
Abstract
Background
Hormone-dependent promoters are very efficient in transgene expression. Plasmid-based reporter assays have identified regulatory sequences of the Ovalbumin promoter that are involved in response to estrogen and have shown that the deletion of the steroid-dependent regulatory element (SDRE) and negative regulatory element (NRE) leads to a steroid-independent expression of a reporter. However, the functional roles of these regulatory elements within the native genomic context of the Ovalbumin promoter have not been evaluated.
Results
In this study, we show that the negative effects of the NRE element on the Ovalbumin gene can be counteracted by CRISPR interference. We also show that the CRISPR-mediated deletion of SDRE and NRE promoter elements in a non-oviduct cell can lead to the significant expression of the Ovalbumin gene. In addition, the targeted knock-in of a transgene reporter in the Ovalbumin coding region and its expression confirms that the truncated promoter of the Ovalbumin gene can be efficiently used for an estrogen-independent expression of a foreign gene.
Conclusions
The methodology applied in this paper allowed the study of promoter regulatory sequences in their native nuclear organization.
Similar content being viewed by others


Background
Avian expression systems represent desirable platforms for the production of recombinant human proteins. Production in chicken cells offers significant advantages over other systems, including providing human-like glycosylation on target proteins [1]. In the early attempts to produce foreign proteins in avian systems, viral vectors containing a constitutive promoter, such as a cytomegalovirus (CMV) promoter, were utilized to drive expression [2, 3]. However, the utilization of these constitutive/strong promoters had several disadvantages including variations in protein expression levels, improper folding of the protein product, promoter silencing possibilities, and toxicity arising from their expression in a broad range of tissues [4,5,6]. Thus, there has been an increasing trend toward the use of regulated promoters. Among these, native hormone-dependent promoters have been demonstrated to be efficient in transgene expression. One such example is the Ovalbumin (OVA) promoter, which has been used in cultured primary oviduct cells or transgenic chickens for the production of exogenous proteins [7,8,9,10,11,12,13,14,15,16,17].
Seven members of the chicken clade B serpins have orthologues in the human genome. However, the Ovalbumin gene (SerpinB14) and two of its paralogues, the Ovalbumin-related protein Y (SERPINB14B) and the Ovalbumin-related protein X (SERPINB14C) do not have any orthologues in the human genome [18]. The expression of these three genes, in contrast to other serpins, is hormone-dependent [19]. Consistent with this, the regulatory elements of these genes do not exhibit significant similarity to the regulatory regions of other serpins. The sequences of the steroid-dependent regulatory element (SDRE; − 900 to − 732) and negative regulatory element (NRE; − 308 to − 88) are unique to the chicken OVA gene. Several plasmid-based reporter assays have been used to elucidate the role of the regulatory elements within the OVA promoter [20,21,22,23,24,25,26,27,28,29,30,31]. These studies have demonstrated that the presence of a proximal promoter (− 87 to + 9) is sufficient for steroid-independent expression. The deletion of the SDRE and NRE, along with the linker between them, in the chicken OVA promoter resulted in increased activity of the reporter gene [24, 27, 28]. However, it is important to identify additional distant regulatory elements that are associated with the oviduct-specific function of the OVA promoter [10].
To induce the expression of exogenous genes in plasmid constructs, researchers have utilized different 5' and 3' flanking regions of the chicken OVA promoter. Some studies suggest that incorporating the two key regulatory elements, SDRE, and NRE, present within the chicken OVA promoter is sufficient to achieve oviduct-specific expression of a therapeutic protein [8, 16]. These two regulatory elements are critical for the appropriate regulation of the OVA gene expression [24, 25, 32,33,34]. The SDRE is essential for the response to steroid hormones, including estrogen, progesterone, androgen, and glucocorticoids [21]. On the other hand, the NRE acts as a bifunctional element. It collaborates with SDRE to activate OVA gene expression in the presence of steroids in the oviduct tissue while repressing OVA gene transcription in the absence of steroids in both oviduct and non-oviduct cells [24, 27,28,29]. It has been demonstrated that a specific element within the NRE, known as the COUP adjacent repressor (CAR) element (-130 to -100), plays a major role in mediating the repressive activities of the NRE [28, 29]. Another negative element within the NRE is a ubiquitous silencer (-239 to -220), which leads to a reduction in transcriptional activity by approximately three-fold and acts as a genuine transcriptional silencer since it is capable of repressing a heterologous promoter in an orientation-independent manner [27].
In an attempt to improve the expression level of the transgene in a non-native genomic site or a plasmid construct, additional regulatory sequences comprising the OVA exon 1, intron 1, and the beginning of exon 2 were incorporated into the promoter construct [8]. Zhu et al. utilized 7.5 kb and 15 kb of the 5' flanking region, as well as 15.5 kb of the 3' flanking region from the OVA gene to direct ex-situ transgene expression [7]. Despite containing all known oviduct-specific regulatory elements, the ectopic expression of the transgene was detected in non-oviduct tissues of the chimeric chicken when utilizing these regions. [7]. In other studies, the estrogen-responsive enhancer element (ERE, located approximately 3.3 kb upstream of the transcription start site in the genome [35]) was incorporated into the construct containing the OVA promoter [8, 9]. However, the results of the study did not demonstrate any increase in the level of recombinant protein produced in transgenic chickens [8]. Herron et al. reintroduced an additional regulatory sequence between the ERE and SDRE elements in their targeting construct to enhance the expression level of protein in the egg white [15]. The OVA promoter, ranging from 1.35 kb to 3.0 kb in length, which has been used in most of the ex-situ studies so far, contains five main conserved sites that have been identified in chicken and other avian species [36]. These studies were unable to evaluate the functions of these regulatory elements within a genomic context, where additional factors such as trans-acting regulatory elements, the chromosomal structure of the gene locus, and the three-dimensional (3D) nuclear organization [37] are involved.
