
Abstract
Background
Multiple acyl-CoA dehydrogenase deficiency (MADD) is an autosomal recessive disorder resulting from pathogenic variants in three distinct genes, with most of the variants occurring in the electron transfer flavoprotein-ubiquinone oxidoreductase gene (ETFDH). Recent evidence of potential founder variants for MADD in the South African (SA) population, initiated this extensive investigation. As part of the International Centre for Genomic Medicine in Neuromuscular Diseases study, we recruited a cohort of patients diagnosed with MADD from academic medical centres across SA over a three-year period. The aim was to extensively profile the clinical, biochemical, and genomic characteristics of MADD in this understudied population.
Methods
Clinical evaluations and whole exome sequencing were conducted on each patient. Metabolic profiling was performed before and after treatment, where possible. The recessive inheritance and phase of the variants were established via segregation analyses using Sanger sequencing. Lastly, the haplotype and allele frequencies were determined for the two main variants in the four largest SA populations.
Results
Twelve unrelated families (ten of White SA and two of mixed ethnicity) with clinically heterogeneous presentations in 14 affected individuals were observed, and five pathogenic ETFDH variants were identified. Based on disease severity and treatment response, three distinct groups emerged. The most severe and fatal presentations were associated with the homozygous c.[1067G > A];c.[1067G > A] and compound heterozygous c.[976G > C];c.[1067G > A] genotypes, causing MADD types I and I/II, respectively. These, along with three less severe compound heterozygous genotypes (c.[1067G > A];c.[1448C > T], c.[740G > T];c.[1448C > T], and c.[287dupA*];c.[1448C > T]), resulting in MADD types II/III, presented before the age of five years, depending on the time and maintenance of intervention. By contrast, the homozygous c.[1448C > T];c.[1448C > T] genotype, which causes MADD type III, presented later in life. Except for the type I, I/II and II cases, urinary metabolic markers for MADD improved/normalised following treatment with riboflavin and L-carnitine. Furthermore, genetic analyses of the most frequent variants (c.[1067G > A] and c.[1448C > T]) revealed a shared haplotype in the region of ETFDH, with SA population-specific allele frequencies of < 0.00067–0.00084%.
Conclusions
This study reveals the first extensive genotype–phenotype profile of a MADD patient cohort from the diverse and understudied SA population. The pathogenic variants and associated variable phenotypes were characterised, which will enable early screening, genetic counselling, and patient-specific treatment of MADD in this population.
Similar content being viewed by others
Background
Multiple acyl-CoA dehydrogenase deficiency (MADD; ORPHAcode 26791), also known as glutaric aciduria type II (GAII), is an autosomal recessive disorder that occurs at a global and ethnically variable prevalence of approximately 1 in 200,000 births [1]. It is caused by pathogenic variants (at least 400) in the ETFA, ETFB and ETFDH genes, which encode for the alpha and beta subunits of the electron transfer flavoprotein (ETF) and ETF-ubiquinone oxidoreductase (ETFQO; EC:1.5.5.1), respectively. Collectively, ETF and ETFQO are responsible for re-oxidising reduced mitochondrial flavin adenine dinucleotide (FADH2), which in turn sustains mitochondrial fatty acid β-oxidation (FAO), amino acid catabolism and choline metabolism [2, 3].
Clinically, MADD may be divided into three phenotypes. The first and second (MADD types I and II, respectively) are characterised by the neonatal-onset of severe and often fatal symptoms with multi-system involvement (including leukodystrophy, hypotonia, cardiomyopathy, hepatomegaly, and renal abnormalities), as well as hypoglycaemia, hyperammonaemia, and metabolic acidosis (with/without ketosis) [4,5,6]. MADD types I and II may be distinguished from each other by the presence (type I) or absence (type II) of congenital abnormalities and are caused, at essentially equal frequencies, by pathogenic variants in ETFA, ETFB, and ETFDH [5]. By contrast, the third phenotype (MADD type III) has a later-onset and presents with milder/delayed symptoms which are highly heterogeneous. Such symptoms may include recurrent/intermittent episodes of lethargy and vomiting; muscle, cardiac and/or liver involvement; as well as lipid storage myopathy, hypoglycaemia, metabolic acidosis, and/or hyperammonaemia. MADD type III is reported to be caused mainly by ETFDH variants and is also known as riboflavin (Rb)-responsive MADD owing to its amenability to treatment [5, 7]. To estimate the burden of disease, a disease severity scoring system (MADD-DS3; MADD-disease severity 3) may be used [6]. Moreover, disease-severity is often linked to the patient’s response to treatment, which includes a high-caloric diet with fat and protein restriction, the strict avoidance of fasting, and supplementation with L-carnitine and Rb (as first-line treatment) [8, 9].
At present, South Africa (SA) does not have a compulsory newborn screening (NBS) programme. Instead, the biochemical diagnosis of MADD follows (i) a clinical presentation in symptomatic cases, or (ii) an abnormal NBS profile in asymptomatic cases—during which MADD is defined as a secondary condition. Symptomatic patients undergo a thorough metabolic work-up including (i) urinary organic acids, (ii) plasma/serum acylcarnitines, and (iii) plasma/serum amino acids and display a unique metabolic fingerprint as described in a recent review by Mereis et al. [3]. As with other inherited metabolic disorders, final confirmation of the diagnosis is obtained by genetic analysis.
