibroblasts culture failed and MLPA was performed in buccal smear sample showing four copies of the short arm of chromosome 12 (Fig. 1). In another patient, who had fibroblast karyotype, it was also performed MLPA of buccal smear and fibroblast in order to confirm that MLPA was able to diagnosis PKS.

a and b: Histogram showing MLPA analysis of DNA from buccal smear; c: FISH in interphase cells showing 12p specific subtelomeric probe (Aquarius®, Cytocell Cambridge, UK ) hybridization; d: Fibroblast skin karyotype showing isochromosome 12p in G band
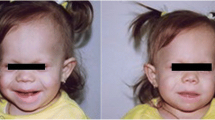
Clinical aspects are shown in Table 2. All patients presented clinical features associated with PKS (Fig. 2). Two of these patients were previously described [12]. Age at diagnosis varied from the prenatal period to 1y10mo.

Patient # 5: dysmorphic features described in PKS: frontotemporal alopecia, hypertelorism, anteverted nostrils, long philtrum with “Pallister lip”, hypopigmented streaks. a: frontal view; b: lateral view
Two patients died: one at 5 days due to respiratory problems and other at 14 years old due to pneumonia. Two patients have lost follow-up.
Current age of six patients ranges from 1y9mo to 19 years.
Discussion
Although typical clinical features of PKS commonly exist, clinicians often do not raise the possibility of this diagnosis. Another factor that makes this diagnosis difficult is the need for cytogenetic analysis of tissues other than peripheral blood.
Even though there is no pathognomonic feature of PKS, there are some typical findings such as coarse facies, prominent forehead, bitemporal alopecia, upslanting palpebral fissures, cleft palate, sparse eyebrows, pigmentary skin anomalies including hyperpigmented and hypopigmented streaks, developmental delay, hypotonia and seizures. It is important to note that some of these findings might not be evident at birth; the coarsening of the face occurs as the child ages [2].
All patients described in our study exhibited typical clinical features of PKS, and parental age at conception was in accordance with that reported in the literature [3].
Cardiac malformations are common in PKS patients described in the literature; the most common malformations are patent foramen ovale, atrial septal defects and patent ductus arteriosus [3, 13]. Two of our patients had aneurysm of the interatrial septum, which had not been previously described in PKS.
Skin anomalies, such as hyperpigmented and hypopigmented streaks, were observed in 6/10 patients. Despite being a frequent finding in PKS patients, such skin anomalies are considered suggestive of PKS and not a mandatory feature.
With regard to the ophthalmological findings, one patient had a diffuse abnormality of retina pigmentation, which has been previously described by Graham et al. This may suggest a retinal pigment mosaicism similar to that described in skin [14].
Gastrointestinal malformations were found less frequently in our cohort than described in the literature, although the malformations were similar to those previously described: displacement of the anus and umbilical hernia. Diaphragmatic hernias, present in 11–29 % of reported cases, were not observed in our patients [3].
Hearing and ophthalmologic impairment were observed less frequently in our population compared to reports in the literature. Only one patient had bilateral hip dislocation.
Compared to the literature, seizures were observed less frequently in our cohort. Because the onset of seizures in PKS patients might occur between the ages of 2 to 5 years, some of our patients might develop seizures later in life. There is no typical seizure pattern in PKS patients [15–17].
Similar to our findings, karyotypes of peripheral blood are usually normal. Wenger et al. showed that only 3 % of cells from peripheral blood exhibited i(12)(p10), whereas analysis of skin and lung tissues that were obtained postmortem exhibited i(12)(p10) in 98.5 % and 97.5 % of the cells, respectively [9]. Liehr et al. described a variety of PKS’s karyotypes: complete trisomy 12 in mosaicism: 47,XX,+12[20 %]/47,XX,+i(12)(p10)[80 %], hexasomy 12p: 48,XX,+i(12)(p10),+i(12)(p10)[16 %], even when PKS phenotype is typical [18]. More studies are necessary to stablish if there is a clinical correlation with the cytogenetics findings.
We used MLPA method for diagnosis with DNA prepared from buccal smear samples in three patients, with the detection of four copies of the short arm of chromosome 12, confirming the diagnosis of PKS. There are few reports of PKS diagnosis using MLPA and DNA from buccal smears in the literature. We recommend this analysis as the first exam, before a skin biopsy.
MLPA assay is a recently developed technique that is able to detect variations in the genes copy number and dosage. This test is a high throughput analysis, allowing up to 96 samples to be handled simultaneously allowing the study of several regions of the human genome in a single reaction [19, 20].
