
Abstract
Background
Zellweger spectrum disorders (ZSD) and X-linked adrenoleukodystrophy (X-ALD) are inherited metabolic diseases characterized by dysfunction of peroxisomes, that are essential for lipid metabolism and redox balance. Oxidative stress has been reported to have a significant role in the pathogenesis of neurodegenerative diseases such as peroxisomal disorders, but little is known on the intracellular activation of Mitogen-activated protein kinases (MAPKs). Strictly related to oxidative stress, a correct autophagic machinery is essential to eliminated oxidized proteins and damaged organelles. The aims of the current study are to investigate a possible implication of MAPK pathways and autophagy impairment as markers and putative therapeutic targets in X-ALD and ZSDs.
Methods
Three patients with ZSD (2 M, 1 F; age range 8–17 years) and five patients with X-ALD (5 M; age range 5- 22 years) were enrolled. A control group included 6 healthy volunteers. To evaluate MAPKs pathway, p-p38 and p-JNK were assessed by western blot analysis on peripheral blood mononuclear cells. LC3II/LC3I ratio was evaluated ad marker of autophagy.
Results
X-ALD and ZSD patients showed elevated p-p38 values on average 2- fold (range 1.21- 2.84) and 3.30-fold (range 1.56- 4.26) higher when compared with controls, respectively. p-JNK expression was on average 12-fold (range 2.20–19.92) and 2.90-fold (range 1.43–4.24) higher in ZSD and X-ALD patients than in controls. All patients had altered autophagic flux as concluded from the reduced LC3II/I ratio.
Conclusions
In our study X-ALD and ZSD patients present an overactivation of MAPK pathways and an inhibition of autophagy. Considering the absence of successful therapies and the growing interest towards new therapies with antioxidants and autophagy inducers, the identification and validation of biomarkers to monitor optimal dosing and biological efficacy of the treatments is of prime interest.
Similar content being viewed by others


Introduction
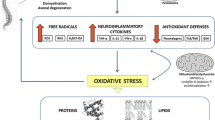
Peroxisomes are ubiquitous intracellular organelles with an essential role in multiple metabolic pathways. They are responsible for saturated very long chain fatty acids (VLCFAs, > C22:0) and branched chain fatty acids (phytanic and pristanic acids) oxidation, degradation of pipecolic acid as well as for the synthesis of bile salts, polyunsaturated fatty acids and plasmalogens [1]. Moreover, they are implicated in hydrogen peroxide (H2O2) detoxification by the enzymatic antioxidant catalase [2].
Peroxisomal disorders are a group of inherited metabolic diseases caused by either a defect in peroxisome biogenesis or a deficiency of one or more peroxisomal enzyme activities [3]. Zellweger Spectrum Disorders (ZSD) include a group of autosomal recessive peroxisome biogenesis disorders (PBD), caused by bi-allelic mutations in different PEX genes which code for a diverse group of proteins called peroxins required for the proper biosynthesis of peroxisomes [4]. The overall incidence is about 1 in 50,000 to 100,000 live births [5]. The main clinical features include dysmorphic features, visual and hearing impairment, cerebral malformations, white matter disorders, hepatomegaly and renal cysts [6, 7].
X-linked adrenoleukodystrophy (X-ALD) represents the most frequent peroxisomal disorder, with an incidence of 1:17,000–1:20,000 [8]. It is due to mutations in the ABCD1 gene (Xq28), coding for a transmembrane protein that imports VLCFAs across the peroxisomal membrane for its degradation [9]. The disorder primarily affects the adrenal cortex and the nervous system. Three core clinical syndromes are usually discriminated which include: the severely progressive form dominated by leukodystrophy (cerebral X-ALD, CALD), the progressive form with myeloneuropathy as major abnormality affecting patients in adulthood (adrenomyeloneuropathy, AMN) and primary adrenocortical insufficiency. Myeloneuropathy is the most common phenotype in affected females [10].
Patients affected by X-ALD and ZSD show increased plasma VLCFAs (mainly C26:0, C24:0/C22:0 ratio and C26:0/C22:0 ratio) and C26:0-Lysophosphatidylcholine (C26:0-lysoPC) [11]. The diagnosis is confirmed by genetic testing. At present, treatment focuses largely on symptomatic or supportive therapies [12]. For cerebral forms of X-ALD, allogeneic hematopoietic stem cell transplantation (HSCT) can be effective in childhood CALD if performed in neurologically asymptomatic boys with early brain magnetic resonance imaging (MRI) lesions [13,14,15].