In SA, as with most understudied populations, pathogenic variant screening for inherited metabolic disorders is less common. Such screening is, however, offered for a small selection of disorders (including glutaric aciduria type I, isovaleric acidaemia, galactosemia, and MPV17-hepatocerebral mitochondrial DNA depletion syndrome) following the comprehensive description of cohorts in literature. Currently, substantial knowledge of the genotype–phenotype and biochemical profiles, and the subsequent response to treatment are not available for MADD in the different SA populations. Prior to this study, a novel pathogenic ETFDH variant (c.[1067G > A], p.[Gly356Glu]) and one previously described pathogenic variant (c.[1448C > T], p.[Pro483Leu]) were reported to result in MADD in the White SA population [10]. In the homozygous state, these variants cause MADD types I and III, respectively, and it was proposed by van der Westhuizen et al. [10] that the prevalence of c.[1067G > A] in this SA population could possibly be the result of a founder effect. In our study, we aimed to address these hypotheses and other limitations by investigating a cohort of 14 recruited MADD patients, under the auspices of the International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD). We extensively characterised the clinical phenotype, biochemical profile, and genetics of MADD in SA, and provide knowledge that will contribute to its timely diagnosis, clinical management, and therapeutic intervention.
Results and discussion
Cohort
Over a period of three years and using a retrospective and prospective recruitment strategy at three different centres across SA, 12 apparently unrelated families (ten of White SA and two of mixed ethnicity) with 14 clinically affected patients (five males and nine females) were recruited. All patients were born to non-consanguineous parents and disease onset ranged from birth to 41 years, with four deaths recorded (at 9 days, 14 days, 3 months, and 23 years, respectively). Where possible, a consistent clinical re-evaluation was conducted according to ICGNMD guidelines; for most of the cases, biological samples acquired during acute metabolic presentation (n = 12/14) and following therapeutic management (n = 13/14) were collected/available and could be re-analysed (for the remainder, the original data were used, where available).
It should be noted that SA patients of African and Indian ethnicity have previously been diagnosed with MADD on a biochemical level at the Centre for Human Metabolomics [North-West University (NWU)] and the NHLS Inherited Metabolic Disease Molecular Laboratory [University of Cape Town (UCT)]—the two SA facilities where metabolic testing is offered as a main service (unpublished data). However, while all possible efforts were made to include patients of these population groups, they were regrettably lost to follow-up prior to the study’s onset and an opportunity for subsequent genetic confirmation did not present itself. Moreover, no new cases were identified in these population groups during this study. We therefore recognise a potential referral bias within the population groups included. It is our hope that this problem can be addressed by metabolic and genetic NBS in the future.
Clinical, biochemical, and genetic features
Overall, the SA MADD cohort displayed heterogeneous clinical presentations (Table 1 and Additional file 1), together with the characteristic diagnostic urinary metabolites associated with MADD (Table 2 and Additional file 2). While plasma/serum acylcarnitine and amino acid findings are typically used for diagnosis, this matrix was limited for the majority of the patients (sample collection and analyses were conducted over a 30-year period) and consequently, the levels of these metabolites in the urine are reported instead. The variants identified, their location in ETFQO, and their pathogenicity, according to the American College of Medical Genetics and Genomics (ACMG) criteria, are described in Table 3 [11]. The elucidation of these variants in the SA populations now enable rapid screening methods which were not available before, thereby allowing the timely confirmation of the disorder (in hours) in symptomatic patients. This prompt identification of MADD-related variants can be used to aid the reproductive choices of carrier/affected patients and is especially valuable for prenatal testing and the testing of neonates who may require immediate therapy following birth (i.e., those affected with MADD type I/II). In all recruited cases, a resolved genotype–phenotype result was obtained. Three distinct groups were observed, based on disease severity and response to treatment, as discussed below.
Severe, Rb-unresponsive MADD patient group
The first group included three patients (P6, P8 and P12), who presented with the hallmark features of neonatal-onset MADD. Clinically, their symptoms were severe and progressive, and patients were metabolically and phenotypically unresponsive to treatment with Rb. The first case was a male neonate (P6), born prematurely (at 38 weeks) to parents of White SA ethnicity, who presented within the first week of life with acute metabolic decompensation. Key clinical features included congenital cardiac abnormalities (pulmonary stenosis, a patent foramen ovale with a right to left shunt, and a large atrial septum aneurysm) and convulsions with early neonatal death on day 9 of life. Metabolic profiling indicated the typical MADD biochemical fingerprint [12], including dicarboxylic aciduria, increased 2-hydroxyglutaric acid, elevated glycine conjugates (short-, short-branched-, and medium-chain-related), increased disease-associated acylcarnitine conjugates, as well the characteristic increase in sarcosine on the amino acid profile. Both FAO and branched-chain amino acid catabolism were affected, correlating with previous studies on severe MADD cases [6]. The patient displayed a MADD-DS3 score of 30, further supporting a diagnosis of MADD type I. A then novel homozygous variant—c.[1067G > A] (p.[Gly356Glu])—with in silico and structural evidence of pathogenicity (ACMG classification: likely pathogenic) was identified in this neonate [10].
The second patient was a female infant (P8), born at term to parents of mixed ethnicity, who presented at birth with metabolic acidosis, hypotonia and feeding difficulties. Disease-specific metabolic markers were similar to those of P6, and the MADD-DS3 score of 21 was high. The patient succumbed at the age of three months. The homozygous c.[1067G > A] variant was subsequently identified; however, based on the lack of any reported congenital features (absent post-mortem examination), a diagnosis of MADD type I/II was given. The third case was a female neonate (P12), born at term to parents of mixed ethnicity. A urinary organic acid profile typical of MADD was confirmed by the hospital that made the diagnosis (no residual urine collected before or after treatment was available to re-analyse for the purpose of this study). Clinical features included metabolic acidosis, hypoglycaemia, hyperammonaemia, pancytopaenia, acute kidney injury, hyponatremia, and hypocalcaemia, and the patient succumbed at 14 days of age. Based on the absence of any reported congenital features (no post-mortem examination) and the high MADD-DS3 score of 24, a diagnosis of MADD type I/II was given. Whole exome sequencing (WES) and segregation analysis revealed that the patient was compound heterozygous for c.[976G > C];c.[1067G > A] (p.[Gly326Arg];p.[Gly356Glu]). Variant c.[976G > C] is classified as a likely pathogenic variant of unknown significance according to the ACMG criteria and, to our knowledge, has been reported to occur in only one late-onset case of Chinese ethnicity by Xi et al. [13] as compound heterozygous with the common variant, c.[250G > A] (p.[Ala84Thr]; ACMG classification: pathogenic). By contrast, the c.[1067G > A] variant has only been encountered in the SA population to date [10].
