
Abstract
Background
Glutaric acidemia type 1 (GA1) is a rare autosomal recessive inherited metabolic disorder caused by variants in the gene encoding the enzyme glutaryl-CoA dehydrogenase (GCDH). The estimated prevalence of GA1 and the mutational spectrum of the GCDH gene vary widely according to race and region. The aim of this study was to assess the acylcarnitine profiles and genetic characteristics of patients with GA1 in Fujian Province, southeastern China.
Results
From January 2014 to December 2022, a total of 1,151,069 newborns (631,016 males and 520,053 females) were screened using MS/MS in six newborn screening (NBS) centers in Fujian Province and recruited for this study. Through NBS, 18 newborns (13 females and 5 males) were diagnosed with GA1. Thus, the estimated incidence of GA1 was 1 in 63,948 newborns in Fujian province. In addition, 17 patients with GA1 were recruited after clinical diagnosis. All but one patient with GA1 had a remarkable increase in glutarylcarnitine (C5DC) concentrations. The results of urinary organic acid analyses in 33 patients showed that the concentration of glutaric acid (GA) increased in all patients. The levels of C5DC and GA in patients identified via NBS were higher than those in patients identified via clinical diagnosis (P < 0.05). A total of 71 variants of 70 alleles were detected in patients with GA1, with 19 different pathogenic variants identified. The three most prevalent variants represented 73.23% of the total and were c.1244-2 A > C, p.(?) (63.38%), c.1261G > A, p.Ala421Thr (5.63%), and c.406G > T, p.Gly136Cys (4.22%). The most abundant genotype observed was c.[1244-2 A > C]; [1244-2 A > C] (18/35, 52.43%) and its phenotype corresponded to high excretors (HE, GA > 100 mmol/mol Cr).
Conclusions
In conclusion, we investigated the biochemical and molecular features of 35 unrelated patients with GA1. C5DC concentrations in dried blood spots and urinary GA are effective indicators for a GA1 diagnosis. Our study also identified a GCDH variant spectrum in patients with GA1 from Fujian Province, southeastern China. Correlation analysis between genotypes and phenotypes provides preliminary and valuable information for genetic counseling and management.
Similar content being viewed by others

Background
Glutaric acidemia type 1 (GA1; OMIM #231670) is a rare inherited neurometabolic disorder caused by variants of the gene encoding glutaryl-CoA dehydrogenase (GCDH; EC 1.3.8.6) [1]. The GCDH gene is located on chromosome 19p13.2. The defective activity of GCDH hinders the degradation of L-lysine, L-hydroxylysine, and L-tryptophan, resulting in the abnormal accumulation of glutaric acid (GA) and 3-hydroxyglutaric acid (3HGA) in biological fluids and various tissues, especially in the brain [2]. Without medical management, the majority of patients with GA1 experience an acute encephalopathic crisis in the first 3–36 months following an intercurrent febrile illness or surgical intervention, resulting in bilateral striatal damage [3, 4]. The clinical presentation of striatal damage is a complex dystonic movement disorder. To avoid disease complications, patients usually receive timely medical management, including a diet low in lysine and tryptophan, supplementation with l-carnitine, and prompt complication management [5, 6]. Therefore, early diagnosis of GA1 is crucial for improved outcomes. Depending on the concentration of urinary GA, patients with GA1 are classified into two biochemical subgroups: low excretor (LE, GA < 100 mmol/mol Cr) and high excretor (HE, GA > 100 mmol/mol Cr) [7]. Several studies have assumed that HE patients have poorer outcomes than LE patients [8,9,10,11].
