
Abstract
Escherichia coli is a widely used platform for metabolic engineering due to its fast growth and well-established engineering techniques. However, there has been a demand for faster-growing E. coli for higher production of desired substances. Here, to increase the growth of E. coli cells, we optimized the expression level of Hfq protein, which plays an essential role in stress responses. Six variants of the hfq gene with a different ribosome binding site sequence and thereby a different expression level were constructed. When the Hfq expression level was optimized in DH5α, its growth rate was increased by 12.1% and its cell density was also increased by 4.5%. RNA-seq and network analyses revealed the upregulation of stress response genes and metabolic genes, which increases the tolerance against pH changes. When the same strategy was applied to five other E. coli strains (BL21 (DE3), JM109, TOP10, W3110, and MG1655), all their growth rates were increased by 18–94% but not all their densities were increased (− 12 − + 32%). In conclusion, the Hfq expression optimization can increase cell growth rate and probably their cell densities as well. Since the hfq gene is highly conserved across bacterial species, the same strategy could be applied to other bacterial species to construct faster-growing strains.
Similar content being viewed by others


Introduction
The production of chemical substances and proteins from genetically engineered bacteria has been studied for decades for practical use [1], and metabolic engineering has proven the essential role of bacteria engineering and its practical usefulness in the industry [2,3,4]. For instance, recent advances in metabolic engineering make it possible to engineer bacteria or yeast cells for high and cost-efficient production of recombinant therapeutic proteins such as insulin and growth hormones [5,6,7] and biofuels such as alcohols, fatty acid methyl, and ethyl esters [8,9,10]. Regarding recombinant protein production, there have been several strategies to increase protein titers. High copy-number plasmids have been used to produce larger amounts of desired proteins [11, 12], but high copy-number plasmids often place a metabolic burden on host cells, which eventually decreases cell growth, reduces plasmid stability [13], and consequently decreases protein productivity [14, 15]. Optimization of promoter strength has been also used to obtain a high protein titer. Depending on the properties or activities of desired proteins, toxicity, e.g., transcriptional optimization often increases overall protein titers [16,17,18,19]. Recently, the increase in bacterial growth rate or cell density has gained great interest to achieve higher production of proteins [20]. Several strategies for improving the cell density of Escherichia coli are summarized in Table 1.
In this study, we developed an E. coli strain that had a higher growth rate by optimizing the expression level of Hfq protein. Hfq protein was originally discovered as a host factor for phage Qβ (Hfq) replication in E. coli [34]. This chaperone, Hfq protein, is an abundant 11 kDa protein that forms a hexameric ring, can be found in more than 50% of bacterial species [35], and present widely in proteobacteria and firmicutes [36, 37]. There have been several studies indicating that Hfq promotes the ability of host cells to resist stresses such as oxidative stress and pH stress [38]. In addition, the role of Hfq in the control of growth-related genes has also been reported [39, 40]. Therefore, we hypothesized that the optimization of Hfq expression could increase bacterial growth rate.
Here, we designed diverse ribosome-binding site (RBS) sequences of the hfq gene to fine-tune its expression level by using a computational RBS design tool, RBSDesigner [41], or random screening. A conventional approach to control gene expression in E. coli is to use an inducible promoter or a synthetic promoter. The native promoter of hfq gene was replaced with an inducible promoter such as lac promoter and then its expression level can be controlled by the concentration of its inducer, isopropyl β-d-1-thiogalactopyranoside (IPTG) [42,43,44]. However, hfq transcription is driven from several promoters including three σ32-dependent heat shock promoters within the miaA open reading frame, PmutLHS, PmiaAHS, and P1hfqHS, and four σ70-dependent promoters, PmutL, PmiaA, P2hfq, and P3hfq [45]. Thus, replacement of the hfq promoters may result in disruption of cellular regulation systems, and which may lead to unexpected outcomes. Therefore, we designed various RBS sequences to control the level of Hfq protein expression without modifying its promoter. Thereafter, we investigated whether Hfq expression levels affect the growth of various E. coli strains.