Considering the treatment-unresponsive metabolic profile (P6 and P12) and rapid clinical deterioration of patients P6, P8 and P12, it may be inferred that c.[1067G > A] is a highly pathogenic variant. Its presence on both alleles, or its bi-allelic combination with another variant affecting the same protein domain (ubiquinone-binding domain) of ETFQO, appears to lead to insufficient enzymatic compensation for adequate ETFQO activity. While it is evident that L-carnitine supplementation facilitated the formation of disease specific acylcarnitine conjugates, we hypothesise that the homozygous c.[1067G > A];c.[1067G > A] and compound heterozygous c.[976G > C];c.[1067G > A] genotypes result in an ETFQO protein of which the folding cannot be sufficiently rescued/stabilised by Rb treatment.
Moderate, variably Rb-responsive MADD patient group
The second group included eight patients who presented with moderate, heterogeneous phenotypes, all showing a varying response to treatment. The onset of symptoms was observed in the neonatal period (P1), infancy (P2, P7, P9 and P10), as well as childhood (P3, P11 and P13), and all patients displayed the characteristic clinical features of MADD. These included metabolic decompensation (n = 5), muscle weakness (n = 4), muscle pain (n = 3), hypotonia (n = 5), neck flexor weakness (n = 5), susceptibility to fatigue (n = 2), restrictive ventilatory defect (n = 1), gastrointestinal involvement (n = 6), elevated creatine kinase (CK) (n = 2), recurrent infections (n = 2), lethargy (n = 2), cognitive disability (n = 2), delayed gross motor development (n = 2), migraine/paroxysmal headache (n = 3), seizures (n = 2), coma (n = 3), skeletal involvement (n = 2), and liver dysfunction (n = 4). Uncommon symptoms included ketosis at the time of metabolic crisis (n = 5) and Beevor’s sign (n = 1). Owing to the availability of data, the baseline urine organic acids of only five of the eight patients are reported, and the data of P7 represent the urinary organic acids present upon considerable decompensation near the time of demise.Footnote 1 At first presentation, patients P1, P7 and P9–P11 displayed an increase (to a variable extent) in the diagnostic urine organic acid markers associated with MADD, albeit less pronounced than that of P6, P8 and P12. Most of these patients had increased concentrations of urinary glutaric acid, ethylmalonic acid, dicarboxylic acids, and 2-hydroxyglutaric acid. The excretion of N-hexanoylglycine and, to a variable extent, branched-chain-related glycine conjugates were mostly observed. Moreover, all eight patients displayed increased disease-associated urinary acylcarnitines and elevated sarcosine. The biomarker assessment correlated with previous observations in moderate cases where FAO seems to be initially/mostly affected and branched-chain amino acid catabolism is influenced to a lesser extent [6]. It is important to note that the metabolic profiling was greatly dependent on the time of sample collection and that P3 and P9 had received L-carnitine treatment from an early stage in their lives due to the prior diagnosis of a sibling with MADD. Apart from P7, the clinical symptoms, together with most of the urine metabolites, improved upon dietary adjustment in combination with treatment with L-carnitine (P9 and P10), L-carnitine and Rb (P2, P3, P11 and P13), or L-carnitine, Rb, and coenzyme Q10 (P1). Carnitine conjugation indicated that accumulating acyl-CoAs were being detoxified via the carnitine transportation system, which likely explains the less prominent MADD organic acid signature observed. By contrast, the metabolic response to treatment of P7—who succumbed to a stroke at the age of 23 years—was more comparable to that of severe MADD, a finding that correlated with the severity of the clinical presentation as summarised in Table 1 and Additional file 1.
WES and segregation analysis by Sanger sequencing revealed four compound heterozygous variants in this group of White SA-ethnicity patients. These included: (i) c.[740G > T];c.[1448C > T] (p.[Gly247Val];p.[Pro483Leu]) in P13, (ii) c.[287dupA*];c.[1448C > T] (p.[Asp97Glyfs*24];p.[Pro483Leu]) in siblings P2 and P3, and (iii) c.[1067G > A];c.[1448C > T] (p.[Gly356Glu];p.[Pro483Leu]) in P1, P7, P9, P10, and P11. The novel c.[287dupA*] variant affects the third exon of ETFDH, leading to a premature stop codon, and is classified as likely pathogenic according to the ACMG criteria. The c.[740G > T] variant shares the same classification as c.[287dupA*], and has been reported only once before as a compound heterozygous variant along with the likely pathogenic c.[389A > T] (p.[Asp130Val]) variant in a late-onset MADD case of Chinese ethnicity [14]. All variants identified in this group encode for highly conserved amino acids.