In both ZSD and X-ALD, the central nervous system (CNS) is one of the main tissues affected and the lack of effective and curative treatment may probably reflect the fact that the pathophysiology of the brain injury in these disorders is poorly known.
The CNS is highly susceptible to oxidative damage due to the relatively low activity of antioxidant defenses, high iron content, high lipid content and high oxygen consumption [16]. Oxidative stress and inflammation may also have a significative role in the pathogenesis of peroxisomal disorders. Peroxisomes do produce intracellular H2O2 by virtue of the fact that peroxisomes contain multiple oxidases which turn molecular oxygen into H2O2 and their metabolism is highly connected to mitochondrial metabolism in the regulation of cellular redox homeostasis [17, 18]. Furthermore, the excess of VLCFAs are incorporated into complex lipids of cell membranes, which may contribute to the activation of inflammatory responses and oxidative stress [6, 19]. Mitochondrial dysfunction and excessive reactive oxygen species (ROS) production have been described in both X-ALD and ZSD [20,21,22].
Because of the very short half-life and rapid reactivity of ROS, there is currently no reliable method or technology to directly measure its level. Consequently, redox levels are mostly indirectly reflected by measuring antioxidant capacity (e.g. superoxide dismutase, catalase, glutathione peroxidase), or markers of oxidative damage to DNA, proteins and lipids [23].
To our knowledge, there are no studies on intracellular pathways activated by ROS, specifically on the activation of Mitogen-activated protein kinases (MAPKs). For this reason, we decided to investigate the possible implication of MAPK pathways as markers and putative therapeutic targets in X-ALD and ZSD. Given the close link between oxidative stress/MAPKs and autophagy, we also investigated autophagy impairment, previously described in X-ALD but understudied in ZSD.
Material and methods
Study population
Three patients with ZSD and five patients with X-ALD were enrolled. All caregivers gave informed consent to participate in the study which was conducted in accordance with the principles of the Declaration of Helsinki. The biochemical diagnosis was established by increased levels of plasma VLCFAs, measured by gas chromatography, and confirmed by molecular examination. C26:0-lysoPC was measured by LC- MS/MS on dried blood spot (Tables 1 and 2).
The 3 patients with ZSDs, 1 female and 2 males, were aged between 8 and 17 years (Table 3). All had intellectual disability, hearing loss, and visual loss. Patient 2 additionally had epilepsy, patients 1 and 3 hepatomegaly. All patients were supplemented with vitamin D and calcium. Patient P2 was treated with antiepileptics (carbamazepine and clobazam) and had been supplementing with vitamin E (200 mg/day), coenzyme Q 10 (200 mg/day) and docosahexanoic acid (DHA) (800 mg/day) since the age of 6.5 years. Patients with adrenoleukodystrophy, all males, ranged in age from 5 to 22 years. Patient’s characteristics are summarized in Table 4. Patients P4 and P5 and patients P6 and P7 were two pairs of brothers. All but one patient (P8) had Addison's disease and were on replacement therapy with hydrocortisone.
Patients P6, P7 and P8 had CALD with abnormal brain MRI. Patients P4 and P5 had normal MRI at the time of the study, but P5 developed white matter abnormalities at cerebral MRI one year later. All presented normal cognitive performance, except P8, who had moderate cognitive disability (IQ 55).
At the time of sample collection, two patients with X-ALD (P7 and P8) had received hematopoietic stem cell transplantation (HSCT) for 1 month and 2 years, respectively.
All X-ALD patients were treated with a mixture of glycerol trioleate/glycerol trierucate (GTO/GTE) containing alfa-lipoic acid, L-glutathione and vitamin E.
Six healthy adult volunteers (26–32 years) were recruited to set up a control group.
Sample collection
A blood sample was obtained by venous puncture with heparinized vials. Whole blood was centrifuged at 3000 rpm, plasma was used for routine biochemical analysis and measure of plasma VLCFAs. The cell pellet, which is usually discarded, was used to isolate peripheral blood mononuclear cells (PBMCs) by density gradient centrifugation on Ficoll (purchased from Cytiva). The pellet thus obtained was frozen at − 80 °C until analysis.