Material and methods
Bacterial strains, plasmids, and antibiotics
Bacterial strains and plasmids used in this study are listed in Table S1. The media used for E. coli culture were Luria-Bertani broth (10 g Bacto Tryptone, 5 g yeast extract, and 10 g NaCl per L) [46] and cells were cultured in 250-mL Erlenmeyer flasks at 37 °C shaking incubator. Antibiotic was added to reach the following final concentration: chloramphenicol, 25 μg/mL.
Various hfq RBS sequences and plasmid construction
Different E. coli hfq variants with a variety of translation efficiencies were designed by RBSDesigner. The variants 1–4 were designed by RBSDesigner. To achieve a higher expression level, randomly generated RBS sequences were screened, and variants 5 and 6 were selected. Sequences of primers and genes are shown in Table S2.
Briefly, the amplified hfq gene from E. coli DH5α strain using HfqF_AatII and HfqR_XhoI primers was cloned into the corresponding restriction sites in pSC101 plasmid. A strong transcriptional terminator T1/TE was cloned downstream of the inserted hfq sequence. The hfq variant plasmids were constructed by inverse PCR on the pSC-WT hfq template. pSC-hfq-x-gfp had the gfp gene cloned downstream of the Hfq coding sequence using SpeI/XhoI and thereby their coding sequences were fused. To increase the structural flexibility between Hfq and GFP proteins, a stretch of Gly and Ser residues (“GS” linker) was used to connect hfq and gfp genes. All constructed RBS sequences of the hfq variants are listed in Table S3.
Growth measurement
The hfq-harboring cells of E. coli DH5α and hfq-deleted DH5α were cultured in a 250-mL flask, and their optical densities (OD600) were measured every 2 h. The growth of other E. coli strains was measured by using Biotek Synergy H1 plate reader (Winooski, VT, USA) at OD600 every 1 h for 24 h. All experiments were carried out in triplicate. E. coli cells harboring an empty vector were used as a control.
Growth rate (μ) was determined by the equation below [47]:
where (t2 – t1) denotes the interval between two sampling points at t1 and t2, and OD1 and OD2 denote the optical densities of cells sampled at times t1 and t2, respectively.
Fluorescence measurement
To confirm the expression level of hfq variants, the gfp gene was cloned downstream of the hfq coding sequence as a fusion protein. The fluorescence intensity of GFP was measured by Guava Easycyte HT BG Flow Cytometer (Millipore, Billerica, MA, USA) to determine the expression level of hfq variants. For fluorescence measurement, harvest cells were washed and resuspended in 1× phosphate-buffered saline (PBS) buffer at pH 7.4 (Sigma-Aldrich, St. Louis, USA), and subsequently diluted 1000 times with 1× PBS buffer. All experiments were performed in triplicate.
mRNA-seq analysis
To investigate the effect of hfq variants on cellular gene expressions, RNA-seq analysis was conducted. When the hfq variant 4 was introduced into hfq-deleted DH5α, it achieved the highest cell density and comparably high growth rate. The mRNAs in the cells were prepared for the analysis. mRNAs extracted from hfq-deleted DH5α and wild-type DH5α were used as a control. Three samples from individual cultures at stationary phase were used for RNA-seq analysis.
The number of reads for each gene was determined using HTSeq [48]. To reduce gene length bias, Reads Per Kilobase Million (RPKM) of each gene were calculated by dividing the total number of read count aligned to a gene by 1,000,000 and by the length of the gene in kilobase [49].
To identify differentially expressed genes (DEGs), genes were filtered as the following criteria: |log2(fold change)| > 2; p-value < 0.05; and normalized read count ≥ 10. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment of DEGs were analyzed by the Database for Annotation, Visualization and Integrated Discovery (DAVID) [50]. Enriched GO terms and KEGG pathways were selected by a p-value < 0.05.
The protein-protein interaction (PPI) network of DEGs was constructed using the STRING database [51] and Cytoscape [52]. Highly interconnected clusters were identified using Molecular Complex Detection (MCODE) [53].