Once again, the MADD-DS3 scores confirmed that the disease burden increases when the c.[1067G > A] variant is present. This finding is corroborated by the level of steady state ETFQO protein in skin fibroblasts of P5 (c.[1448C > T];c.[1448C > T]) and P9 (c.[1067G > A];c.[1448C > T]), which show a 55% and 73% decrease, respectively, in comparison to fibroblasts from healthy controls (Additional file 3). Similarly, van der Westhuizen et al. [10] reported an 83% decrease in the steady state level of ETFQO in muscle from a patient with a compound heterozygous c.[1067G > A];c.[1448C > T] genotype, when compared to a healthy control. By contrast to the severe, Rb-unresponsive MADD patient group, four of the five patients affected by the c.[1067G > A] variant (P9, P10, P11 and P1) in this group were found to be very amenable to treatment. It is, therefore, reasonable to conclude that when c.[1067G > A] is encountered as a compound heterozygous variant along with a variant which affects a different protein domain of ETFQO (e.g., c.[1448C > T]), sufficient enzymatic compensation occurs which may allow for adequate ETFQO activity. Based on the onset of disease, a diagnosis of either MADD type II (P1) or type III (P2, P3, P7, P9, P10, P11 and P14) was given to the patients in this group.
Mild, Rb-responsive MADD patient group
The final group included three patients (P4, P5 and P14) of White SA ethnicity, who presented later in life with mild and non-progressive (treatment-related) phenotypes. Clinically, their symptoms were heterogenous, but to some extent characteristic of MADD. The disease presentation included metabolic decompensation (n = 2), muscle weakness (n = 3), neck flexor weakness (n = 2), gastrointestinal involvement as the disease progressed (n = 1), elevated CK (n = 2), lethargy (n = 1), encephalopathy (n = 1), cerebral white matter abnormalities (n = 1), and liver dysfunction (n = 1), with two patients exhibiting ketosis. Initially, statin-induced myositis was suspected in P5 until hepatic features, including lipid deposits on the liver biopsy and raised transaminases prompted a metabolic work-up. P5 and P14 displayed urine metabolites associated with MADD, including dicarboxylic aciduria, raised 2-hydroxyglutaric acid (P5), acylglycine conjugates (with less prominent branched-chain-related conjugates), short- and medium-chain acylcarnitines as well as the presence of sarcosine on the amino acid profile. As indicated earlier in the moderate MADD group, FAO tends to be more affected compared to the branched-chain amino acid catabolism in the less severe cases [6], which correlated with our findings. Literature shows that plasma/serum acylcarnitine profiling is typically most informative when diagnosing a late-onset MADD case, as organic acid profiling may only be remarkable at the time of a metabolic crisis or catabolic status induced by fasting. However, acylcarnitine assessments have been inconclusive in some cases, particularly if the patient has insufficient free carnitine available to promote conjugation [5, 12]. The latter has also led to false negative NBS results in mild MADD cases, as reported by Lin et al. [15]. In our study, P14 showed a free carnitine concentration below the limit of detection and normal butyryl/isobutyryl-, isovaleryl- and glutarylcarnitine levels in plasma/serum (results not shown). Interestingly, this patient presented with severe ketosis, as well as a prominent increase in 2-hydroxyglutaric acid and cis-4-decenedioic acid with unremarkable increases in glutaric acid and ethylmalonic acid. These inconsistent urinary organic acid findings corroborate the observations of Goodman et al. [16] and support their suggestion to discontinue the use of the term “Glutaric aciduria type II,” as it may be diagnostically misleading. In addition, our data indicate that mild cases of MADD may be missed or incorrectly diagnosed (e.g., as a different fatty acid oxidation disorder) on a biochemical level, depending on the metabolic (i.e., anabolic/catabolic status due to the time of sample collection) and systemic free carnitine status of the patient. Consequently, this study emphasises the metabolite variation within this disease group and advocates for repeat testing if the clinical presentation and routine chemistry are suggestive of MADD.
Following dietary adjustment and treatment with L-carnitine, Rb and coenzyme Q10 (P5 and P14), the clinical and biochemical aberrations of P14 essentially normalised. P4, however, presented with only increased ethylmalonic acid. This patient, the mother of P2 and P3, underwent metabolic testing following the children’s diagnosis and was on L-carnitine treatment at the time of sample collection. Clinical-biochemical improvements, specifically after therapeutic intervention, have been observed in several cases of late-onset MADD, and our clinical and metabolic results (although only three cases were included) strongly correlate with previous investigations [5, 17].
Genetic analyses revealed that all three patients had a homozygous genotype for the known pathogenic c.[1448C > T] (p.[Pro483Leu]) variant. This genotype (c.[1448C > T]; c.[1448C > T]) has been reported in numerous other cases that, similarly to this group, presented with adult-onset, Rb-responsive MADD [10, 17]. The Rb-responsive nature of this variant is further corroborated in a study by Cornelius et al. [18], in which it was shown that ETFQO activity in c.[1448C > T]-modified HEK293 cells could be restored from ~ 45 to ~ 85% (corresponding to an increase of ~ 50 to ~ 80% steady-state ETFQO) when moderately Rb-deficient cultures were treated with a saturating concentration of Rb. Therefore, considering the literature, together with the time of onset, severity and treatment response of this group, a diagnosis of MADD type III was given to P4, P5 and P14, despite moderate MADD-DS3 scores.
Allele frequency spectrum and haplotypes
To determine the allele frequency of the variants identified, PCR–RFLP analysis was used to screen the two most frequently occurring variants, c.[1067G > A] and c.[1448C > T], in the four largest population groups in SA. The study yielded no homozygous or heterozygous genotypes for any of the variants in any of the population groups assessed so that the allele frequencies of the population groups investigated were calculated as < 0.00067% (African, White SA, and Indian ethnicity) or < 0.00084% (mixed ethnicity) (Table 3). All five variants identified in the cohort were subsequently compared to gnomAD (v2.1.1) [19] and the Human Heredity and Health in Africa (H3Africa) project [20]. Of these variants, only two were recorded on gnomAD, displaying exome allele frequencies of < 0.0001% (c.[740G > T] and c.[1448C > T]); hitherto, none of the variants has been identified in the H3Africa data. The absence of the variants in the above-mentioned population databases, together with the frequency at which they were identified in the SA cohort, indicates with high probability the pathogenicity of these five variants and supports their causative role in MADD.