The experimental work presented here provides the first evaluation of OVA regulatory elements within their genomic context, where the trans-acting regulatory elements can exert their effects, leading to the OVA gene upregulation. As previously shown by Dougherty et al. [20], the repressor activity associated with the CAR site is mediated by the binding of interferon regulatory factors to this site. Based on this knowledge, we reasoned that binding of dCas9 to the negative regulatory regions would hinder the binding of proteins to the CAR and Silencer regions. Consequently, in the absence of these proteins, the negative regulatory sequences would not be able to exert their inhibitory effect on the expression of the OVA gene. Therefore, we hypothesized that the spatial occupancy of the CAR and silencer regions may serve as a physical barrier, preventing the binding proteins to access these sites.. Using the DF1 fibroblast cell line, we first showed that CRISPR interference (CRISPRi) exerted on certain regulatory elements of the promoter results in the upregulation of the OVA expression. Second, by deleting the OVA distal promoter elements including SDRE and NRE via dual sgRNA CRISPR/Cas9-mediated excision, we observed an increased expression of the OVA gene. Finally, we evaluated the activity of a foreign gene within this modified region by integrating a transgene reporter under the control of the engineered promoter via CRISPR HDR (homology-directed repair). Our findings indicate that the targeted modification and engineering of the promoter have led to a significant upregulation of the OVA gene in the absence of estrogen activation. The methodology applied here overcomes the limitation of cloned promoters, where the promoter regulatory sequences have to be taken out of their native spatial nuclear organization into a plasmid for further evaluation.
Results
CRISPR interference of the regulatory sequences in the Ovalbumin promoter
The previous plasmid-based reporter assays have shown that the SDRE and NRE regulatory elements are important for the promoter activity of the OVA gene. Deletion of these two elements, as well as the linker in between, results in an increased reporter gene activity in an estrogen-independent manner [24, 27, 28]. We hypothesized that it is the negative effects of the NRE element in the distal promoter that keep the OVA gene transcriptionally inactive in the absence of estrogen in non-oviduct cells (Fig. 1A). We transfected DF1 fibroblast cells with plasmids encoding dCas9, as well as CAR and silencer sgRNAs, which targeted the CAR, and silencer sequences of the NRE element, respectively (Tables 1 and 2). Three days after transfection, we were able to detect the transcription of the OVA gene (Fig. 1B), while transfection of dCas9 without sgRNA (pdCas9-X) did not result in transcriptional activation. Our RT-qPCR results showed that the expression of OVA in DF1 fibroblast cells subjected to CRISPR interference with two sgRNAs was more than 100-fold and significantly higher (p < 0.05) than that in wild-type DF1 cells (Fig. 1C and S1). These experiments indicated that the negative effects of the NRE element on the OVA gene can be counteracted by CRISPR interference. We reasoned that one possible mechanism for the negative effects of the NRE element on the transcription of the OVA gene could be exerted by regulatory RNAs originating from the distal promoter. However, using PCR or hemi-nested PCR, we were not able to identify any RNA transcripts that might originate from the NRE element (Fig. S2, Table 1).

CRISPRi-mediated activation of the Ovalbumin promoter in DF1 cells. A The schematic representation of the promoter and coding region of the OVA gene in DF1 cells. Two regulatory elements of SDRE and NRE are shown in the distal promoter. The bottom panel shows binding sites for two guide RNAs (Silencer-gRNA and CAR-gRNA) that bind the silencer and CAR regions in the NRE element, respectively. SDRE, steroid-dependent regulatory element; NRE, negative regulatory element; CAR, COUP-adjacent repressor site; COUP, Chicken OVA upstream promoter; TATA, TATA box; TSS, transcription start site; dCas9, Catalytically dead Cas9. The enlarged inset in the lower section of panel A shows the location and orientation of PAM regions and protospacers for the two regulatory regions of ‘silencer’ and ‘CAR’. B The left panel shows agarose gel electrophoresis for analysis of the RT-PCR products which were amplified by primers P8 and P9 (for OVA, Fig. 2A and Table 1). The right panel shows agarose gel electrophoresis for analysis of the RT-PCR products which were amplified by P10 and P11 (for GAPDH, Table 1). RNA was extracted from DF1 cells which were transfected with CRISPRi vectors that target the NRE element at CAR, Silencer, both CAR and silencer sequences, and pdCas9-X as the negative control. The expected amplicon size for OVA was 179 bp, and for GAPDH was 187 bp. WT, wild-type; Magnum, hormonally-activated tissue of magnum from a 35-week-old laying hen; M, DNA size marker; NTC, no template control. C Upregulation of the OVA mRNA in CRISPRi-modified DF1 cells was assessed by RT-qPCR. Upon transfection with CRISPRi vectors that target the NRE element at CAR, Silencer, and both CAR and silencer sequences, an increment in the OVA gene expression level was determined. The transcript levels for OVA in the hormonally-activated tissue of the magnum (from a 35-week-old laying hen) show the highest level of expression. The gene expression ratio for the OVA over GAPDH was calculated by the Pfaffl method of relative quantification [38]. For each group of CRISPRi-DF1 cells, three biological replicates were used. Each biological replicate was read as three technical replicates. The Mann–Whitney assay was used to analyze significant statistical differences between groups. The asterisk (*) indicates statistical differences with p values < 0.05
Deletion of the distal elements in the Ovalbumin gene promoter induces the expression of the Ovalbumin mRNA
An alternative mechanism for the effects of the distal promoter on gene transcription could be mediated by intra-chromosomal contacts (loops) that bring together the distal regulatory segments to the core promoter [37]. Previous studies have shown that the cloned proximal segment of the OVA promoter lacking the major regulatory elements of SDRE and NRE, can significantly increase (up to 17-fold) the chloramphenicol acetyltransferase (CAT) gene activity on a plasmid construct in LMH cells (a chicken hepatoma cell line) and chicken primary oviduct cells, and this increase occurs in an estrogen-independent manner [27, 29]. Thus, we asked whether the deletion of the SDRE and NRE elements from the native promoter would be able to increase the transcription of the OVA gene in a non-oviduct cell. To this end, we used the CRISPR excision strategy to delete the SDRE and NRE elements from the OVA promoter in DF1 fibroblast cells (Fig. 2A). To confirm this deletion, these cells (DF1 +/OVA Pro ∆) were subjected to genomic PCR and Sanger sequencing (Figs. 2B and C). Then, individual cells were grown in three 96 well plates to acquire correctly edited isogenic clones for subsequent expansion and validation of gene expression.