Immunoblotting
PBMC extracts were resuspended in RIPA buffer (phosphate-buffered saline PBS with 1% NP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate SDS) supplemented with 1% protease inhibitors (Thermo Fisher), disrupted by vortexing and centrifuged at 1300 rpm for 30 min at 4 °C. Protein concentration was measured by BCA (bicinchoninic acid) assay according to manufacturer’s instructions (PierceTM BCA Protein Assay kit, Thermo Fisher). Equal amounts (20 ug of protein) of extracts were heat-denatured for 5 min at 95 °C and subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (4–15% polyacrylamide). PBMC extracts from healthy subjects were run in parallel for comparison. Proteins were transferred to a nitrocellulose membrane, that was incubated with blocking solution (Everyblot, Bio-Rad) for 5 min. Then, it was incubated with the primary antibody (anti-p-p38, anti-p-JNK, anti-LC3 rabbit polyclonal antibodies) overnight at 4 °C and subsequently with secondary antibodies (horseradish peroxidase HRP conjugated antirabbit IgG) for 1 h. Immunoreactive proteins were detected by chemiluminescence (ECL, Bio-Rad). The GAPDH peptide allowed comparison of different samples. All reagents (blocking solution, precast gels, nitrocellulose membranes, TBS-Tween 20, ECL) and instruments (Transblot, Chemidoc) were from Bio-Rad. Primary antibodies were from Cell Signaling (p-JNK, p-p38) or Biorad (LC3, GAPDH) and used with a dilution of 1:1000. Secondary antibodies were from Biorad and used with a dilution of 1:2000. Quantitative analysis of band intensity was performed using Image Lab.
Results
MAPK pathways
In patients with ZSD, the expression of p-p38 was on overage 3.30-fold (range 1.56–4.26) higher than in controls. In particular, it was about fourfold higher in patients 1 and 3, respectively, and only 1.56 times in patient 2 who was in chronic therapy with vitamin E, coenzyme Q10, DHA. In patients with X-ALD the expression of p-p38 was on average 2 times greater than controls (range 1.21–2.84) (Fig. 1). No differences were observed according to the phenotypic subtype and treatment (transplant vs. no-transplant).

p-p38 is upregulated in PBMC of ZSDs and X-ALD patients. Representative immunoblots for p-p38 in control (CTL) and in ZSD and X-ALD patients. Protein levels are normalized respect to GAPDH. The histograms below show the p-p38 levels relative to CTL
The expression of p-JNK was on average 12-fold higher in ZSD patients (range 2.20–19.92) than in controls. It was 12 and 20 times greater in patients 1 and 3 respectively, and only 2 times greater in patient 2, who is on chronic therapy with vitamin E, coenzyme Q10, DHA. In patients with X-ALD it was on average 2.90- fold (range 1.43–4.24) greater than controls (Fig. 2). Again, no differences were noticed based on the phenotypic subtype or the treatment (transplant vs. no-transplant). There was a significant correlation between p-p38 and p-JNK values (r = 0.86).

p-JNK is upregulated in PBMC of ZSDs and X-ALD patients. Representative immunoblots for p-JNK in control (CTL) and in ZSD and X-ALD patients. Protein levels are normalized respect to GAPDH. The histograms below show the p-JNK levels relative to CTL
Autophagy
To test cells for autophagic activity, LC3 immunoblot analysis is most widely used. Endogenous LC3 is detected as two bands: one represents LC3-I, which is cytosolic (16 kDa), and the other LC3-II, which is conjugated with phosphatidylethanolamine and is present on isolation membranes and autophagosomes (14 kDa). The amount of LC3-II has been found to correlate with the number of autophagosomes, thus representing a good indicator of autophagosome formation [24].
We studied the autophagic flux by measuring LC3II/I ratio. In ZSDs patients, the values were reduced by 10% if compared with healthy controls without significant differences among the patients. A reduction by 40% was noticed in patients with X-ALD (Fig. 3). No significative differences were noticed based on phenotypic subtypes or treatment (transplant vs no transplant).

LC3. Autophagy is downregulated in PBMC of ZSDs and X-ALD patients. Representative immunoblots for LC3-I and LC3-II in control (CTL) and in X-ALD and ZSDs patients. Protein levels are normalized respect to GAPDH. The histograms below show the LC3-II/LC3-I ratio levels relative to CTL
No correlation with the MAPK levels emerged.