Due to this capability, MLPA can be applied in the molecular diagnosis of several syndromes since the duplication of entire genomic regions causes a disease due to the presence of extra copies of the genes, while complete or partial deletion can produce a completely different phenotypic effect [19].
In fact, MLPA assay is one of the most widely used techniques for the molecular investigation of microdeletion/microduplication syndromes. The use of this technique has important advantages such as relative simplicity of approach, low cost, rapid turnaround, ease of multiplexing to permit high confidence in the results, high accuracy of copy number estimation, and the potential for combination of copy number analysis with other applications, such as methylation detection or SNP genotyping [19, 20].
MLPA use target sequences, between 50–80 nucleotides, that applied to DNA extracted from a buccal swab allow identifying genomic aberrations, easier than detected by FISH. Over 300 kits are so far commercially available, dedicated to the study of several diseases [19].
Finally compared to array CGH (Comparative Genomic Hybridization) and FISH (Fluorescent In situ Hybridization) whose have been previously described as alternatives to diagnose PKS, MLPA technique is less expensive and technically uncomplicated method [19].
MLPA on buccal smears is also non-invasive and yields a shorter turnaround time for results compared to a fibroblast karyotype. A limitation of MLPA is that mosaicism in low level may not be detected, so a normal MLPA result cannot rule out PKS diagnosis [20].
Conclusion
A precocious PKS diagnosis is important to optimally manage the disease and to provide genetic counseling.
MLPA on buccal smears is a good and non-invasive method to detect extra copies of 12p and should be considered as the first exam, before a skin biopsy for a fibroblast karyotype is performed.
Thus we highlight the importance of associate cytogenomic approaches in clinical genetics, to provide additional information relevant to confirm the diagnosis, for patient management and genetic counseling.
Methods and patients
This is a descriptive study based on the medical records of ten patients with a confirmed diagnosis of PKS who were followed in the Genetics Unit of the Instituto da Criança (Brazil) from 1990 to 2014. The study was approved by the by the ethics committee of the HCFMUSP (University of São Paulo-CAPPesq) and informed consent form was obtained from all families.
Cytogenomic analysis
Peripheral blood karyotype was performed using a standard phytohemagglutinin-stimulated lymphocyte culture method followed by G-banding. Twenty metaphase cells were analyzed for all patients except one, who died at 5 days of age. Also, we performed a skin biopsy for fibroblast karyotype and the long-term closed flask fibroblast explant cultures were set up according to the protocol adapted from Rooney [21]. Additional Fluorescence in situ hybridization (FISH) with chromosome 12p specific subtelomeric probe (Aquarius®, Cytocell Cambridge, UK) according to the technique of Pinkel [22], with minor modifications was carried out on interphase nuclei of subcultured fibroblasts.
In three of the patients buccal smear was collected with the Oragene Kit (Ottawa, Canada) and DNA was extracted using the QIAamp DNA Blood Midi Kit 250 (QIAGEN, Valencia, California) in order to perform Multiplex Ligation-dependent Probe Amplification (MLPA-MRC Holland, Amsterdam, The Netherlands) assay.
MLPA with kits SALSA P070 and SALSA P036 that contain subtelomeric probes, RBBP2 and SLC6A12 respectively, determine the DNA copy number variations in 12p region and can provide a definitive diagnosis.
Consent
Written informed consent was obtained from the parents of the patients.
References
Liehr T. Small supernumerary marker chromosomes. 2015. http://ssmc-tl.com/sSMC.html [accessed 03/16/2015].
Schinzel A. Tetrasomy 12p (Pallister-Killian syndrome). J Med Genet. 1991;28:122–5.
Wilkens A, Liu H, Park K, Campbell LB, Jackson M, Kostanecka A, et al. Novel clinical manifestations in Pallister Killian syndrome: comprehensive evaluation of 59 affected individuals and review of previously reported cases. Am J Med Genet Part A. 2012;158A:3002–17.
Kostanecka A, Close LB, Izumi K, Krantz ID, Pipan M. Developmental and behavioral characteristics of individuals with Pallister-Killian syndrome. Am J Med Genet Part A. 2012;158A:3018–25.
Genevieve D, Cormier-Daire V, Sanlaville D, Faivre L, Gosset P, Allart L, et al. Mild phenotype in a 15 year old boy with Pallister-Killian syndrome. Am J Med Genet Part A. 2003;116A:90–3.
Vogel I, Lyngbye T, Nielsen A, Pedersen S, Hertz JM. Pallister-Killian syndrome in a girl with mild developmental delay and mosaicism for hexasomy 12p. Am J Med Genet Part A. 2009;149A:510–4.