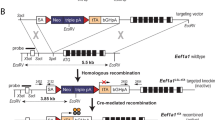
Design and validation of the targeted deletion of Ovalbumin distal promoter elements in DF1 cells. A The schematic representation of CRISPR/Cas9 mediated deletion strategy of the OVA promoter in DF1 cells. The top diagram shows the wild-type (WT) chicken OVA locus. The two guide RNA (SDRE-gRNA and NRE-gRNA) binding sites are shown. The NRE- and SDRE- gRNAs target two positions downstream of NRE (downstream of CAR) and upstream of SDRE, respectively. The bottom diagram shows the locus after CRISPR-mediated deletion of the distal OVA promoter in DF1 cells (DF1+/OVA Pro ∆ cell). The PCR primers used for the assessment of deletion (P5 to P7), and the OVA gene expression (P8 and P9, used in Figs. 1 and 3) are shown as small red arrows. B Two-step genomic PCR to confirm the deletion of the distal promoter of the OVA gene. In the first PCR (using P5 and P7 primers, Table 1), an amplicon of 1310 bp was amplified from the wild-type (WT) allele (In the first PCR, amplicon of ~ 370 bp were not detected from the promoter-deleted (DF1∆) alleles). In a hemi-nested PCR (using P5 and P6 primers), amplicons of 1256 bp and ~ 316 bp were amplified from the wild-type and promoter-deleted (DF1∆) alleles, respectively. C Alignment of the representative sequences of the wild-type (WT DF1) and promoter-deleted (DF1∆) sequences determined by Sanger sequencing. The gRNA-binding sites are shown in blue, and the PAM regions are shown in green letters. WT, wild-type; DF1 ∆, DF1 cells knockout for the distal OVA promoter (DF1 +/OVA Pro ∆); NHEJ, non-homologous end-joining; ERE, estrogen-responsive enhancer element; TSSL, tissue-specific silencer-like element; SDRE, steroid-dependent regulatory element; NRE, negative regulatory element; CAR, COUP-adjacent repressor site; COUP, Chicken OVA upstream promoter; TATA, TATA box; TSS, transcription start site; P, primer. M, DNA size marker; NTC, no template control
Three isogenic DF1 +/OVA Pro ∆ clones with the confirmed deletion of SDRE and NRE elements were cultured in vitro without estrogen and were analyzed for the expression of the OVA gene by RT-qPCR. The transcript levels of the OVA gene in the DF1 +/OVA Pro ∆ cells increased more than 104-fold compared to that in the wild-type DF1 cell (p < 0.01) (Fig. 3). The transcript levels of the OVA gene in the hormonally-activated tissue of the magnum from the 35-week-old laying hen were 107-fold higher compared to that in the wild-type DF1 cells (Figs. 3A and B, Fig. S3, Table 1).

Gene expression ratio for Ovalbumin transcript in DF1+/OVA Pro ∆ cells. A Agarose gel (2%) electrophoresis for analysis of the RT-PCR products amplified by primers P8 and P9 (for OVA, Fig. 1), and P10 and P11 (for GAPDH). The expected amplicon size for OVA and GAPDH are 179 bp and 187 bp, respectively. WT, wild-type; DF1 ∆, distal OVA promoter knockout DF1 cells (DF1 +/OVA Pro ∆); M, DNA size marker; NTC, no template control; RT, reverse transcriptase. The full-length gel electrophoresis images are shown in Fig. S3. B Upregulation of the OVA mRNA in DF1 +/OVA Pro ∆cells was assessed by RT-qPCR. Upon deletion of the distal OVA promoter, an increased level of expression of the OVA gene was determined (DF1∆). The transcript levels of OVA for these samples (Three isogenic DF1 +/OVA Pro ∆ clones) were ~ 104-fold higher than the OVA transcript levels in the wild-type DF1 (WT DF1). The transcript levels for OVA in the hormonally-activated tissue of the magnum (from a 35-week-old laying hen) show the highest level of expression. The gene expression ratio for the OVA over GAPDH was calculated by the Pfaffl method of relative quantification [38]. The Mann–Whitney assay was used to analyze significant statistical differences between the WT-DF1 group and DF1∆ and magnum groups. * and ** show statistical differences with p values < 0.05 and < 0.01, respectively
A fluorescent genomic reporter is activated under the control of the Ovalbumin promoter with the deletion of distal elements
Next, we asked whether the OVA gene promoter with the deletion of its distal elements in the DF1 +/OVA Pro ∆ cells can activate a foreign transgene. For this purpose, we designed a reporter construct containing a promoterless DsRed2 (IRES-DsRed2-HSV TK polyA-CMV promoter-PuroR-IRES2-EGFP-SV40 polyA) and inserted it into exon 2 of the OVA gene, 125 bp after ATG codon, using CRISPR HDR (Fig. 4A). In these cells (DF1 +/OVA Pro ∆−Tg (promoterless dsRed)), the insertion of the reporter was confirmed by genomic PCR, Sanger sequencing, and fluorescence microscopy for GFP (Figs. 4B, C). The promoterless DsRed2 reporter, under the function of a distally-deleted OVA promoter, became activated, and its red fluorescence was visualized using fluorescence microscopy (Fig. 4C). However, when the promoterless reporter was inserted at the same region in the OVA locus of the wild-type DF1 cells, it did not result in red fluorescence (Fig. S4). This experiment confirmed that non-oviduct chicken cells with the deletion of distal elements in their OVA promoter can express an inserted transgene in an estrogen-independent manner. The wild-type DF1 cells did not show any transcriptional activity for the OVA gene (Fig. 3).

Activation of transgene expression in DF1 +/OVA Pro ∆−Tg (promoterless dsRed) cells. A The schematic representation of CRISPR HDR mediated knockin strategy in DF1+/OVA Pro ∆ cells. The top diagram shows the donor vector that was designed to have a promoterless DsRed2 and a CMV-Puro-EGFP cassette flanked by left and right homology arms. The OVA E2 indicates the gRNA-binding site on exon 2 of the OVA (+ 174 to + 1784) gene. The bottom diagram shows the allele after CRISPR-HDR insertion of the reporter cassette (DF1 +/OVA Pro ∆−Tg (promoterless dsRed)). PCR primers (P12 and P13) were used for the assessment of the CRISPR-HDR insertion of the promoterless DsRed2 in DF1 +/OVA Pro ∆−Tg (promoterless dsRed) cells. B Genomic PCR analysis of the targeted gene knock-in DF1 +/OVA Pro ∆−Tg (promoterless dsRed) cells. For the assessment of the CRISPR-HDR insertion of the promoterless DsRed2 in DF1 ∆−Tg cells, primers (P12 and P13) were used to amplify a 2569 bp amplicon. The insertion-specific PCR products of DF1 ∆−Tg cells were sequenced by Sanger sequencing and aligned to the donor plasmid (used as a DNA repair template during transfection). C Fluorescence microscopy of DF1 ∆−Tg cells indicating DsRed2 expression under the control of the endogenous truncated OVA promoter, Magnification: 20X. DF1∆, DF1 cells knock-out for distal OVA promoter (DF1 +/OVA Pro ∆); DF1 ∆−Tg cells, promoterless DsRed2 knockin DF1 cells (DF1 +/OVA Pro ∆−Tg (promoterless dsRed)); HDR, homology-directed repair; M, DNA size marker; WT, wild-type; NTC, no template control
Discussion
In this study, we have shown that the negative effects of the NRE element on the OVA gene can be counteracted to some extent by CRISPR interference (Fig. 1). We have also demonstrated that the deletion of the distal OVA promoter in DF1 cells (DF1 +/OVA Pro ∆) leads to the induction of the OVA gene expression (Figs. 2 and 3). In addition, the insertion of a promoterless reporter in these cells (DF1 +/OVA Pro ∆−Tg (promoterless dsRed)) resulted in the expression of the fluorescent reporter protein (DsRed2) (Fig. 4), indicating that a chicken non-oviduct cell line with the deletion of distal promoter sequences can serve as a model for steroid-independent expression of a transgene driven by the endogenous OVA promoter.