Discussion
Oxidative stress has been reported to have a significant role in the pathogenesis of peroxisomal disorders such as other neurodegenerative diseases. Lit is known on the intracellular activation of Mitogen-activated protein kinases (MAPKs) pathways in the peroxisomal disorders. Our results demonstrate that X-ALD and ZSD patients present an abnormal activation of MAPK pathway, showing higher values in ZSDs versus X-ALD patients.
MAPKs compose a family of protein kinases that play an essential role in relaying intracellular and extracellular signals, including ROS and inflammatory cytokines, to the nucleus via a cascade of phosphorylation events. There are three well-defined subgroups of MAPKs: the extracellular signal regulated kinases (ERKs), the c-Jun N-terminal kinases (JNKs), and the p38 MAPKs. Diverse cellular functions are regulated by MAPK signaling and the deleterious consequences of sustained activation of MAPK pathways may include uncontrolled proliferation, and unscheduled cell death. p38 and p-JNK activation has been implicated in the pathogenesis of several neurodegenerative diseases other than peroxisomal disorders [2, 25].
In addition to the inflammatory mediators, H2O2, produced by peroxisomes, activates MAPK pathways via activation of growth factor receptors and the oxidative modification of intracellular kinases. For example, thioredoxin becomes oxidized and disassociates from ASK-1 protein (MAP3K), leading to activation of JNK and p38 pathways. Moreover, intracellular H2O2 accumulation inactivates MAPKs phosphatases by oxidation of their catalytic cysteine, which leads to sustained activation of pathway [26].
Peroxisomes do not only generate ROS, but also interact with mitochondria, which may lead to further oxidative stress [23]. In particular, VLCFA accumulation may induce mitochondrial inner membrane depolarization and permeability transformation by replacing the lateral chains of phospholipid bilayers, which would disturb the proper functioning of oxidative phosphorylation system [2, 23, 27].
Finally, peroxisomal antioxidant defenses, enzymatic (catalase) and non-enzymatic (plasmalogen, DHA), can be compromised in peroxisomal disorders.
In patient P2, antioxidant treatment with vitamin E, DHA and Coenzyme Q10 appeared to reduce the activation of MAPK pathway. In fact, in the untreated ZSD patients (P1 and P3) the value of p-p38 was about 4 -fold higher than controls, while in P2 it was only 1.56. Accordingly, the value of p-JNK was about 12-fold higher in untreated ZSD patients than controls, but only twice as high in patient 2.
Our data showed that the induction of the p38 and JNK mediated MAPK pathways is attenuated by antioxidants, indicating the possibility for a new therapeutic option in ZSD patients and a possible use of MAPKs in PBMCs as biomarkers.
In our experience, the HSCT appears to have little effect on oxidative stress. Rockenbach et al. [16] demonstrated that parameters of oxidative stress in plasma of 4 X-ALD patients were reduced after HSCT, without achieving normalization. A limitation of our study is the small amount of time which has occurred since the transplant was performed in patient 7 and the lack of longitudinal data (pre-and post HSCT). Nevertheless, we can confirm the absence of a true normalization of MAPK activation after HSCT.
To maintain homeostasis in various internal and external stress responses and to remove oxidized products and damaged organelles, including oxidative stress, a correct autophagic machinery is essential [28]. Autophagy has been observed to be downregulated in X-ALD mouse model and human fibroblasts and could contribute to neurodegeneration [29, 30]. Our X-ALD patients showed a defective autophagy (LC3-II/LC3-I ratio), in accordance with what has been previously described. Moreover, we have demonstrated the same defect in our ZSD patients. Autophagic activity can be directly (e.g., through oxidative modification of autophagy-related proteins) or indirectly (e.g., through oxidative modification of transcription factors or signaling proteins, including MAPK) modulated by H2O2 [31]. Indeed, regarding PEX5-mediated pexophagy, H2O2 production can decrease the intracellular levels of the PEX5-ubiquitin, determining impairment of pexophagy itself [32]. Other factors besides oxidative stress can be involved in the blocking of autophagy. It has been demonstrated that VLCFAs accumulation destabilizes and increases the viscosity in model membranes [33]. This process can lead to a defect in the fusion of autophagosomes and lysosomes and impaired autophagy, as demonstrated in human fibroblasts [29, 34].