Haplotyping was performed on those patients from whom sufficient DNA could be obtained. Upon recruitment, patients and their families were invited to self-report their ethnicity and region of birth. Consequently, haplotyping was performed on ten patients of White SA ethnicity (P1–P6, P9–P11 and P13) and one patient of mixed ethnicity (P8) born in various geographical regions across SA (including the central, north-eastern, north-western, south-eastern, and south-western provinces).
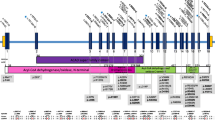
Apart from the siblings, P2 and P3, and their mother, P4, all patients were found to be unrelated down to the second degree. DNA samples from those patients harbouring the c.[1067G > A] variant (P1, P6, P8 and P9–P11), displayed variable lengths of a shared haplotype on one allele, with a minimal overlapping region of 7.2 Mb (Fig. 1). Of these samples, the two with a homozygous c.[1067G > A];c.[1067G > A] genotype (P6 and P8) exhibited homozygosity for the shared haplotype in the region of ETFDH. Similar results were obtained for the DNA samples of those patients harbouring the c.[1448C > T] variant [P1, P4 (including P2 and P3 due to their first-degree relation), P5, P9–P11 and P13], with a minimal overlapping region of 4 Mb (Fig. 2). Again, the two patients who had a homozygous c.[1448C > T];c.[1448C > T] genotype (P4 and P5), displayed homozygosity for the shared haplotype in the ETFDH region. These findings suggest the presence of two separate founder haplotypes on which the c.[1067G > A] and c.[1448C > T] variants arose, with that of the former in the White SA population. However, without access to additional control data from the same population(s), it is currently not possible to estimate the haplotype frequency with confidence.

Shared haplotype of patients affected by ETFDH c.[1067G > A]. All samples listed show variable lengths of a shared haplotype on a single allele (grey) with a minimal overlapping region of 7.2 Mb, and samples P6 and P8 exhibit homozygosity (c.[1067G > A];c.[1067G > A]) for the shared haplotype (blue). The shared region includes the genes listed. These results indicate a founder for the c.[1067G > A] variant in the White SA population. Abbreviations: ETFDH electron transfer flavoprotein-ubiquinone oxidoreductase gene, GT genotype

Shared haplotype of patients affected by ETFDH c.[1448C > T]. All samples listed show variable lengths of a shared haplotype on a single allele (grey) with a minimal overlapping region of 4 Mb, and samples P4 and P5 exhibit homozygosity (c.[1448C > T];c.[1448C > T]) for the shared haplotype (blue). The shared region includes the genes listed. These results indicate a founder for the c.[1448C > T] variant. Abbreviations: ETFDH electron transfer flavoprotein-ubiquinone oxidoreductase gene, GT genotype
Conclusion
This study provides the first extensive clinical and biochemical profiles, along with the genetic aetiology, of MADD in the diverse and understudied SA population. Within the cohort investigated, we describe one novel (c.[287dupA*]) and four previously identified causal variants in ETFDH, with an autosomal recessive inheritance pattern. In addition, the data support the suspicion that MADD is more prevalent in the White population of SA [10]. We also demonstrate a distinct shared haplotype for each of the two most common variants (c.[1067G > A] and c.[1448C > T]), suggesting the existence of a founder for each, with that of c.[1067G > A] likely having arisen in the White SA population. Furthermore, we show that, depending on the variant(s) involved, it is possible to anticipate the clinical progression and treatment response of the patient, which underscores the need for genetic confirmation in SA. It is our belief, that this genotype–phenotype correlation of MADD in the SA population will assist physicians—who rarely encounter this disorder—to recognise and treat MADD in a more efficient manner. In addition, our study provides background for the subsequent genetic counselling of families and patient-specific treatment of local MADD cases. Altogether, the data reported support a policy of including MADD in the NBS program of SA.
Methods
Cohort selection and sampling
Following informed consent/assent, the patients in this study were enrolled as part of the ICGNMD study with ethical approval numbers 19/LO/1796 (HRA and HCRW), NWU-00966-19-A1 and NWU-00966-19-A1-01 (NWU), 296/2019 (University of Pretoria; UP), B19/01/002 (Stellenbosch University; SU), and 605/2020 (UCT).
Probands and their affected/unaffected first-degree relatives were recruited either retrospectively or prospectively, based on a clinical and metabolic diagnosis of MADD, via one of three SA academic, state-funded hospitals: Steve Biko Academic Hospital, Tygerberg Hospital, and Red Cross War Memorial Children's Hospital. For (i) genetic, (ii) protein, and (iii) metabolic analyses, the following samples were obtained: (i) whole blood, saliva (living patients), or urine (deceased patients from whom no blood or saliva was available), (ii) primary skin fibroblasts (P5 and P9), as well as (iii) urine collected during the first metabolic presentation (P1, P4–P6, P8–P12 and P14)/during metabolic decompensation (P7), and after therapeutic management (P1–P11, P13 and P14).
Clinical and biochemical investigations
Patients were extensively evaluated by paediatric or adult neurologists according to protocols set forth by the ICGNMD. This included obtaining relevant demographic information, family history, medical history, and current treatment (Table 1). Moreover, deep phenotypic data (Additional file 1) were collected during a comprehensive clinical re-assessment of each proband.
SDS-PAGE and Western blot analysis of primary skin fibroblasts, established from two adult patients (P5 and P9) and two healthy controls (matched in age, gender, and ethnicity to the patients), were performed to evaluate the steady state level of ETFQO. Immunoblotting was conducted using primary antibodies at 1:1,000 directed against ETFQO (ab131376; Abcam, Cambridge, UK) and β-actin (as housekeeping gene; ab6276; Abcam), and an HRP-conjugated secondary antibody at 1:10,000 (ab97023; Abcam). Findings are shown in Additional file 3.