The tissue-specific OVA promoter has been identified as a novel candidate for the large-scale production of pharmaceutical proteins. It has been effectively employed in the synthesis of several therapeutic proteins [7,8,9,10,11,12,13,14,15,16,17]. Although the regulatory elements in the OVA promoter have been fairly well characterized [20,21,22,23,24,25,26,27,28,29,30,31, 35], it is not clear which regulatory sequences are sufficient and efficient enough to induce oviduct-specific expression of exogenous genes. Previous studies demonstrated that deletion of the SDRE and NRE, along with the linker between them, increased chloramphenicol acetyltransferase (CAT) activity on a plasmid [24, 27, 28]. These studies indicated that the cooperation between multiple distal regulatory and promoter-proximal regions confers oviduct-specific OVA expression. Deletion of regulatory elements upstream of − 80 abolished the tissue-specific expression of OVA in primary oviduct cell cultures, while basal expression was increased to levels comparable to those seen after estrogen induction in genes that contain an SDRE [24, 27, 30]. Additionally, a few reports have shown that the expression of the reporter CAT gene was induced by the OVA proximal promoter (− 87 to + 9) in primary oviduct cells and non-oviduct cell cultures, such as LMH/2A (Table 3) [25, 27,28,29,30, 39, 40].
In this investigation, we studied the role of regulatory elements of the OVA promoter in their natural genomic context. CRISPRi was performed on two regulatory sequences of the NRE element, CAR and silencer, using dCas9 and manually selected sgRNAs (Fig. 1A). This resulted in a likely counteracting effect exerted on the NRE element, which, in turn, increased the transcription of the OVA RNA (Fig. 1B). Researchers have used various strategies including biochemical methods [41], crystallographic methods [41, 42], and atomic force microscopy [43] to identify the length of Cas9/dCas9 footprint on the DNA template. Zhang et al., [44] used a single-molecule approach to measure the footprint and determined the length of the DNA over which Cas9 binding likely affects the binding of another protein. To ensure interference of binding proteins with CAR and Silencer, we manually selected sgRNAs that specifically target these sites. Our CRISPR interference experiment confirmed the negative role of both CAR and silencer in OVA gene expression. The expression of OVA subjected to CRISPR interference with two sgRNAs was significantly higher and more than 100-fold (p < 0.05) than that in the wild-type DF1 cells. In the next set of experiments, we decided to knock out these regulatory sequences to examine their potential impact on the transcription of the OVA gene. Our findings demonstrated that the in situ deletions of the distal OVA promoter led to the upregulation of OVA transcript in DF1 cells. Our RT-qPCR analysis, following the deletion of the distal OVA promoter which includes the SDRE and the NRE, in the DF1+/OVA Pro ∆ cells, revealed a significant increase of approximately104-fold in OVA transcript levels compared to wild-type DF1 cells (Fig. 3). This finding strongly supports our hypothesis that negative regulatory elements have a highly effective role in controlling OVA expression. Furthermore, based on the same results, the magnum tissue exhibited transcript levels approximately 103-fold higher than the DF1+/OVA Pro ∆ cells, indicating that positive regulatory signals, including estrogen, can further boost the expression. We found that the deletion of a 962-bp region (− 1044 to − 82 bp) containing the distal promoter elements resulted in a significant reduction in the tissue-restricted and hormone-dependent expression of the OVA gene. It has been reported that the chicken OVA upstream promoter (COUP) site (− 85 to − 73) represses basal OVA expression in the absence of steroids and is required for its induction by steroids [30]. Although previous reports have shown that the deletion of the COUP site in OvCAT constructs increases transcriptional activity in the absence of the NRE and confirms its repressive role on basal gene expression, our data clearly show that even in the presence of the COUP site, transcriptional activity is increased when the NRE is absent. Muramatsu et al. demonstrated that the sequence from − 3200 to − 2800 acts as a tissue-specific silencer-like (TSSL) element, repressing the expression of OVA gene in non-oviduct tissue [40]. Although our experiment with DF1+/OVA Pro ∆ cells did not detect the effect of TSSL element in repressing the OVA gene expression, it remains unclear whether this TSSL element causes tissue-specific repression in other tissues or if universal transcription factors bind to it in all tissues except the oviduct. This finding suggests that the opposing effect of the COUP site on transcriptional activity depends on the native genomic context and, perhaps, other regulatory elements are brought together in a spatial configuration by chromatin loops (Fig. 5).

A schematic model depicting the mechanism of increased expression of the Ovalbumin gene in different cell types in steroid-dependent and –independent manners. The main induction for the expression of the OVA gene in oviduct cells is estrogen that by binding to the SDRE region overcomes the inhibitory circuits exerted by the tissue-specific silencer-like element (TSSL), and negative regulatory element (NRE). The CRISPR/CAS-mediated deletion of the regulatory sequences of the OVA distal promoter (SDRE, NRE, and the linker in between) leads to the expression of the OVA gene in DF1 cells. The CRISPR-mediated interference of regulatory sequences of the NRE element as well leads to an increased expression of the OVA gene in DF1 cells
In our DF1+/OVA Pro ∆ cells, although the core promoter elements (TATA box and the initiator element, INR) that contain sufficient information for the initiation of transcription have remained intact, we cannot rule out the potential regulatory role of alternative promoters in the genome [45]. Kodama et al. identified several TATA-like and other promoter motifs located at a position around − 1800 bp [10]. Bradshaw et al. demonstrated that the region from − 1094 to − 1125 (− 1100), in the presence of an NF-1-like protein, functions as a steroid hormone-independent enhancer and increases OVA gene transcription [46]. A nuclear factor-1-like factor binds to a far upstream OVA enhancer [46]. Our results support the notion that the transcriptional regulation of the OVA gene is not determined only by promoter regions, but may involve multiple regulatory elements in the local genomic context that work in the three-dimensional organization of the locus [47, 48] (Fig. 5). This three-dimensional organization of the OVA locus in the oviduct cells, which might be dependent on the nuclear positioning of chromosomes and/or the architecture of chromatin within chromosome territory [37] can establish a structural scaffold for interaction between enhancer-promoter, enhancer-enhancer, promoter-promoter, and superenhancer elements. These kinds of interactions may be further promoted and changed by the activity of specific transcription factors, signaling pathways, hormones, and developmental stages [49,50,51,52,53]. The overall output from these interactions might result in the transcriptional activation of the OVA locus. We hypothesize that in non-oviduct differentiated cells, a specific repressive chromatin organization is established as well, which is perturbed by CRISPRi and the excision of the distal promoter using CRISPR-Cas9 (Fig. 5), leading to the upregulation of the OVA gene.