Study limitations
Peroxisomal disorders are rare and clinically heterogeneous. Our clinical population is diverse in regard of its clinical phenotype and treatment received. This allows us to study the effect of therapies in single patients, but studies on larger populations are required in the future.
A limitation of our study is the lack of longitudinal data that may reflect the evolution of disease and the impact of the therapies. Longitudinal studies on early- treated patients will allow to better evaluate the correlation between said biomarkers, the clinical phenotype and the efficacy of the therapies.
Conclusion
Our study demonstrates a greater activation of MAPK pathways and an inhibition of autophagy in patients with Zellweger spectrum disorders and X-linked adrenoleukodystrophy. Considering the absence of successful therapies and the growing interest towards new therapies with antioxidants and autophagy inducers, the identification and validation of biomarkers to monitor optimal dosing and biological efficacy of the treatments is of prime interest. Further studies on larger and earlier treated population are warranted.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- AMN:
-
Adrenomyeloneuropathy
- C26:0-lysoPC:
-
C26:0-Lysophosphatidylcholine
- CALD:
-
Cerebral X-ALD
- CNS:
-
Central nervous system
- DHA:
-
Docosahexaenoic acid
- GTO/GTE:
-
Glycerol trioleate/glycerol trierucate
- HSCT:
-
Hematopoietic stem cell transplantation
- IQ:
-
Intelligence quotient
- LC–MS/MS:
-
Liquid chromatography tandem mass spectrometry
- MAPK:
-
Mitogen-activated protein kinases
- MRI:
-
Magnetic resonance imaging
- ROS:
-
Reactive oxygen species
- PBD:
-
Peroxisome biogenesis disorders
- PBMC:
-
Peripheral blood mononuclear cells
- VLCFA:
-
Very long chain fatty acid
- X-ALD:
-
X-linked adrenoleukodystrophy
- ZSD:
-
Zellweger spectrum disorders
References
Wanders RJA, Vaz FM, Ferdinandusse S, Kemp S, Ebberink MS, Waterham HR. Laboratory diagnosis of peroxisomal disorders in the -omics era and the continued importance of biomarkers and biochemical studies. J Inborn Errors Metab Screen. 2018;6:232640981881028. https://doi.org/10.1177/2326409818810285.
Fourcade S, Ferrer I, Pujol A. Oxidative stress, mitochondrial and proteostasis malfunction in adrenoleukodystrophy: a paradigm for axonal degeneration. Free Radic Biol Med. 2015;88:18–29. https://doi.org/10.1016/j.freeradbiomed.2015.05.041.
Kawada Y, Khan M, Sharma AK, Ratnayake DB, Dobashi K, Asayama K, Moser HW, Contreras MA, Singh I. Inhibition of peroxisomal functions due to oxidative imbalance induced by mistargeting of catalase to cytoplasm is restored by vitamin E treatment in skin fibroblasts from zellweger syndrome-like patients. Mol Genet Metab. 2004;83(4):297–305. https://doi.org/10.1016/j.ymgme.2004.07.012.
Argyriou C, D’Agostino MD, Braverman N. Peroxisome biogenesis disorders. Transl Sci Rare Dis. 2016;1(2):111–44. https://doi.org/10.3233/TRD-160003.
Elumalai V, Pasrija D. Zellweger Syndrome. 2023 Feb 15. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan
Crane DI. Revisiting the neuropathogenesis of Zellweger syndrome. Neurochem Int. 2014;69:1–8. https://doi.org/10.1016/j.neuint.2014.02.007.
Faust PL, Banka D, Siriratsivawong R, Ng VG, Wikander TM. Peroxisome biogenesis disorders: the role of peroxisomes and metabolic dysfunction in developing brain. J Inherit Metab Dis. 2005;28(3):369–83. https://doi.org/10.1007/s10545-005-7059-y.
Moser HW, Mahmood A, Raymond GV. X-linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3(3):140–51. https://doi.org/10.1038/ncpneuro0421.
Kemp S, Wanders R. Biochemical aspects of X-linked adrenoleukodystrophy: biochemical aspects of X-ALD. Brain Pathol. 2009;20(4):831–7. https://doi.org/10.1111/j.1750-3639.2010.00391.x.