Stalker HJ, Gray BA, Bent-Williams A, Zori RT. High cognitive functioning and behavioral phenotype in Pallister-Killian syndrome. Am J Med Genet Part A. 2006;140A:1950–4.
Shen JD, Liang DS, Zhou ZM, Xia Y, Long ZG, Wu LQ. Pallister-Killian syndrome: meiosis II non-disjunction may be the first step in the formation of isochromosome 12p. Chin Med J (Engl). 2010;123(23):3482–5.
Wenger SL, Boone LY, Steele MW. Mosaicism in Pallister i(12p) syndrome. Am J Med Genet. 1990;35:523–5.
Leube B, Majewski F, Gebauer J, Royer-Pokora B. Clinical, cytogenetic, and molecular observations in a patient with Pallister-Killian-syndrome with an unusual karyotype. Am J Med Genet A. 2003;123A:296–300.
Cormier-Daire V, Le Merrer M, Gigarel N, Morichon N, Prieur M, Lyonnet S, et al. Prezygotic origin of the isochromosome 12p in Pallister-Killian syndrome. Am J Med Genet. 1997;69:166–8.
Sugayama SM, Bertola DR, Albano LMJ, Manissadjian A, Bittencourt EA, Borovik C, et al. Síndrome de Pallister Killian ou tetrassomia do braço curto do cromossomo 12 em mosaico: relato de dois casos diagnosticados pela hibridização in situ por fluorescência (FISH). Pediatria (São Paulo). 2000;22:255–63.
Tilton RK, Wilkens A, Krantz ID, Izumi K. Cardiac manifestations of Pallister-Killian syndrome. Am J Med Genet Part A. 2014;164A:1130–5.
Graham W, Brown SM, Shah F, Tonk VS, Kukolich MK. Retinal pigment mosaicism in Pallister-Killian syndrome (mosaic tetrasomy 12p). Arch Ophthalmol. 1999;117:1648–9.
Candee MS, Carey JC, Krantz ID, Filloux FM. Seizure characteristics in Pallister-Killian syndrome. Am J Med Genet Part A. 2012;158A:3026–32.
Filloux FM, Carey JC, Krantz ID, Ekstrand JJ, Candee MS. Occurrence and clinical features of epileptic and non-epileptic paroxysmal events in five children with Pallister-Killian syndrome. Eur J Med Genet. 2012;55:367–73.
Giordano L, Viri M, Borgatti R, Lodi M, Accorsi P, Faravelli F, et al. Seizures and EEG patterns in Pallister-Killian syndrome: 13 new Italian patients. Eur J Paediatr Neurol. 2012;16:636–41.
Liehr T, Wegner RD, Stumm M, Joksie G, Polityko A, Kosyakova N, et al. Pallister-Killian syndrome: rare phenotypic features and variable karyotypes. Balk J Med Gen. 2008;11:65–7.
Dutra RL, Honjo RS, Kulikowski LD, Fonseca FM, Pieri PC, Jehee FS, et al. Copy number variation in Williams-Beuren syndrome: suitable diagnostic strategy for developing countries. BMC Res Notes. 2012;5:13.
Van Opstal D, Boter M, Noomen P, Srebniak M, Hamers G, Galjaard RJ. Multiplex ligation dependent probe amplification (MLPA) for rapid distinction between unique sequence positive and negative marker chromosomes in prenatal diagnosis. Mol Cytogenet. 2011;4:2.
Rooney D. Human cytogenetics constitutional analysis. 3rd ed. Oxford, UK: Oxford University Press; 2001.
Pinkel D, Straume T, Gray JW. Cytogenetic analysis using quantitative, high-sensitivity, fluorescence hybridization. Proc Natl Acad Sci U S A. 1986;83:2934–8.
Acknowledgments
We thank all patients evaluated in this paper and FAPESP grants numbers (2009/53105-9) and (2011/16664-0) and CNPq (302618/2013) for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LSAC was responsible for clinical evaluation, records review, literature review, writing and review of the manuscript; ACZT participated in laboratory work and writing the manuscript; MMM, RLD did laboratory work; ATD did laboratory work and reviewed the manuscript; LDK was responsible for laboratory and manuscript supervision; RSH, DRB and CAK participated in clinical evaluation, review and supervision of the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Costa, L.S.d.A., Zandona-Teixeira, A.C., Montenegro, M.M. et al. Cytogenomic delineation and clinical follow-up of 10 Brazilian patients with Pallister-Killian syndrome. Mol Cytogenet 8, 43 (2015). https://doi.org/10.1186/s13039-015-0142-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13039-015-0142-7