Conclusions
Our study overcomes the limitation of previous studies that relied on cloned promoters, where the promoter regulatory sequences have to be taken out of their cis context and spatial organization into a plasmid. The utilization of CRISPR technology enabled us to precisely interfere with and delete the negative regulatory sequences of the OVA gene promoter directly within the chicken cell's native genomic context. We demonstrate that the expression of a transgene can be driven in a hormonally independent manner through the function of the OVA gene promoter and associated endogenous regulatory elements.
Methods
Plasmid construction
The CRISPR design tool (http://crispr.mit.edu/) was employed to identify sgRNA binding sites within the OVA promoter and coding region for the deletion and insertion of the distal promoter and reporter gene, respectively. However, for our CRISPR interference strategy, we manually selected the sgRNAs and analyzed them regarding their off-target binding and secondary structure. Different plasmids were constructed using routine subcloning techniques. To perform CRISPRi, three plasmids were created: pdCas9_silencer-gRNA (encoding sgRNA for targeting the − 241 to − 222 region), pdCas9_CAR-gRNA (encoding sgRNA for targeting the − 129 to − 110 region), and pdCas9-X (with no sgRNA) (Table 2). The CRISPRi vectors were generated by modifying the plasmid pdCas9-DNMT3A-EGFP (#71,666) using standard techniques. The plasmid was digested with BamHI and BsrGI to remove a 1726 bp fragment that included the DNMT3A catalytic domain. The resulting overhangs were filled in, and the plasmid was self-ligated using T4 DNA ligase, resulting in a plasmid named 71666delta. The CAR- and Silencer-gRNA were then subcloned into the BbsI-digested region of this plasmid for CRISPRi.
For CRISPR excision, two plasmids (px459-14 & px459-15) were constructed, expressing Cas9 and sgRNAs targeting the OVA distal promoter. The designed sgRNAs were subcloned into the BbsI-digested region of the px459 plasmid for this purpose. To perform CRISPR HDR, another plasmid named px459-6 was created. It contained Cas9 and a sgRNA targeting the OVA exon 2. The designed sgRNA was also subcloned into the BbsI-digested region of the px459 plasmid.
The donor vector (pHD_4520) was generated by ligating a 556 bp fragment of the OVA gene (beginning of exon 2) representing the 5' homology arm, and a 526 bp fragment of the OVA gene, representing the 3' homology arm. To create the donor vector, an initial base vector containing an EGFP reporter gene and necessary restriction sites for subsequent subcloning was synthesized (Table 2). Detailed plasmid maps displaying the specific components can be found in Fig. S7.
CRISPR interference of the negative regulatory elements of the Ovalbumin gene in cultured DF1 cells
DF1 cells were cultured as recommended by the ATCC. The cells were transfected into four groups: group one was transfected with pdCas9_silencer-gRNA, targeting the silencer; group two was transfected with pdCas9_CAR-gRNA, targeting the CAR; group three was transfected with pdCas9_silencer-gRNA and pdCas9_CAR-gRNA; and the control group was transfected with pdCas9-X (with no sgRNA) (Table 2). Lipofectamine 3000 (Invitrogen, USA) was used for transfections as previously described [54]. Briefly, 0.5 μg from each plasmid was diluted with 50 μl OPTI-MEM + GlutaMax (Thermo Fisher Scientific, USA), mixed with 1 μl Lipofectamine 3000 reagent, and then incubated with 0.1–0.15 × 106 DF1 cells for 4 h. Subsequently, the cells were cultured in 500 μl of an antibiotic-free DMEM-F12 culture medium (Thermo Fisher Scientific) and incubated for 24 h at 38 °C in a 7.5% CO2 environment. The medium was replaced with fresh medium containing penicillin and streptomycin antibiotics 24 h after transfection. Transfected cells were passaged for subsequent assays for three days.
The effects of CRISPRi on the expression of the OVA gene were analyzed by RT-PCR. From DF1 cells subjected to CRISPRi and the positive control magnum tissue (from a 35-week-old laying hen), total RNA was isolated using the Total RNA Isolation Kit (DENAzist Asia, Iran). After checking the quality and quantity of isolated RNA using gel electrophoresis and a spectrophotometer (Epoch 2, BioTek Instruments Inc., USA), total RNA was reverse transcribed using MMLV reverse transcriptase and random hexamer primer (Thermo Fisher Scientific, USA). The complementary DNA for OVA and GAPDH transcripts was subjected to PCR amplification using Taq DNA Polymerase 2 × Master Mix RED (Ampliqon, Denmark) and specific primers (Table 1). The amplification steps included an initial 95 °C for 3 min, followed by 35 cycles of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 10 s, with the final elongation step at 72 °C for 10 min.
To investigate the presence of regulatory RNAs that might transcribe from the distal promoter, different primers (Table 1) were designed and used in the PCR or hemi-nested PCR amplification reactions on cDNA which was generated from wild-type DF1 total RNA.
Targeted deletion of Ovalbumin promoter in cultured DF1 cells
To perform CRISPR excision with dual sgRNAs on the OVA promoter, DF1 cells were transfected with pX459-14 and pX459-15 (Table 2) using Lipofectamine 3000 (Invitrogen, USA), The transfectd DF1 cells were exposed to puromycin dihydrochloride (2.5 μg/ml; Sigma-Aldrich, USA) for 3 days. DF1 cells after antibiotic exposure were expanded for 2 to 3 weeks. A mixed population of these cells was initially screened using genomic PCR to confirm the deletion of the OVA distal promoter in a fraction of cells. Genomic DNA was extracted from wild-type and knockout DF1 cells (DF1 +/OVA Pro ∆) using the Genomic DNA Extraction Kit (DENAzist Asia Co., Iran). Gene-targeting events were detected by a single-round or nested PCR using the designed primers (Table 1) and Taq DNA polymerase master mix RED (Ampliqon, Denmark), and confirmed by Sanger sequencing of the amplicons (Genomin Co., Iran). After single-cell isolation and clonal expansion, three clones of knockout DF1 cells with the deletion of the distal OVA promoter (DF1 +/OVA Pro ∆) were confirmed using genomic PCR. These three clones were analyzed for the expression of OVA by RT-qPCR.