Results and discussion
Role of Hfq protein in cell growth
There have been reports that the host factor for phage Qβ (Hfq) is closely related to cell growth [39]. For example, Hfq protein is associated with stress resistance in E. coli [38] and it showed to be crucial for cell survival under nutrient limitation [54]. Hfq protein is also known as a global regulator and is involved in post-transcriptional regulation by facilitating the interaction between small regulatory RNAs (sRNAs) and mRNAs [55, 56], and regulations of RNA stability [57, 58]. Hfq protein also controls the activity of several proteins involved in mRNA turnover by directly or indirectly interacting with RNase E [59], polynucleotide phosphorylase, and poly(A) polymerase [60,61,62,63]. Therefore, we hypothesized that increased Hfq protein expression may promote cell growth by enhancing resistance to environmental stresses. However, since over-expression of Hfq protein may disrupt cellular physiology by extensive protein expression alteration, which may retard cell growth, we also hypothesized that the expression of Hfq protein should be finely optimized.
Construction of various RBS sequences to diversify the expression level of Hfq protein in E. coli
We constructed six RBS variants of the hfq gene by a computational model and random screening (Table S3) to achieve a desired expression level of Hfq protein (Fig. 1a) and thereby increase cell growth. To avoid the disruption of inherent regulation of hfq transcription, its native promoters were used without modification (Fig. 1a). To maintain a low copy number of the hfq gene, the constructed genes were introduced into the pSC101 plasmid with only < 5 copies in E. coli.

Constructed hfq variants and their expression levels. a Constructed hfq gene structure and RBS variants. Four to six nucleotides upstream of Shine Dalgarno (SD) sequence and eight nucleotides downstream of SD designed or randomly generated. To avoid prolonged transcription downstream of the hfq gene, a transcription terminator was added to the gene construct. b GFP-fused Hfq expression levels. The green fluorescence intensity emitted from E. coli DH5α with a hfq variant was measured. Three replicates and mean ± SD. When tested with each other by t-test, there were no statistically significant differences only between the expression levels of variants 2 and 3 (p-value < 0.05)
To confirm the expression levels of the constructed hfq variants, the GFP coding sequence was fused at the C-terminus of the hfq coding sequence with a stretch of Gly and Ser residues (GS linker). The constructed variants show diverse expression levels of Hfq protein (Fig. 1b). The cells harboring hfq variant 6 displayed the highest expression level, whereas hfq variant 1 demonstrated the lowest expression level, which was 19.5-fold lower than that of hfq variant 6.
Fine optimization of Hfq expression increased E. coli growth
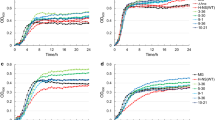
To effectively investigate the difference in cell growth, the hfq variants were introduced into hfq-deleted DH5α cells (Fig. 2a). For comparison, the variants were also introduced into wild-type DH5α cells (Fig. 2b). As shown in Fig. 2a, deletion of hfq gene dramatically reduced growth rate and maximum cell density (optical density measured at 600 nm): the measured growth rate was 0.422 ± 0.005 h− 1 and the highest OD was 3.36. Introduction of the hfq variants recovered the growth rate as well as maximum cell density. Variant 4 achieved the highest cell density (42.2% increase) in hfq-deleted DH5α cells. Though the variant 6 achieved the highest growth rate (94.3% increase), the variant 4 also achieved a comparably high growth rate (83.4% increase) (Fig. 2c and d). Regarding wild-type DH5α cells, since the cells already harbor an inherent hfq gene, the hfq variants could not significantly increase growth (Fig. 2b, c, and d). The growth rate and maximum OD of wild-type DH5α cells were 0.587 ± 0.001 h− 1 and 4.47, respectively. When Hfq expression was optimized in wild-type DH5α cells, the growth rate was increased by 12.1% by the variant 6 and the cell density was increased only by 4.5% by the same variant. These results represent that Hfq expression optimization is able to increase cell growth.