Engelen M, van Ballegoij WJC, Mallack EJ, Van Haren KP, Köhler W, Salsano E, van Trotsenburg ASP, Mochel F, Sevin C, Regelman MO, Tritos NA, Halper A, Lachmann RH, Davison J, Raymond GV, Lund T, Orchard PJ, Kuehl J-S, Lindemans CA, Caruso P, Turk BR, Moser AB, Vaz FM, Ferdinandusse S, Kemp S, Fatemi A, Eichler FS, Huffnagel IC. International recommendations for the diagnosis and management of patients with adrenoleukodystrophy: a consensus-based approach. Neurology. 2022. https://doi.org/10.1212/WNL.0000000000201374.
Jaspers YRJ, Ferdinandusse S, Dijkstra IME, Barendsen RW, van Lenthe H, Kulik W, Engelen M, Goorden SMI, Vaz FM, Kemp S. Comparison of the diagnostic performance of C26:0-lysophosphatidylcholine and very long-chain fatty acids analysis for peroxisomal disorders. Front Cell Dev Biol. 2020;29(8):690. https://doi.org/10.3389/Fcell.2020.00690.PMID:32903870;PMCID:PMC7438929.
Braverman NE, Raymond GV, Rizzo WB, Moser AB, Wilkinson ME, Stone EM, Steinberg SJ, Wangler MF, Rush ET, Hacia JG, Bose M. Peroxisome biogenesis disorders in the zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016;117(3):313–21. https://doi.org/10.1016/j.ymgme.2015.12.009.
Moser AB, Liu Y, Shi X, Schrifl U, Hiebler S, Fatemi A, Braverman NE, Steinberg SJ, Watkins PA. Drug discovery for X-linked adrenoleukodystrophy: an unbiased screen for compounds that lower very long-chain fatty acids. J Cell Biochem. 2021;122(10):1337–49. https://doi.org/10.1002/jcb.30014.
Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, Thrasher AJ, et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N Engl J Med. 2017;377(17):1630–8. https://doi.org/10.1056/NEJMoa1700554.
Tran C, Patel J, Stacy H, Mamak EG, Faghfoury H, Raiman J, Clarke JTR, Blaser S, Mercimek-Mahmutoglu S. Long-term outcome of patients with X-linked adrenoleukodystrophy: a retrospective cohort study. Eur J Paediatr Neurol. 2017;21(4):600–9. https://doi.org/10.1016/j.ejpn.2017.02.006.
Rockenbach FJ, Deon M, Marchese DP, Manfredini V, Mescka C, Ribas GS, Habekost CT, Castro CG, Jardim LB, Vargas CR. The effect of bone marrow transplantation on oxidative stress in x-linked adrenoleukodystrophy. Mol Genet Metab. 2012;106(2):231–6. https://doi.org/10.1016/j.ymgme.2012.03.019.
Petrillo S, D’Amico J, Nicita F, Torda C, Vasco G, Bertini ES, Cappa M, Piemonte F. Antioxidant response in human X-linked adrenoleukodystrophy fibroblasts. Antioxidants. 2022;11(11):2125. https://doi.org/10.3390/antiox11112125.
Ivashchenko O, Van Veldhoven PP, Brees C, Ho Y-S, Terlecky SR, Fransen M. Intraperoxisomal redox balance in mammalian cells: oxidative stress and interorganellar cross-talk. Mol Biol Cell. 2011;22(9):1440–51. https://doi.org/10.1091/mbc.e10-11-0919.
Berger J, Dorninger F, Forss-Petter S, Kunze M. Peroxisomes in brain development and function. Biochim Biophys Acta. 2016;1863(5):934–55. https://doi.org/10.1016/j.bbamcr.2015.12.005.
Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta. 2006;1763(12):1755–66. https://doi.org/10.1016/j.bbamcr.2006.09.006.
Fourcade S, Lopez-Erauskin J, Galino J, Duval C, Naudi A, Jove M, Kemp S, Villarroya F, Ferrer I, Pamplona R, Portero-Otin M, Pujol A. Early oxidative damage underlying neurodegeneration in X-adrenoleukodystrophy. Hum Mol Genet. 2008;17(12):1762–73. https://doi.org/10.1093/hmg/ddn085.
Rahim RS, Chen M, Nourse CC, Meedeniya ACB, Crane DI. Mitochondrial changes and oxidative stress in a mouse model of zellweger syndrome neuropathogenesis. Neuroscience. 2016;334:201–13. https://doi.org/10.1016/j.neuroscience.2016.08.001.