For targeted metabolic analyses, urine organic acids and their glycine conjugates were extracted, derivatised, and analysed using the 7890A gas chromatography system coupled to the 5977A MSD mass spectrometer (Agilent Technologies, California, USA) as described by Erasmus et al. [21] and refined by Reinecke et al. [22]. Data acquisition was facilitated with the 5977 MassHunter Data Acquisition software (B.07.04.2260; Agilent Software); Automated Mass spectral Deconvolution and Identification System software (AMDIS v2.73; National Institute for Standards and Technology) was used to identify the component peaks and perform spectral deconvolution. Underivatised amino acids in urine were analysed using validated liquid chromatography-triple quadrupole mass spectrometry and the MassChrom® Amino Acid Analysis kit (75111; Chromsystems, Gräfelfing, Germany). Samples were prepared via the Microlab STAR Liquid Handling System (Hamilton, Nevada, USA) and analysed on the Infinity 1290 II liquid chromatography system coupled to the 6470 Triple Quadrupole liquid chromatography-mass spectrometer (Agilent Technologies). For urine acylcarnitine analysis, electrospray ionisation-tandem mass spectrometry was performed using the 1290 Infinity II liquid chromatography system coupled to the 6410 Triple Quadrupole liquid chromatography-mass spectrometer (Agilent Technologies) as described by Pitt et al. [23]. A standard mixture of stable acylcarnitine isotopes was used for quantification [24]. Amino acid and acylcarnitine data were acquired and quantified using MassHunter Data Acquisition software and MassHunter QQQ Quantitative software (v10.00; Agilent Software), respectively. All reported processed data are shown in Additional file 2.
Genetic analyses
To identify potential pathogenic variants in the proband DNA, WES was performed at Macrogen Europe (Amsterdam, the Netherlands) using the NovaSeq 6000 HiSeq platform (Illumina Inc., San Diego, USA) and the SureSelect Human All Exon V6 panel (5190-8865; Agilent Technologies). Reads (50 × average depth coverage) were aligned to the Genome Reference Consortium Human Build (GRCh) 38.p13, and variants were annotated according to GRCh38 using the Ensembl Variant Effect Predictor (v108) [25]. High-quality variants were filtered by applying the Genomics’ England PanelApp, “Rhabdomyolysis and metabolic muscle disorders_1.57” panel [26]; the pathogenicity of potential disease-causing variants was evaluated and classified as such, according to the ACMG guidelines [11]. All potential pathogenic variants identified were confirmed and segregation analysis in family members was conducted via Sanger sequencing.
Allele frequency and haplotyping
To estimate the allele (carrier) frequency of the two most encountered variants in the cohort (c.[1067G > A] and c.[1448C > T]), PCR–RFLP analysis was performed as previously described [10] on newborn dried blood spot (DBS) samples of the four largest SA population groups—African, White SA, Indian, and mixed ethnicity. To this end, a total of 2,844 anonymised and randomised DBS samples per variant per population group were used: 594 representatives of mixed ethnicity as well as 750 representatives of each of the following populations: African, White SA and Indian ethnicity. The samples represented an equal distribution of healthy males and females and were kindly provided by the NBS laboratory at the NWU Centre for Human Metabolomics.
Next, we investigated whether these two variants arose due to founder mutations by determining the haplotype(s) of the affected individuals using a GSA v3.0 array (Illumina Inc.). Sample processing was performed at UCL Genomics (UCL Great Ormond Street Institute of Child Health, London, UK), according to the manufacturer's instructions; the resulting raw IDAT files were processed using Genome Studio (v2.0.5; Illumina Inc.) and converted to PLINK (v1.9) format [27]. Samples and variants with a call rate below 90% were excluded from further analysis. Thereafter, samples were compared to determine familial relations and phased using Eagle (v2.4.1) [28]. Finally, haplotype sharing between each sample was determined with Germline2 software (v1.0) [29] and shared regions were visualised using R (v4.2.2).
Availability of data and materials
Previous data and samples were made available by the Centre for Human Metabolomics (NWU), SU, and UCT. New samples were collected with the help of paediatric and adult neurologists via Steve Biko Academic Hospital, Tygerberg Hospital, and Red Cross War Memorial Children’s Hospital. The datasets generated and/or analysed during the current study are not publicly available due to the data sharing policy of the ICGNMD study, but are available from the corresponding author on reasonable request.