The effect of diverse hfq variants on the growth of wild-type DH5α and ∆hfq DH5α cells. Growth curves of ∆hfq DH5α cells (a) and wild-type DH5α cells (b) when an hfq variant was introduced. Relative growth rate increase (c) and relative cell density increase (d) by the hfq variants. To calculate the relative values, the measured values were compared with that of control cells that did not harbor any hfq variants. * denotes p-value < 0.05 and ** denotes p-value < 0.0001
Differentially expressed genes
To investigate the biological effect of Hfq expression on cellular physiology leading to the increase in bacterial growth, we performed RNA-seq analysis to discover up- and down-regulated genes of the three strains (hfq variant 4 in hfq-deleted DH5α, hfq-deleted DH5α, and wild-type DH5α). Hfq protein is an RNA-binding protein, specifically interacts with sRNAs to regulate mRNAs at post-transcription stage. Thus, RNA-seq analysis does not reveal the direct relationship between Hfq protein and transcripts. However, since transcripts encoding for proteins are actual players in cellular physiology, RNA-seq approach is useful to capture which cellular functions are implicated in increased cell growth.
Firstly, we compared the up- and down-regulated genes between hfq variant 4 in hfq-deleted DH5α and hfq-deleted DH5α. It was expected that hfq-deleted cells were an effective control to clearly reveal the underlying mechanisms related to improved cell growth. However, as explained in Text S1, the absence of the global regulator, Hfq protein, globally affected the cellular expression of genes even that are not related to cell growth, and as a result the RNA-seq analysis results misled to an inappropriate conclusion (Fig. S1).
Thus, we compared the expression patterns of hfq variant 4 in hfq-deleted DH5α and wild-type DH5α. The hfq-deleted DH5α cells harboring variant 4 showed a higher growth rate as well as a higher cell density than wild-type DH5α, and therefore wild-type DH5α cells already expressing the inherent hfq gene would be a better control to eliminate the global effect of Hfq protein. From the RNA-seq analysis, we found 110 differentially expressed genes (DEGs) with |log2(fold change)| > 2 and p-value < 0.05. Of the 110 genes, 64 genes were up-regulated and the remaining 46 genes were down-regulated. We performed enrichment analysis of GO terms and KEGG pathways within the DEGs to investigate physiological differences between variant 4 and wild-type DH5α E. coli. The DAVID bioinformatics tool [50] was utilized to identify the functions enriched within DEGs. The enriched terms are listed in Fig. 3a. In addition to the enrichment analyses, we generated a protein-protein interaction network of the DEGs using STRING database [64] and identified densely interconnected clusters. As shown in Fig. 3b, six highly interconnected clusters were identified by using Cytoscape [52] and MCODE [53]. The clusters were functionally similar to the enriched GO terms and KEGG pathways.

RNA-seq analysis results including enriched annotations and dense clusters. (a) Enriched GO terms and KEGG pathways within the DEGs. (b) Dense clusters constructed from the DEGs. Red and blue colors denote up- and down-regulated genes, respectively
As bacterial cells grow in LB broth, alkalinization occurs and cells are exposed to alkaline stress (Fig. S1C) [46]. Compared with wild-type E. coli, the cells harboring the variant 4 showed up-regulated biological processes such as “Arginine catabolic process to succinate” (GO:0019545, p-value = 2.79 × 10− 7), “Arginine catabolic process to glutamate” (GO:0019544, p-value = 2.79 × 10− 7), and “Arginine and proline metabolism” (eco00330, p-value = 9.23 × 10− 4). These processes convert molecules into acids instead of producing alkaline amines, and which could reduce the alkaline stress. Additionally, of the three up-regulated genes (aldB, gabD, and astD) involved in “Aldehyde dehydrogenase (NAD) activity” (GO:0004029, p-value = 7.0 × 10− 3), gabD encodes for a succinic semialdehyde dehydrogenase (SSDH) that plays an important role in γ-aminobutyric acid (GABA) shunt pathway to produce succinate. The astD encodes for succinylglutamic semialdehyde dehydrogenase involved in the arginine catabolism to glutamate. Aldehyde dehydrogenase B (aldB) was previously reported as an NADP+-dependent general detoxifying enzyme [65]. Besides, arginine is an important carbon and nitrogen source during starvation (stationary phase) [66], and the production of glutamate and succinate also fuels the tricarboxylic acid cycle (TCA) for energy generation.