Increased concentrations of glutarylcarnitine (C5DC) levels in dried blood spots (DBS) and GA in urine can be reliably identified in the vast majority of patients with GA1 using tandem mass spectrometry (MS/MS) and gas chromatography/mass spectrometry (GC/MS), respectively. GA1 is a treatable disorder [1], and newborn screening (NBS) programs for GA1 have been implemented in many developed countries [12,13,14]. The incidence of GA1 and characteristics of the variants of GCDH gene have been reported in certain different populations, including the Chinese population [14,15,16,17,18]. However, the prevalence of GA1 ranges from 1/221,053 to 1/52,078 among the populations in different regions of China [18,19,20,21,22]. Limited data are available on the prevalence and mutational spectrum of GCDH in GA1 in China based on large-scale NBS. Although a single-center study on GA1 was conducted in Quanzhou, Fujian Province, the prevalence and genotypes of GA1 in Fujian Province, southeastern China, have not been reported.
In this study, we report the prevalence and mutational spectrum of GCDH in patients with GA1 based on a multicenter and large-scale NBS in Fujian Province, southeastern China. We also report the biochemical features of 18 patients with GA1 identified through NBS and 17 patients with GA1 diagnosed through clinical screening. Our work provides preliminary and valuable information for the genetic counseling and management of these patients.
Results
NBS for GA1
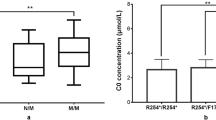
Over 12 years, 1,151,069 newborns were screened, and 265 had elevated concentrations of C5DC at initial NBS, yielding a positivity rate of 0.023%. After repeated testing, 42 newborns with positive results underwent urinary organic acid analysis and genetic testing. Additionally, 17 newborns (12 females and 5 males) were diagnosed with GA1, with a positive predictive value of 6.42% (17/265). Furthermore, one patient (no. 12) had a normal concentration of C5DC and an extremely low free carnitine (C0) level at initial NBS and was ultimately diagnosed with GA1, as shown in Table 1. As a result, the detection incidence of GA1 was 1 in 63,948 (18/1,151,069) newborns in Fujian Province.
Biochemical results and clinical features
A total of 35 unrelated patients with GA1 were investigated, including 18 NBS and 17 clinical patients. The acylcarnitine concentrations showed that all but one patient had remarkably increased C5DC concentrations, with the one anomalous patient showing a normal C5DC level and an extremely low C0 level (3.18 µmol/L) during NBS. The mean C5DC in this cohort of patients was 1.60 ± 1.17 µmol/L. Additionally, 33 patients underwent urinary organic acid analyses. The results showed that the concentration of GA increased in all patients, with 28 and 5 presenting the HE and LE phenotypes, respectively (Table 1). We further compared the differences in C5DC and GA levels between patients identified via NBS and clinical diagnosis. The results showed that the levels of C5DC and GA in NBS patients were higher than those in clinically diagnosed patients (Table 2).
The clinical characteristics of all 17 patients with clinical diagnoses are summarized in Table 3. “Movement disorders” was the most common symptom, followed by seizures and mental retardation. 11 patients (64.71%) had at least one acute encephalopathic crisis, while 6 individuals (35.29%) presented with insidious onset and developed neurological disease in the absence of encephalopathic crises.
Variant spectra of the GCDH gene
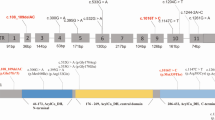
From the 35 patients with GA1, 71 variants of 70 alleles were detected, among which 19 different variants were confirmed, including 15 (78.94%) missense variants, 2 (10.53%) nonsense variants, 1 (5.26%) synonymous variant, and 1 (5.26%) splice-site variant (Table 4). At the amino acid sequence level, the three most prevalent variants accounted for 73.23% of the total: c.1244-2A > C, p.(?) (63.38%), c.1261G > A, p.Ala421Thr (5.63%), and c.406G > T, p.Gly136Cys (4.22%) (Table 4). Except for exons 1 and 3, the variants were relatively evenly distributed in the GCDH gene (Fig. 1).

Nnineteen variants were identified in patients with GA1 from Fujian Province E: exon; I: intron. The number of GCDH transcription version is NM_000159.4
As c.1244-2A > C was the most prevalent variant, we further compared the differences in its frequency between the NBS and clinical diagnosis groups. The results showed that the frequency of c.1244-2A > C did not differ between the two groups (Table 2).