Notes
P2 and P3 (siblings) were diagnosed prior to 1995 (before the storage of electronic records at the contributing diagnostic facility) and did not decompensate following the onset of treatment. Although P7 was diagnosed at the same time, the patient decompensated considerably near the time of her demise. The urinary organic acid data collected during this period of decompensation, while the patient was on Rb and L-carnitine, are reported.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- AMDIS:
-
Automated Mass spectral Deconvolution and Identification System
- CK:
-
Creatine kinase
- CoA:
-
Coenzyme A
- DBS:
-
Dried blood spot
- DNA:
-
Deoxyribonucleic acid
- EC:
-
Enzyme Commission number
- ETF:
-
Electron transfer flavoprotein
- ETFA :
-
Electron transfer flavoprotein subunit alpha (gene)
- ETFB :
-
Electron transfer flavoprotein subunit beta (gene)
- ETFDH :
-
Electron transfer flavoprotein-ubiquinone oxidoreductase (gene)
- ETFQO:
-
Electron transfer flavoprotein-ubiquinone oxidoreductase
- FADH2 :
-
Flavin adenine dinucleotide (reduced)
- FAO:
-
Mitochondrial fatty acid β-oxidation
- GAII:
-
Glutaric aciduria type II
- gnomAD:
-
Genome Aggregation Database
- GRCh:
-
Genome Reference Consortium Human Build
- H3Africa:
-
Human Heredity & Health in Africa
- HCRW:
-
Health and Care Research Wales
- HEK293:
-
Human embryonic kidney 293 cell line
- HRA:
-
Health Research Authority
- HRP:
-
Horse radish peroxidase
- ICGNMD:
-
International Centre for Genomic Medicine in Neuromuscular Diseases
- MADD:
-
Multiple acyl-CoA dehydrogenase deficiency
- MADD-DS3:
-
Multiple acyl-CoA dehydrogenase deficiency-disease severity 3
- Mb:
-
Megabase(s)
- MPV17:
-
Mitochondrial inner membrane protein MPV17
- MRC:
-
Medical Research Council
- n:
-
Number of patients
- NBS:
-
Newborn screening
- NHLS:
-
National Health Laboratory Service
- NHS:
-
National Health Service
- NIHR:
-
National Institute for Health and Care Research
- NRF:
-
National Research Foundation
- NWU:
-
North-West University
- NWU-HREC:
-
North-West University-Health Research Ethics Committee
- P and number:
-
Patient
- PCR-RFLP:
-
Polymerase chain reaction-restriction fragment length
- Rb:
-
Riboflavin
- SA:
-
South Africa(n)
- SAMRC:
-
South African Medical Research Council
- SDS-PAGE:
-
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SU:
-
Stellenbosch University
- UCL:
-
University College London
- UCT:
-
University of Cape Town
- UK:
-
United Kingdom
- UP:
-
University of Pretoria
- USA:
-
United States of America
- WES:
-
Whole exome sequencing
References
Orphanet. Multiple acyl-CoA dehydrogenase deficiency (ORPHAcode:26791). https://www.orpha.net/. Accessed 14 Apr 2023.
Ghisla S, Thorpe C. Acyl-CoA dehydrogenases. A mechanistic overview. Eur J Biochem. 2004;271:494–508.
Mereis M, Wanders RJA, Schoonen M, Dercksen M, Smuts I, van der Westhuizen FH. Disorders of flavin adenine dinucleotide metabolism: MADD and related deficiencies. Int J Biochem Cell Biol. 2021;132:105899.
Frerman FE, Goodman SI. Chapter 103: defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric academia type. In: Valle D, Beaudet AL, Vogelstein B, editors. The online metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2004.
Grünert SC. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme a dehydrogenase deficiency. Orphanet J Rare Dis. 2014;9:117.
van Rijt WJ, Ferdinandusse S, Giannopoulos P, Ruiter JPN, de Boer L, Bosch AM, et al. Prediction of disease severity in multiple acyl-CoA dehydrogenase deficiency: a retrospective and laboratory cohort study. J Inherit Metab Dis. 2019;42:878–89.
Yıldız Y, Talim B, Haliloglu G, Topaloglu H, Akçören Z, Dursun A, Tokatlı A. Determinants of riboflavin responsiveness in multiple Acyl-CoA dehydrogenase deficiency. Pediatr Neurol. 2019;99:69–75. https://doi.org/10.1016/j.pediatrneurol.2019.06.015.
Mosegaard S, Dipace G, Bross P, Carlsen J, Gregersen N, Olsen RKJ. Riboflavin deficiency-implications for general human health and inborn errors of metabolism. Int J Mol Sci. 2020;21:3847.
Gordon N. Glutaric aciduria types I and II. Brain Dev. 2006;28:136–40.
van der Westhuizen FH, Smuts I, Honey E, Louw R, Schoonen M, Jonck LM, et al. A novel mutation in ETFDH manifesting as severe neonatal-onset multiple acyl-CoA dehydrogenase deficiency. J Neurol Sci. 2018;384:121–5.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Ruiz-Sala P, Peña-Quintana L. Biochemical markers for the diagnosis of mitochondrial fatty acid oxidation diseases. J Clin Med. 2021;10:4855.
Xi J, Wen B, Lin J, Zhu W, Luo S, Zhao C, et al. Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2014;37:399–404.
Hong DJ, Zhu M, Zhu ZJ, Cong L, Zhong SS, Liu L, et al. Clinical and muscle magnetic resonance image findings in patients with late-onset multiple acyl-CoA dehydrogenase deficiency. Chin Med J (Engl). 2019;132:275–84.
Lin Y, Zhang W, Chen Z, Lin C, Lin W, Fu Q, et al. Newborn screening and molecular features of patients with multiple acyl-CoA dehydrogenase deficiency in Quanzhou, China. J Pediatr Endocrinol Metab. 2021;34:649–52.
Goodman SI, McCabe ER, Fennessey PV, Mace JW. Multiple acyl-CoA dehydrogenase deficiency (glutaric aciduria type II) with transient hypersarcosinemia and sarcosinuria; possible inherited deficiency of an electron transfer flavoprotein. Pediatr Res. 1980;14(1):12–7.
Gempel K, Topaloglu H, Talim B, Schneiderat P, Schoser BG, Hans VH, et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain. 2007;130(Pt 8):2037–44.
Cornelius N, Frerman FE, Corydon TJ, Palmfeldt J, Bross P, Gregersen N, et al. Molecular mechanisms of riboflavin responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency. Hum Mol Genet. 2012;21:3435–48.
gnomAD. https://gnomad.broadinstitute.org/. Accessed 14 Apr 2023.
H3Africa. https://agvd-dev.h3abionet.org. Accessed 14 Apr 2023.
Erasmus C, Mienie LJ, Reinecke CJ, Wadman SK. Organic aciduria in late-onset biotin-responsive multiple carboxylase deficiency. J Inherit Metab Dis. 1985;8(Suppl 2):105–6.