The processes “Butanoate metabolism” (eco00650, p-value = 3.4 × 10− 3) and “Pentose and glucuronate interconversions” (eco00040, p-value = 7.9 × 10− 3) are carbohydrate metabolism pathways in E. coli. All the genes involved in “Butanoate metabolism” encode for the enzymes that catalyze the synthesis of metabolites such as fumarate (catalyzed by frdA, frdD, gabD), pyruvate (catalyzed by dmlA), and acetyl-CoA (catalyzed by yqeF), which eventually enter the TCA cycle. “Pentose and glucuronate interconversions” are responsible for the production of L-xylonate and L-lyxonate (non-oxidative branch), degradation of L-arabinose to form D-xylulose-5-phosphate, eventually entering the pentose phosphate pathway (PPP). The PPP is one of the primary sources for the generation of NADPH (reduced nicotinamide adenine dinucleotide phosphate), which is an essential electron donor, and serves as the reducing power for the biosynthesis of all major cell components [67, 68]. In addition, several processes related to cold and heat shock responses were down-regulated in the cells harboring the variant 4: “Response to cold” (GO:0009409, p-value = 1.3 × 10− 5) and “Response to heat” (GO:0009408, p-value = 5.5 × 10− 3). Since the cells were grown at an isothermal condition, 37 °C, the genes involved in temperature-dependent processes seemed to be down-regulated for better performance of cells.
Overall, GO and KEGG enrichment analyses and network analysis indicate that the variant 4 increased the growth of E. coli by producing more acidic molecules to reduce alkaline stress and by directing nutrient sources to the TCA cycle to generate energy and provide metabolites to essential pathways.
Application of hfq variants to promote the growth of other E. coli strains
In this study, optimization of Hfq protein expression was able to increase cell growth and it opens a new way to enhance bacterial cell growth. To prove the practical applicability of the strategy, we applied the constructed variants to optimize Hfq expression in various E. coli strains: one B strain (BL21 (DE3)) and four K12-derivative strains (JM109, TOP10, W3110, and MG1655). The B strain is widely used in industry to produce proteins, and the K12-derivative strains are commonly used for molecular biology studies.
Hfq expression optimization was able to improve growth rate in all strains (Fig. 4a), but not all strains showed improved cell density by the variants (Fig. 4b). For example, the growth rate of the industrial strain BL21 (DE3) was improved by 87.2% by variant 4 and this variant also increased cell density by 32.1%. For JM109, the variants 2 and 4 increased a growth rate by 67.2 and 80.6%, and also increased a cell density by 31.6 and 22.9%, respectively. For W3110, only variants of a higher expression level could enhance both cell growth rate and density. For MG1655, the variant 6 increased a growth rate by 23.1%, but its cell density was decreased by 4.8%. For TOP10 and MG1655, their cell densities were decreased by the variants though their growth rates were improved.

Cell density and growth rate of various E. coli strains harboring an hfq variant. The six hfq variants were introduced into the five wild-type E. coli strains and their relative increases of growth rate (a) and cell density (b) are shown. The relative values denote the percent increase or decrease compared with respective control cells harboring no hfq variants. * denotes p-value < 0.05 and ** denotes p-value < 0.0001
Conclusion
This study proves a strong correlation between Hfq expression and E. coli growth and that the optimization of Hfq expression improves E. coli growth via regulation of several pathways participating in cellular stress tolerance. Since the hfq protein is widely conserved in bacterial species, the strategy could be applied to other bacterial strains to enhance cell growth. This approach would be beneficial for constructing cell factories with slow-growing bacteria.
References
Schein CH. Production of soluble recombinant proteins in bacteria. Nat Biotechnol. 1989;7(11):1141–9.
Quianzon CC, Cheikh I. History of insulin. J Community Hosp Intern Med Perspect. 2012;2:2.
Sanchez-Garcia L, Martin L, Mangues R, Ferrer-Miralles N, Vazquez E, Villaverde A. Recombinant pharmaceuticals from microbial cells: a 2015 update. Microb Cell Factories. 2016;15:33.
Jozala AF, Geraldes DC, Tundisi LL, Feitosa VA, Breyer CA, Cardoso SL, et al. Biopharmaceuticals from microorganisms: from production to purification. Braz J Microbiol. 2016;47(Suppl 1):51–63.
Rezaei M, Zarkesh-Esfahani SH. Optimization of production of recombinant human growth hormone in Escherichia coli. J Res Med Sci. 2012;17(7):681–5.