Genotype of GA1 patients
Out of the 35 patients, 97.14% (34/35) carried biallelic variants and were either compound heterozygous (n = 15) or homozygous (n = 19), while the other patient (no. 33) carried a triallelic variant, with one variant originating from the mother and the other two originating from the father (Table 1). At the amino acid level, 13 distinct combinations were found in 35 patients, and the most abundant genotypes observed were c.[1244-2A > C];[1244-2A > C] (18/35, 52.43%) and c.[1244-2A > C];[1261G > A] (4/35, 11.43%) (Table 1). The phenotype of the patients with the genotype (c.[1244-2A > C];[1244-2A > C]) was HE.
Discussion
Early detection and timely intervention for inborn metabolic disorders through NBS are crucial for preventing adverse clinical symptoms in affected individuals. With the widespread application of MS/MS, GC/MS, and NGS, patients with GA1 have been diagnosed timely [18, 23, 24]. The estimated prevalence of GA1 varies widely depending on race and region, ranging from 1:125,000 to 1:250 newborns [25,26,27,28,29]. Based on a national cross-sectional survey of 7 million NBS results from mainland China from 2016 to 2017, the estimated incidence of GA1 was 1:147,900 [30]. The exact prevalence of GA1 in Fujian Province, southeastern China, remains unknown. In this study, 18 patients with GA1 were identified via 1.15 million NBS results, and the incidence was approximately 1 in 63,948 births in Fujian Province. This incidence is much higher than that in Zhejiang Province (1:221,053) [18] and Jining City (1:171,411) in eastern China [20].
In this study, 35 unrelated patients with GA1 were identified, including 18 patients who underwent NBS and 17 who received a clinical diagnosis. All but one patient showed remarkably increased C5DC concentrations. The one anomalous patient was diagnosed with GA1 and presented normal C5CD levels and a much lower C0 level during NBS, which was suggestive of primary carnitine deficiency (PCD) or maternal PCD. However, the SLC22A5 genetic mutation was not detected in patients via multi-gene-targeted NGS, although the GCDH genetic mutation was unexpectedly detected. Thus, it is noteworthy that, although NBS is a cost-effective strategy for identifying GA1, false negative results may still occur, especially in patients with the LE phenotype. Therefore, Newborns with persistently low C0 concentrations should be monitored for the possibility of GA1. Two patients with NBS did not undergo urine organic acid analyses because their urine could not be collected. Moreover, the increased GA concentration in 33 patients in this cohort suggests that urinary GA is an effective indicator for the diagnosis of GA1. Of note, GA1 patients with LE may have normal GA concentrations [18, 29]. Interestingly, the levels of C5DC and GA in the NBS patients were higher than those in the clinical patients, which is inconsistent with the results of a previous study [31]. We assumed that some factors led to the differences in the levels of C5DC and GA1 between the two groups. Firstly, patients with a clinical diagnosis may experience vomiting and eating difficulties that lead to lower C5DC and GA concentrations. Secondly, there may be differences in the levels of acylcarnitine among different ages, and the ages of patients with clinical diagnoses were significantly higher than those from the NBS. Additionally, the treatment that clinical patients were receiving was different from the nutrition the newborns were receiving.
More than 300 (confirmed or likely) pathogenic GCDH variants have been detected (http://www.hgmd.cf.ac.uk; data collected on January 1, 2023). The high-frequency region of variants and mutational spectrum of GCDH vary widely by race and region. Previous reports indicated that exon 11 is the most frequently mutated region in India and Brazil, accounting for approximately 50% of variants [32, 33], while another study showed that exon 8 is the most commonly mutated region in Chinese patients [31]. However, except for exons 1, 3, and 7, the variants were relatively evenly distributed in the GCDH gene in this study. The c.1204C > T (p.Arg402Trp) mutation is highly prevalent in European and Caucasian patients [34, 35], whereas c.914C > T (p.Ser305Leu) is a common mutation in Japanese patients [36]. Furthermore, 19 distinct variants were detected in this study, two-thirds of which were investigated only once, revealing a high degree of genetic heterogeneity among the patients with GA1 in Fujian Province. The following three variants accounted for 73.23% of the total variants among the patients with GA1: c.1244-2A > C, p.(?) (63.38%), c.1261G > A, p.Ala421Thr (5.63%), and c.406G > T, p.Gly136Cys (4.22%). Consistent with the results of previous studies [19, 31], the c.1244-2A > C variant was the predominant variant in the Chinese population. Furthermore, the c.1244-2A > C variant was more prevalent in Fujian Province than in Zhejiang Province [18].