Yu J, Chen T, Guo X, Zafar MI, Li H, Wang Z, Zheng J. The role of oxidative stress and inflammation in X-link adrenoleukodystrophy. Front Nutr. 2022;9:864358. https://doi.org/10.3389/fnut.2022.864358.
Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–5. https://doi.org/10.4161/auto.4600.
Falcicchia C, Tozzi F, Arancio O, Watterson DM, Origlia N. Involvement of P38 MAPK in synaptic function and dysfunction. Int J Mol Sci. 2020;21(16):5624. https://doi.org/10.3390/ijms21165624.
Son Y, Cheong Y-K, Kim N-H, Chung H-T, Kang DG, Pae H-O. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J Signal Transduct. 2011;2011:1–6. https://doi.org/10.1155/2011/792639.
López-Erauskin J, Galino J, Ruiz M, Cuezva JM, Fabregat I, Cacabelos D, Boada J, Martínez J, Ferrer I, Pamplona R, Villarroya F, Portero-Otín M, Fourcade S, Pujol A. Impaired mitochondrial oxidative phosphorylation in the peroxisomal disease X-linked adrenoleukodystrophy. Hum Mol Genet. 2013;22(16):3296–305. https://doi.org/10.1093/hmg/ddt186.
Cho DH, Kim YS, Jo DS, Choe SK, Jo EK. Pexophagy: molecular mechanisms and implications for health and diseases. Mol Cells. 2018;41(1):55–64. https://doi.org/10.14348/molcells.2018.2245.
Launay N, Aguado C, Fourcade S, Ruiz M, Grau L, Riera J, Guilera C, Giròs M, Ferrer I, Knecht E, Pujol A. Autophagy induction halts axonal degeneration in a mouse model of X-adrenoleukodystrophy. Acta Neuropathol. 2015;129(3):399–415. https://doi.org/10.1007/s00401-014-1378-8.
Wu JJ, Quijano C, Chen E, Liu H, Cao L, Fergusson MM, Rovira II, Gutkind S, Daniels MP, Komatsu M, Finkel T. Mitochondrial dysfunction and oxidative stress mediate the physiological impairment induced by the disruption of autophagy. Aging. 2009;1(4):425–37. https://doi.org/10.18632/aging.100038.
Li H, Lismont C, Revenco I, Hussein MAF, Costa CF, Fransen M. The peroxisome-autophagy redox connection: a double-edged sword? Front Cell Dev Biol. 2021;9:814047. https://doi.org/10.3389/fcell.2021.814047.
Lismont C, Nordgren M, Brees C, Knoops B, Van Veldhoven PP, Fransen M. Peroxisomes as modulators of cellular protein thiol oxidation: a new model system. Antioxid Redox Signal. 2019;30(1):22–39. https://doi.org/10.1089/ars.2017.6997.
Ho JK, Moser H, Kishimoto Y, Hamilton J. Interactions of a very long chain fatty acid with model membranes and serum albumin. Implications for the pathogenesis of adrenoleukodystrophy. J Clin Invest. 1995;96(3):1455–63. https://doi.org/10.1172/JCI118182.
Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24(8):3052–65. https://doi.org/10.1096/fj.09-144519.
Acknowledgements
We thank patients and their families for the participation to the study.
Funding
Open access funding provided by Università degli Studi di Padova. Part of the work was funded by Cometa A.S.M.M.E.–Associazione Studio Malattie Metaboliche Ereditarie—ONLUS, Padova Italy.
Author information
Authors and Affiliations
Contributions
Conceptualization VG and AB; Methodology VG, PM, EP, MES, Investigation, VG, DG, CC, AC, AP, CL, LS, Writing—original draft, VG and DG.; Writing—review & editing, VG, DG, AC, AB, RJAW; supervision, AB, RJAW. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was conducted according to the guidelines of the Declaration of Helsinki. The informed consent was obtained by all parents of patients.
Consent for publication
The consent for publication was obtained by all parents of patients.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gragnaniello, V., Gueraldi, D., Puma, A. et al. Abnormal activation of MAPKs pathways and inhibition of autophagy in a group of patients with Zellweger spectrum disorders and X-linked adrenoleukodystrophy. Orphanet J Rare Dis 18, 358 (2023). https://doi.org/10.1186/s13023-023-02940-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-023-02940-x