Ferrer-Miralles N, Domingo-Espín J, Corchero JL, Vázquez E, Villaverde A. Microbial factories for recombinant pharmaceuticals. Microb Cell Factories. 2009;8(1):17.
Baeshen NA, Baeshen MN, Sheikh A, Bora RS, Ahmed MM, Ramadan HA, et al. Cell factories for insulin production. Microb Cell Factories. 2014;13:141.
Yoshimoto H, Fukushige T, Yonezawa T, Sone H. Genetic and physiological analysis of branched-chain alcohols and isoamyl acetate production in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2002;1:501–8.
Lee SK, Chou H, Ham TS, Lee TS, Keasling JD. Metabolic engineering of microorganisms for biofuels production: from bugs to synthetic biology to fuels. Curr Opin Biotechnol. 2008;19(6):556–63.
Kalscheuer R, Stölting T, Steinbüchel A. Microdiesel: Escherichia coli engineered for fuel production. Microbiology. 2006;152:2529–36.
Ramos CR, Abreu PA, Nascimento AL, Ho PL. A high-copy T7 Escherichia coli expression vector for the production of recombinant proteins with a minimal N-terminal his-tagged fusion peptide. Braz J Med Biol Res. 2004;37(8):1103–9.
Son YJ, Ryu AJ, Li L, Han NS, Jeong KJ. Development of a high-copy plasmid for enhanced production of recombinant proteins in Leuconostoc citreum. Microb Cell Factories. 2016;15(1):12.
Glick BR. Metabolic load and heterologous gene expression. Biotechnol Adv. 1995;13(2):247–61.
Bentley WE, Mirjalili N, Andersen DC, Davis RH, Kompala DS. Plasmid-encoded protein: the principal factor in the “metabolic burden” associated with recombinant bacteria. Biotechnol Bioeng. 1990;35(7):668–81.
Birnbaum S, Bailey JE. Plasmid presence changes the relative levels of many host cell proteins and ribosome components in recombinant Escherichia coli. Biotechnol Bioeng. 1991;37(8):736–45.
Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol. 1986;189(1):113–30.
Tolentino GJ, Meng SY, Bennett GN, San KY. A pH-regulated promoter for the expression of recombinant proteins in Escherichia coli. Biotechnol Lett. 1992;14(3):157–62.
Tegel H, Ottosson J, Hober S. Enhancing the protein production levels in Escherichia coli with a strong promoter. FEBS J. 2011;278(5):729–39.
Briand L, Marcion G, Kriznik A, Heydel JM, Artur Y, Garrido C, et al. A self-inducible heterologous protein expression system in Escherichia coli. Sci Rep. 2016;6:33037.
Shiloach J, Fass R. Growing E. coli to high cell density-a historical perspective on method development. Biotechnol Adv. 2005;23(5):345–57.
Matsui T, Yokota H, Sato S, Mukataka S, Takahashi J. Pressurized culture of Escherichia coli for a high-concentration. Agr Biol Chem Tokyo. 1989;53(8):2115–20.
Korz DJ, Rinas U, Hellmuth K, Sanders EA, Deckwer WD. Simple fed-batch technique for high cell density cultivation of Escherichia coli. J Biotechnol. 1995;39(1):59–65.
Markl H, Zenneck C, Dubach AC, Ogbonna JC. Cultivation of Escherichia coli to high cell densities in a dialysis reactor. Appl Microbiol Biotechnol. 1993;39(1):48–52.
Nakano K, Rischke M, Sato S, Markl H. Influence of acetic acid on the growth of Escherichia coli K12 during high-cell-density cultivation in a dialysis reactor. Appl Microbiol Biotechnol. 1997;48(5):597–601.
Wong M, Wright M, Woodley JM, Lye GJ. Enhanced recombinant protein synthesis in batch and fed-batch Escherichia coli fermentation based on removal of inhibitory acetate by electrodialysis. J Chem Technol Biotechnol. 2009;84(9):1284–91.
Zawada J, Swartz J. Maintaining rapid growth in moderate-density Escherichia coli fermentations. Biotechnol Bioeng. 2005;89(4):407–15.