Reinecke CJ, Koekemoer G, van der Westhuizen FH, Louw R, Lindeque JZ, Mienie LJ, et al. Metabolomics of urinary organic acids in respiratory chain deficiencies in children. Metabolomics. 2012;8:264–83.
Pitt JJ, Eggington M, Kahler SG. Comprehensive screening of urine samples for inborn errors of metabolism by electrospray tandem mass spectrometry. Clin Chem. 2002;48(11):1970–80.
Swanepoel A, Bester J, Kruger Y, Davoren E, du Preez I. The effect of combined oral contraceptives containing drospirenone and ethinylestradiol on serum levels of amino acids and acylcarnitines. Metabolomics. 2021;17(9):75.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–70.
Martin AR, Williams E, Foulger RE, Leigh S, Daugherty LC, Niblock O, et al. PanelApp crowdsources expert knowledge to establish consensus diagnostic gene panels. Nat Genet. 2019;51:1560–5.
PLINK. https://www.cog-genomics.org/plink/1.9. Accessed 1 June 2023.
Eagle. https://alkesgroup.broadinstitute.org/Eagle/. Accessed 1 June 2023.
Nait Saada J, Kalantzis G, Shyr D, Cooper F, Robinson M, Gusev A, Palamara PF. Identity-by-descent detection across 487,409 British samples reveals fine scale population structure and ultra-rare variant associations. Nat Commun. 2020;11:6130.
Acknowledgements
We would like to thank the patients and their families, as well as all the referring physicians for their assistance. We would also like to acknowledge the staff of the Centre for Human Metabolomics, NWU (Potchefstroom, South Africa), Central Analytical Facilities, SU (Stellenbosch, South Africa), Macrogen Europe (Amsterdam, The Netherlands), and UCL Genomics (UCL Great Ormond Street Institute of Child Health, London, UK) for service provision, including data acquisition and -analysis. This work was supported by a Medical Research Council (MRC) strategic award to establish an International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD; MR/S005021/1), the National Research Foundation (NRF) of South Africa (121311 and PMDS22080748827) as well as the South African Medical Research Council (SAMRC).
Funding
Open access funding provided by North-West University. This work was supported by a Medical Research Council (MRC) strategic award to establish an International Centre for Genomic Medicine in Neuromuscular Diseases (ICGNMD; MR/S005021/1). MB is supported in part by the National Research Foundation (NRF) of South Africa (121311 and PMDS22080748827) as well as the South African Medical Research Council (SAMRC). RM and RWT are funded by the Wellcome Centre for Mitochondrial Research (203105/Z/16/Z), the Mitochondrial Disease Patient Cohort (UK) (G0800674), the Medical Research Council International Centre for Genomic Medicine in Neuromuscular Disease (MR/S005021/1), the Lily Foundation, the UK NIHR Biomedical Research Centre for Ageing and Age-related Disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust, and the UK NHS Highly Specialised Service for Rare Mitochondrial Disorders of Adults and Children. RWT also receives support from the MRC (MR/W019027/1), Mito Foundation, and the Pathological Society (UK). Funders had no role in the study design, data collection and analysis, data interpretation, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
MB: Conceptualisation, Design, Data Acquisition, Data Analysis, Data Interpretation, Article Drafting, Article Revision; IS: Conceptualisation, Design, Data Acquisition, Data Analysis, Data Interpretation, Article Drafting, Article Revision; MD: Data Acquisition, Data Analysis, Data Interpretation, Article Drafting, Article Revision; MS: Data Acquisition, Data Analysis, Data Interpretation, Article Drafting, Article Revision; BCV: Data Acquisition, Data Analysis; GvdW: Data Acquisition, Data Analysis, Data Interpretation; CS: Data Acquisition, Data Analysis, Data Interpretation; KN: Data Acquisition, Data Analysis, Data Interpretation; FH: Data Acquisition, Data Analysis, Data Interpretation; SM: Data Acquisition, Data Analysis, Data Interpretation; RM: Data Interpretation; RWT: Data Interpretation; KP: Data Acquisition, Data Analysis, Data Interpretation; MRF: Data Acquisition, Data Analysis, Data Interpretation; JV: Data Acquisition, Data Analysis, Data Interpretation, Article Drafting; The ICGNMD Consortium: Data Acquisition, Data Analysis, Data Interpretation, Article Revision; RJAW: Design, Data Interpretation, Article Drafting, Article Revision; FHvdW: Conceptualisation, Design, Data Acquisition, Data Analysis, Data Interpretation, Article Drafting, Article Revision. The submitted work has been approved by all contributing authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The patients in this study were enrolled as part of the ICGNMD study with ethical approval numbers 19/LO/1796 [Health Research Authority (HRA) and Health and Care Research Wales (HCRW)], NWU-00966-19-A1 and NWU-00966-19-A1-01 [Health Research Ethics Committee (NWU-HREC) of the Faculty of Health Sciences, North-West University], 296/2019 (Faculty of Health Sciences Research Ethics Committee, University of Pretoria), B19/01/002 (Health Research Ethics Committee, Stellenbosch University), and 605/2020 (Faculty of Health Sciences Human Research Ethics Committee, University of Cape Town), following informed consent/assent.
Consent for publication
The informed consent/assent accounts for the publication of our research data are available upon request.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
For The ICGNMD Consortium in author group, see list of Consortium Members at https://www.ucl.ac.uk/genomic-medicine-neuromuscular-diseases/global-contributor-list.
Supplementary Information
Additional file 1
. Additional Clinical Information.
Additional file 2
. Additional Metabolic Information.
Additional file 3
. Additional Structural Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bisschoff, M., Smuts, I., Dercksen, M. et al. Clinical, biochemical, and genetic spectrum of MADD in a South African cohort: an ICGNMD study. Orphanet J Rare Dis 19, 15 (2024). https://doi.org/10.1186/s13023-023-03014-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-023-03014-8