Analysis of Ovalbumin expression in DF1 cells with the deletion of distal Ovalbumin promoter
Total RNA was isolated from the magnum tissue (from a 35-week-old laying hen), wild-type DF1 cells and DF1 cells knockout for distal OVA promoter (DF1 +/OVA Pro ∆) using the Total RNA Isolation Kit (DENAzist Asia, Iran). Total RNA was subjected to quality and quantity analysis, and reverse transcription using MMLV reverse transcriptase and random hexamer primers (Thermo Fisher Scientific, USA). Each quantitative PCR reaction contained 1 × SYBR Green Real-time PCR Master Mix (Thermo Scientific, USA), 2μl cDNA template, and each primer (Table 1) at 500nM in a 20μl reaction volume, which was performed in a Rotor-Gene Q real-time PCR cycler (Qiagen, USA). To amplify complementary DNA for OVA and GAPDH transcripts, the amplification steps were: 95 C for 15min, followed by 35 cycles of 95 C for 30 s, 58 C for 30 s, and 72 C for 30s. To acquire melting curves, the temperature was increased in steps of 0.2 C for 5s from 55 °C to 95 C. PCR products after clean-up with the PCR Clean-up Kit (DENAzist Asia Co., Iran), were subjected to Sanger sequencing (Genomin Co., Iran). (Fig. S5).
Different qPCR reactions were performed to adjust the reaction temperature, find the best concentration of primers, and optimize the amplification and melting curves (Fig. S3). Complementary DNA from the magnum of the 35-week-old hen was serially diluted and subjected to qPCR to make standard curves (Fig. S6). Each dilution was subjected to real-time readings in triplicate. To make a standard curve (Fig. S6), the log10 of cDNA concentration for the OVA and GAPDH genes were plotted against the cycle threshold (Ct) numbers. We used the equation of E = (10–1/slope-1) × 100% to calculate the reaction efficiency. The gene expression ratio for the OVA gene over GAPDH was calculated for the magnum, wild-type DF1, and DF1 cell with deletion of distal OVA promoter using the Pfaffl method of relative quantification [38].
Targeted knock-in of a reporter in DF1 cells with the deletion of distal Ovalbumin promoter
DF1 +/OVA Pro ∆ cells were transfected with pX459_6 and pHD_4520 (donor vector) using Lipofectamine 3000 (Invitrogen, USA), as described above. The cells 48h after transfection were subjected to antibiotic selection with puromycin dihydrochloride (2.5 μg/ml; Sigma-Aldrich, USA). To confirm the knock-in of the reporter construct (DsRed2-CMV-Puro-IRES-EGFP), genomic PCR and Sanger sequencing (Genomin Co., Iran) were performed. Cells with the inserted reporter and deleted OVA promoter (DF1 +/OVA Pro ∆−Tg (promoterless dsRed)) were observed and photographed by fluorescence microscopy (Nikon Eclipse Ts2R, Japan) two weeks after transfection.
Availability of data and materials
The data supporting the conclusions of this article are included within the article (and its additional files) and are also available from the corresponding author upon request.
Abbreviations
- CRISPR-HDR:
-
CRISPR-mediated homology-directed repair
- ERE:
-
Estrogen-responsive enhancer element
- SDRE:
-
Steroid-dependent regulatory element
- NRE:
-
Negative regulatory element
- CAR:
-
COUP-TF adjacent repressor
- TSSL:
-
Tissue-specific silencer-like element
- COUP:
-
Chicken Ovalbumin upstream promoter
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- sgRNA:
-
Small guide RNA
- PAM:
-
Protospacer adjacent motif
- dCas9:
-
Endonuclease deficient Cas9, Catalytically dead Cas9
- OVA :
-
Ovalbumin Gene
- CAT:
-
Chloramphenicol acetyltransferase
- CRISPRi:
-
CRISPR interference
- OvCAT:
-
Truncated OVA promoter-CAT reporter
- INR:
-
Initiator element
- NHEJ:
-
Non-homologous end-joining
- TSS:
-
Transcription start site
- NTC:
-
No template control
- DF1∆:
-
Promoter-deleted alleles
- TATA:
-
TATA box
- WT:
-
Wild-type
- DF1+/ OVA Pro ∆ cell:
-
Monoallelic CRISPR-mediated deletion of the distal OVA promoter in DF1 cells
- DF1+/OVA Pro Δ-Tg (promoterless dsRed) :
-
Promoterless DsRed2 knockin DF1 cells
References
Dehdilani N, Yousefi Taemeh S, Goshayeshi L, Dehghani H. Genetically engineered birds; pre-CRISPR and CRISPR era†. Biol Reprod. 2022;106:24–46. https://doi.org/10.1093/biolre/ioab196.
Harvey AJ, Speksnijder G, Baugh LR, Morris JA, Ivarie R. Expression of exogenous protein in the egg white of transgenic chickens. Nat Biotechnol. 2002;20:396–9. https://doi.org/10.1038/nbt0402-396.
Sato N, Matsuda K, Sakuma C, Foster DN, Oppenheim RW, Yaginuma H. Regulated gene expression in the chicken embryo by using replication-competent retroviral vectors. J Virol. 2002;76:1980–5. https://doi.org/10.1128/JVI.76.4.1980-1985.2002.
Thaisuchat H, Baumann M, Pontiller J, Hesse F, Ernst W. Identification of a novel temperature sensitive promoter in CHO cells. BMC Biotechnol. 2011;11:51. https://doi.org/10.1186/1472-6750-11-51.
Liu Z, Tyo KEJ, Martínez JL, Petranovic D, Nielsen J. Different expression systems for production of recombinant proteins in Saccharomyces cerevisiae. Biotechnol Bioeng. 2012;109:1259–68. https://doi.org/10.1002/bit.24409.
Brooks AR, Harkins RN, Wang P, Qian HS, Liu P, Rubanyi GM. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J Gene Med. 2004;6:395–404. https://doi.org/10.1002/jgm.516.
Zhu L, van de Lavoir M-C, Albanese J, Beenhouwer DO, Cardarelli PM, Cuison S, et al. Production of human monoclonal antibody in eggs of chimeric chickens. Nat Biotechnol. 2005;23:1159–69. https://doi.org/10.1038/nbt1132.