Riesenberg D. High-cell-density cultivation of Escherichia coli. Curr Opin Biotechnol. 1991;2(3):380–4.
Bauer KA, Ben-Bassat A, Dawson M, de la Puente VT, Neway JO. Improved expression of human interleukin-2 in high-cell-density fermentor cultures of Escherichia coli K-12 by a phosphotransacetylase mutant. Appl Environ Microbiol. 1990;56(5):1296–302.
Farmer WR, Liao JC. Reduction of aerobic acetate production by Escherichia coli. Appl Environ Microbiol. 1997;63(8):3205–10.
Kim JY, Cha HJ. Down-regulation of acetate pathway through antisense strategy in Escherichia coli: improved foreign protein production. Biotechnol Bioeng. 2003;83(7):841–53.
Nakamura T, Tanaka M, Maruyama A, Higashi Y, Kurusu Y. A nonconserved carboxy-terminal segment of GroEL contributes to reaction temperature. Biosci Biotechnol Biochem. 2004;68(12):2498–504.
Gupta P, Aggarwal N, Batra P, Mishra S, Chaudhuri TK. Co-expression of chaperonin GroEL/GroES enhances in vivo folding of yeast mitochondrial aconitase and alters the growth characteristics of Escherichia coli. Int J Biochem Cell Biol. 2006;38(11):1975–85.
Lee JH, Heo MA, Seo JH, Kim JH, Kim BG, Lee SG. Improving the growth rate of Escherichia coli DH5alpha at low temperature through engineering of GroEL/S chaperone system. Biotechnol Bioeng. 2008;99(3):515–20.
Kajitani M, Ishihama A. Identification and sequence determination of the host factor gene for bacteriophage Q beta. Nucleic Acids Res. 1991;19(5):1063–6.
Sun X, Zhulin I, Wartell RM. Predicted structure and phyletic distribution of the RNA-binding protein Hfq. Nucleic Acids Res. 2002;30(17):3662–71.
Sittka A, Pfeiffer V, Tedin K, Vogel J. The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol Microbiol. 2007;63(1):193–217.
Dambach M, Irnov I, Winkler WC. Association of RNAs with Bacillus subtilis Hfq. PLoS One. 2013;8(2):e55156.
Chao Y, Vogel J. The role of Hfq in bacterial pathogens. Curr Opin Microbiol. 2010;13(1):24–33.
Ali Azam T, Iwata A, Nishimura A, Ueda S, Ishihama A. Growth phase-dependent variation in protein composition of the Escherichia coli nucleoid. J Bacteriol. 1999;181(20):6361–70.
Kajitani M, Kato A, Wada A, Inokuchi Y, Ishihama A. Regulation of the Escherichia coli hfq gene encoding the host factor for phage Q beta. J Bacteriol. 1994;176(2):531–4.
Na D, Lee D. RBSDesigner: software for designing synthetic ribosome binding sites that yields a desired level of protein expression. Bioinformatics. 2010;26(20):2633–4.
Morita T, Aiba H. Mechanism and physiological significance of autoregulation of the Escherichia coli hfq gene. RNA. 2019;25(2):264–76.
Bhatt S, Egan M, Ramirez J, Xander C, Jenkins V, Muche S, et al. Hfq and three Hfq-dependent small regulatory RNAs-MgrR, RyhB and McaS-coregulate the locus of enterocyte effacement in enteropathogenic Escherichia coli. Pathog Dis. 2017;75(1):ftw113.
Vassilieva IM, Rouzanov MV, Zelinskaya NV, Moll I, Blasi U, Garber MB. Cloning, purification, and crystallization of a bacterial gene expression regulator-Hfq protein from Escherichia coli. Biochemistry (Mosc). 2002;67(11):1293–7.
Tsui HC, Feng G, Winkler ME. Transcription of the mutL repair, miaA tRNA modification, hfq pleiotropic regulator, and hflA region protease genes of Escherichia coli K-12 from clustered Eσ32-specific promoters during heat shock. J Bacteriol. 1996;178(19):5719–31.
Sezonov G, Joseleau-Petit D, D'Ari R. Escherichia coli physiology in Luria-Bertani broth. J Bacteriol. 2007;189(23):8746–9.