The most prevalent genotype was c.[1244-2A > C];[1244-2A > C] (52.43%), followed by c.[1244-2A > C];[1261G > A] (11.43%), which is inconsistent with an earlier finding [18]. The phenotype of the patients with the genotype (c.[1244-2A > C];[1244-2A > C]) was HE. Compared with LE patients, HE patients show an increased risk of extrastriatal abnormalities [8] and subdural hemorrhage [9], larger macrocephaly [10], and poorer cognitive outcomes [11]. These results provide preliminary and valuable data regarding the correlation between the genotype and phenotype of GA1.
Conclusions
In conclusion, this study investigated the biochemical and molecular features of 35 unrelated patients with GA1, including 18 patients identified via NBS and 17 patients identified via clinical diagnosis. The estimated incidence of GA1 was 1 in 63,948 newborns in Fujian Province. The concentrations of C5DC in DBS and urinary GA are effective indicators for diagnosing GA1. The levels of C5DC and GA in the NBS patients were higher than those in the clinical patients. The c.1244-2 A > C variant was the most prevalent GCDH variant. The most prevalent genotype was c.[1244-2A > C];[1244-2A > C] (52.43%). The phenotype of the patients with the genotype (c.[1244-2A > C];[1244-2A > C]) was HE, which can provide preliminary and valuable data for the correlation between genotypes and phenotypes.
Methods
Study population
From January 2014 to December 2022, 1,151,069 newborns (520,053 females and 631,016 males) were recruited for this study based on screening using MS/MS in six NBS centers in Fujian Province. In addition, 17 patients with GA1 were recruited based on clinical diagnoses.
NBS and biochemical analysis
The workflow of NBS is based on a previously described procedure [9]. Briefly, DBS samples were collected and transported via a cold-chain transportation system to the corresponding NBS center. All six NBS centers used a unified experimental platform. The acylcarnitine concentrations in the DBS were quantitated using an ACQUITY TQD MS/MS (Waters) with a NeoBase™ MS/MS Kit (PerkinElmer, Turku, Finland). The cutoff value of C5DC was set at the 99.5th (0.05th) percentile. When the C5DC level of newborns exceeded this threshold, they underwent repeated testing. Urine samples were collected and analyzed for urine organic acids, including GA and 3HGA, using GC/MS (QP2010, Shimadzu Corp.). If the patients tested positive on the second screen, they underwent genetic analysis as a confirmatory test. Other laboratory tests were also performed to evaluate the patient’s status, including blood gas, ammonia, lactic acid, glucose, and liver and kidney function assessments.
Genotype analysis
Genomic DNA was isolated from DBS using a QIAamp DNA Mini Kit (Tiangen Biotech, China) according to the manufacturer’s instructions. Target next-generation sequencing was performed to detect a target sequencing panel of 94 genes (including GCDH) related to inborn metabolic errors, as previously described [37]. Briefly, the target region sequences were enriched and purified. Thereafter, a sequencing library was constructed, and sequencing and data analysis were performed using the Illumina NextSeq 500 platform and NextSeq 500 Reporter, respectively. The detected variants were confirmed using Sanger sequencing. The variants of GCDH were classified according to the guidelines of the American College of Medical Genetics and Genomics (https://clinicalgenome.org/).
Statistical analyses
Continuous variables were normally distributed and expressed as mean ± standard deviation. All categorical data are expressed as proportions. The t-test and chi-square test of variance were used to compare differences between the different GA1 subgroups. All statistical analyses were performed using the GraphPad Prism software (GraphPad, version 7.0). P value < 0.05 was considered statistically significant.