Lillico SG, Sherman A, McGrew MJ, Robertson CD, Smith J, Haslam C, et al. Oviduct-specific expression of two therapeutic proteins in transgenic hens. Proc Natl Acad Sci. 2007;104:1771–6. https://doi.org/10.1073/pnas.0610401104.
Kwon MS, Koo BC, Kim D, Nam YH, Cui X-S, Kim N-H, et al. Generation of transgenic chickens expressing the human erythropoietin (hEPO) gene in an oviduct-specific manner: production of transgenic chicken eggs containing human erythropoietin in egg whites. PLoS ONE. 2018;13:e0194721. https://doi.org/10.1371/journal.pone.0194721.
Kodama D, Nishimiya D, Nishijima K, Okino Y, Inayoshi Y, Kojima Y, et al. Chicken oviduct-specific expression of transgene by a hybrid ovalbumin enhancer and the Tet expression system. J Biosci Bioeng. 2012;113:146–53. https://doi.org/10.1016/j.jbiosc.2011.10.006.
Byun SJ, Kim SW, Kim KW, Kim JS, Hwang IS, Chung HK, et al. Oviduct-specific enhanced green fluorescent protein expression in transgenic chickens. Biosci Biotechnol Biochem. 2011;75:646–9. https://doi.org/10.1271/bbb.100721.
Cao D, Wu H, Li Q, Sun Y, Liu T, Fei J, et al. Expression of recombinant human lysozyme in egg whites of transgenic hens. PLoS ONE. 2015;10:e0118626. https://doi.org/10.1371/journal.pone.0118626.
Liu T, Wu H, Cao D, Li Q, Zhang Y, Li N, et al. Oviduct-specific expression of human neutrophil defensin 4 in lentivirally generated transgenic chickens. PLoS ONE. 2015;10:e0127922. https://doi.org/10.1371/journal.pone.0127922.
Park TS, Lee HG, Moon JK, Lee HJ, Yoon JW, Yun BNR, et al. Deposition of bioactive human epidermal growth factor in the egg white of transgenic hens using an oviduct-specific minisynthetic promoter. FASEB J. 2015;29:2386–96. https://doi.org/10.1096/fj.14-264739.
Herron LR, Pridans C, Turnbull ML, Smith N, Lillico S, Sherman A, et al. A chicken bioreactor for efficient production of functional cytokines. BMC Biotechnol. 2018;18:993. https://doi.org/10.1186/s12896-018-0495-1.
Kwon SC, Choi JW, Jang H-J, Shin SS, Lee SK, Park TS, et al. Production of biofunctional recombinant human interleukin 1 receptor antagonist (rhIL1RN) from transgenic quail egg white1. Biol Reprod. 2010;82:1057–64. https://doi.org/10.1095/biolreprod.109.081687.
Oishi I, Yoshii K, Miyahara D, Tagami T. Efficient production of human interferon beta in the white of eggs from ovalbumin gene–targeted hens. Sci Rep. 2018;8:297. https://doi.org/10.1038/s41598-018-28438-2.
Benarafa C, Remold-O’Donnell E. The ovalbumin serpins revisited: perspective from the chicken genome of clade B serpin evolution in vertebrates. Proc Natl Acad Sci. 2005;102:11367–72. https://doi.org/10.1073/pnas.0502934102.
Dombre C, Guyot N, Moreau T, Monget P, Da Silva M, Gautron J, Réhault-Godbert S. Egg serpins: the chicken and/or the egg dilemma. Semin Cell Dev Biol. 2017;62:120–32. https://doi.org/10.1016/j.semcdb.2016.08.019.
Dougherty DC, Park H-M, Sanders MM. Interferon regulatory factors (IRFs) repress transcription of the chicken ovalbumin gene. Gene. 2009;439:63–70. https://doi.org/10.1016/j.gene.2009.03.016.
Schimke RT, McKnight GS, Shapiro DJ, Sullivan D, Palacios R. Hormonal regulation of ovalbumin synthesis in the chick oviduct. Recent Prog Horm Res. 1975;31:175–211. https://doi.org/10.1016/b978-0-12-571131-9.50009-8.
Kaye JS, Pratt-Kaye S, Bellard M, Dretzen G, Bellard F, Chambon P. Steroid hormone dependence of four DNase I-hypersensitive regions located within the 7000-bp 5′-flanking segment of the ovalbumin gene. EMBO J. 1986;5:277–85. https://doi.org/10.1002/j.1460-2075.1986.tb04210.x.
Kaye JS, Bellard M, Dretzen G, Bellard F, Chambon P. A close association between sites of DNase I hypersensitivity and sites of enhanced cleavage by micrococcal nuclease in the 5′-flanking region of the actively transcribed ovalbumin gene. EMBO J. 1984;3:1137–44. https://doi.org/10.1002/j.1460-2075.1984.tb01942.x.
Sanders MM, McKnight GS. Positive and negative regulatory elements control the steroid-responsive ovalbumin promoter. Biochemistry. 1988;27:6550–7. https://doi.org/10.1021/bi00417a053.
Schweers LA, Frank DE, Weigel NL, Sanders MM. The steroid-dependent regulatory element in the ovalbumin gene does not function as a typical steroid response element. J Biol Chem. 1990;265:7590–5. https://doi.org/10.1016/S0021-9258(19)39155-0.
Wang L-H, Tsai SY, Cook RG, Beattie WG, Tsai M-J, O’Malley BW. COUP transcription factor is a member of the steroid receptor superfamily. Nature. 1989;340:163–6. https://doi.org/10.1038/340163a0.
Haecker SA, Muramatsu T, Sensenbaugh KR, Sanders MM. Repression of the ovalbumin gene involves multiple negative elements including a ubiquitous transcriptional silencer. Mol Endocrinol. 1995;9:1113–26. https://doi.org/10.1210/mend.9.9.7491104.
Sensenbaugh KR, Sanders MM. Multiple promoter elements including a novel repressor site modulate expression of the chick ovalbumin gene. DNA Cell Biol. 1999;18:147–56. https://doi.org/10.1089/104454999315538.
Monroe DG, Sanders MM. The COUP-adjacent repressor (CAR) element participates in the tissue-specific expression of the ovalbumin gene. Biochim Biophys Acta Gene Struct Exp. 2000;1517:27–32. https://doi.org/10.1016/s0167-4781(00)00241-4.