Widdel F. Theory and measurement of bacterial growth. Di dalam Grundpraktikum Mikrobiologie. 2007;4(11):1–11.
Anders S, Pyl PT, Huber W. HTSeq-a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8.
Huangda W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–8.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC bioinformatics. 2003;4:2.
Gottesman S, McCullen CA, Guillier M, Vanderpool CK, Majdalani N, Benhammou J, et al. Small RNA regulators and the bacterial response to stress. Cold Spring Harb Perspect Med. 2006;71:1.
Brennan RG, Link TM. Hfq structure, function and ligand binding. Curr Opin Microbiol. 2007;10(2):125–33.
Valentin-Hansen P, Eriksen M, Udesen C. The bacterial Sm-like protein Hfq: a key player in RNA transactions. Mol Microbiol. 2004;51(6):1525–33.
Caron MP, Lafontaine DA, Masse E. Small RNA-mediated regulation at the level of transcript stability. RNA Biol. 2010;7(2):140–4.
Hernández-Arranz S, Sánchez-Hevia D, Rojo F, Moreno R. Effect of Crc and Hfq proteins on the transcription, processing, and stability of the Pseudomonas putida CrcZ sRNA. RNA (New York, NY). 2016;22(12):1902–17.
Vytvytska O, Jakobsen JS, Balcunaite G, Andersen JS, Baccarini M, von Gabain A. Host factor I, Hfq, binds to Escherichia coli ompA mRNA in a growth rate-dependent fashion and regulates its stability. Proc Natl Acad Sci U S A. 1998;95(24):14118–23.
Folichon M, Arluison V, Pellegrini O, Huntzinger E, Regnier P, Hajnsdorf E. The poly(a) binding protein Hfq protects RNA from RNase E and exoribonucleolytic degradation. Nucleic Acids Res. 2003;31(24):7302–10.
Vargas-Blanco DA, Shell SS. Regulation of mRNA stability during bacterial stress responses. Front Microbiol. 2020;11:2111.
Hajnsdorf E, Régnier P. Host factor Hfq of Escherichia coli stimulates elongation of poly(a) tails by poly(a) polymerase I. Proc Natl Acad Sci U S A. 2000;97(4):1501–5.
Folichon M, Allemand F, Régnier P, Hajnsdorf E. Stimulation of poly(a) synthesis by Escherichia coli poly(a) polymerase I is correlated with Hfq binding to poly(a) tails. FEBS J. 2005;272(2):454–63.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52.
Ho KK, Weiner H. Isolation and characterization of an aldehyde dehydrogenase encoded by the aldB gene of Escherichia coli. J Bacteriol. 2005;187(3):1067–73.
Neidhardt FC. Escherichia coli and Salmonella. Typhimurium Cell Mol Biol. 1987.
Christodoulou D, Link H, Fuhrer T, Kochanowski K, Gerosa L, Sauer U. Reserve flux capacity in the pentose phosphate pathway enables Escherichia coli’s rapid response to oxidative stress. Cell Syst. 2018;6(5):569–78.
Kohen R, Nyska A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol. 2002;30(6):620–50.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant, funded by the Korean government (MSIT) (No. NRF-2018R1A5A1025077). This research was supported by the Chung-Ang University Young Scientist Scholarship in 2020.
Author information
Authors and Affiliations
Contributions
PV and DN designed the study, wrote and edited the manuscript. PV conducted all experiments of this study. JR conducted some experiments. HL supervised the study and assisted in drafting the manuscript. DN participated in data analysis and interpretation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The content of the manuscript has been approved by all the authors.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Bacterial strains and plasmids used in this study. Table S2. Oligonucleotide primers and PCR templates used in this study. Table S3. Constructed hfq variants. Text S1. RNA-seq analysis results when hfq-deleted cell was used as a control. Figure S1. RNA-seq analysis results and investigation of the association of acid resistance with cell growth.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
VO, P.N.L., LEE, HM., REN, J. et al. Optimized expression of Hfq protein increases Escherichia coli growth. J Biol Eng 15, 7 (2021). https://doi.org/10.1186/s13036-021-00260-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13036-021-00260-x