Park H-M, Haecker SE, Hagen SG, Sanders MM. COUP-TF plays a dual role in the regulation of the ovalbumin gene. Biochemistry. 2000;39:8537–45. https://doi.org/10.1021/bi0005862.
Dougherty DC, Sanders MM. Estrogen action: revitalization of the chick oviduct model. Trends Endocrinol Metab. 2005;16:414–9. https://doi.org/10.1016/j.tem.2005.09.001.
Gaub MP, Dierich A, Astinotti D, Touitou I, Chambon P. The chicken ovalbumin promoter is under negative control which is relieved by steroid hormones. EMBO J. 1987;6:2313–20.
Schweers LA, Sanders MM. A protein with a binding specificity similar to NF-kappa B binds to a steroid-dependent regulatory element in the ovalbumin gene. J Biol Chem. 1991;266:10490–7.
Nordstrom LA, Dean DM, Sanders MM. A complex array of double-stranded and single-stranded DNA-binding proteins mediates induction of the ovalbumin gene by steroid hormones. J Biol Chem. 1993;268:13193–202.
Kato S, Tora L, Yamauchi J, Masushige S, Bellard M, Chambon P. A far upstream estrogen response element of the ovalbumin gene contains several half-palindromic 5′-TGACC-3′ motifs acting synergistically. Cell. 1992;68:731–42. https://doi.org/10.1016/0092-8674(92)90148-6.
Woodfint RM, Hamlin E, Lee K. Avian bioreactor systems: a review. Mol Biotechnol. 2018;60:975–83. https://doi.org/10.1007/s12033-018-0128-x.
Dehghani H. Regulation of chromatin organization in cell stemness: the emerging role of long non-coding RNAs. Stem Cell Rev and Rep. 2021;58:229. https://doi.org/10.1007/s12015-021-10209-8.
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. https://doi.org/10.1093/nar/29.9.e45.
Dean DM, Jones PS, Sanders MM. Regulation of the chicken ovalbumin gene by estrogen and corticosterone requires a novel DNA element that binds a labile protein, Chirp-1. Mol Cell Biol. 1996;16:2015–24. https://doi.org/10.1128/MCB.16.5.2015.
Muramatsu T, Imai T, Park HM, Watanabe H, Nakamura A, Okumura J. Gene gun-mediated in vivo analysis of tissue-specific repression of gene transcription driven by the chicken ovalbumin promoter in the liver and oviduct of laying hens. Mol Cell Biochem. 1998;185:27–32. https://doi.org/10.1023/a:1016507900718.
Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. https://doi.org/10.1126/science.1247997.
Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–49. https://doi.org/10.1016/j.cell.2014.02.001.
Josephs EA, Kocak DD, Fitzgibbon CJ, McMenemy J, Gersbach CA, Marszalek PE. Structure and specificity of the RNA-guided endonuclease Cas9 during DNA interrogation, target binding and cleavage. Nucleic Acids Res. 2015;43:8924–41. https://doi.org/10.1093/nar/gkv892.
Zhang Q, Wen F, Zhang S, Jin J, Bi L, Lu Y, et al. The post-PAM interaction of RNA-guided spCas9 with DNA dictates its target binding and dissociation. Sci Adv. 2019;5:eaaw9807. https://doi.org/10.1126/sciadv.aaw9807.
Ayoubi TA, Van De Ven WJ. Regulation of gene expression by alternative promoters. FASEB j. 1996;10:453–60.
Bradshaw MS, Tsai MJ, O’Malley BW. A far upstream ovalbumin enhancer binds nuclear factor-1-like factor. J Biol Chem. 1988;263:8485–90.
Gibcus JH, Dekker J. The context of gene expression regulation. F1000 Biol Rep. 2012;4:8. https://doi.org/10.3410/B4-8.
Andersson R, Sandelin A. Determinants of enhancer and promoter activities of regulatory elements. Nat Rev Genet. 2020;21:71–87. https://doi.org/10.1038/s41576-019-0173-8.
Dehghani H, Reith C, Hahnel AC. Subcellular localization of protein kinase C delta and epsilon affects transcriptional and post-transcriptional processes in four-cell mouse embryos. Reproduction. 2005;130:453–65. https://doi.org/10.1530/rep.1.00572.
Dehghani H, Narisawa S, Milln JL, Hahnel AC. Effects of disruption of the embryonic alkaline phosphatase gene on preimplantation development of the mouse. Dev Dyn. 2000;217:440–8. https://doi.org/10.1002/(SICI)1097-0177(200004)217:4%3c440::AID-DVDY11%3e3.0.CO;2-1.
Es-Haghi M, Bassami M, Dehghani H. Construction and quantitative validation of chicken CXCR4 expression reporter. Mol Biotechnol. 2016;58:202–11. https://doi.org/10.1007/s12033-016-9917-2.
Es-Haghi M, Soltanian S, Dehghani H. Perspective: cooperation of Nanog, NF-κΒ, and CXCR4 in a regulatory network for directed migration of cancer stem cells. Tumour Biol. 2016;37:1559–65. https://doi.org/10.1007/s13277-015-4690-6.
Dehghani H, Hahnel AC. Expression profile of protein kinase C isozymes in preimplantation mouse development. Reproduction. 2005;130:441–51. https://doi.org/10.1530/rep.1.00571.
Abu-Bonsrah KD, Zhang D, Newgreen DF. CRISPR/Cas9 targets chicken embryonic somatic cells in vitro and in vivo and generates phenotypic abnormalities. Sci Rep. 2016;6:34524. https://doi.org/10.1038/srep34524.
Acknowledgements
We dedicate this work to our deceased colleague, Dr. Mohammad Reza Bassami, for his help to conceptualize the early stages of this study.
Funding
This study was financially supported by DENAzist Asia Co., Mashhad, Iran. Sara Yousefi Taemeh was partially supported by a grant from the Ferdowsi University of Mashhad (No. FUM-1400–10489).
Author information
Authors and Affiliations
Contributions
SYT: Conceptualization, Investigation, Methodology, Formal analysis, Visualization, Writing—original draft. ND: Investigation, Methodology. LG: Investigation, Methodology. SRG: Methodology, Writing—review & editing. JM: Thesis adviser. BP: Resources, Writing—review & editing, HD: Conceptualization, Supervision, Visualization, Resources, Writing—original draft, Writing—review & editing, Project administration, Funding acquisition.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yousefi Taemeh, S., Dehdilani, N., Goshayeshi, L. et al. Study of the regulatory elements of the Ovalbumin gene promoter using CRISPR technology in chicken cells. J Biol Eng 17, 46 (2023). https://doi.org/10.1186/s13036-023-00367-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13036-023-00367-3