Data Availability
All data generated or analyzed during this study are included in the article, further inquiries can be directed to the corresponding author.
Abbreviations
- GA1:
-
Glutaric acidemia type 1
- GCDH:
-
Glutaryl-CoA dehydrogenase
- NBS:
-
Newborn screening
- GA:
-
Glutaric acid
- 3HGA:
-
3-hydroxyglutaric acid
- C0:
-
Free carnitine
- C5DC:
-
Glutarylcarnitine
- DBS:
-
Dried blood spot
- NGS:
-
Next-generation sequencing
- HE:
-
High excretion
- LE:
-
Low excretion
References
Boy N, Mühlhausen C, Maier EM, Ballhausen D, Baumgartner MR, Beblo S et al. Recommendations for diagnosing and managing individuals with glutaric aciduria type 1: third revision. J Inherit Metab Dis. 2022.
Harting I, Boy N, Heringer J, Seitz A, Bendszus M, Pouwels PJW, et al. 1H-MRS in glutaric aciduria type 1: impact of biochemical phenotype and age on the cerebral accumulation of neurotoxic metabolites. J Inherit Metab Dis. 2015;38:829–38.
Heringer J, Boy SPN, Ensenauer R, Assmann B, Zschocke J, Harting I, et al. Use of guidelines improves the neurological outcome in glutaric aciduria type I. Ann Neurol. 2010;68:743–52.
Kölker S, Garbade SF, Greenberg CR, Leonard JV, Saudubray J-M, Ribes A, et al. Natural history, outcome, and treatment efficacy in children and adults with Glutaryl-CoA dehydrogenase Deficiency. Pediatr Res. 2006;59:840–7.
Boy N, Mengler K, Heringer-Seifert J, Hoffmann GF, Garbade SF, Kölker S. Impact of newborn screening and quality of therapy on the neurological outcome in glutaric aciduria type 1: a meta-analysis. Genet Sci. 2021;23:13–21.
Strauss KA, Williams KB, Carson VJ, Poskitt L, Bowser LE, Young M, et al. Glutaric acidemia type 1: treatment and outcome of 168 patients over three decades. Mol Genet Metab. 2020;131:325–40.
Baric I, Wagner L, Feyh P, Liesert M, Buckel W, Hoffmann GF. Sensitivity and specificity of free and total glutaric acid and 3-hydroxyglutaric acid measurements by stable-isotope dilution assays for the diagnosis of glutaric aciduria type I. J Inherit Metab Dis. 1999;22:867–82.
Boy N, Heringer J, Brackmann R, Bodamer O, Seitz A, Kölker S, et al. Extrastriatal changes in patients with late-onset glutaric aciduria type I highlight the risk of long-term neurotoxicity. Orphanet J Rare Dis. 2017;12:77.
Boy N, Mohr A, Garbade SF, Freisinger P, Heringer-Seifert J, Seitz A, et al. Subdural hematoma in glutaric aciduria type 1: high excreters are prone to incidental SDH despite newborn screening. J Inherit Metab Dis. 2021;44:1343–52.
Märtner EMC, Maier EM, Mengler K, Thimm E, Schiergens KA, Marquardt T, et al. Impact of interventional and non-interventional variables on anthropometric long‐term development in glutaric aciduria type 1: a national prospective multi‐centre study. J Inherit Metab Dis. 2021;44:629–38.
Märtner EMC, Thimm E, Guder P, Schiergens KA, Rutsch F, Roloff S, et al. The biochemical subtype is a predictor for cognitive function in glutaric aciduria type 1: a national prospective follow-up study. Sci Rep. 2021;11:19300.
Mak J, Peng G, Le A, Gandotra N, Enns GM, Scharfe C, et al. Validation of a targeted metabolomics panel for improved second-tier newborn screening. J Inherit Metab Dis. 2023;46:194–205.
Märtner EMC, Maier EM, Mengler K, Thimm E, Schiergens KA, Marquardt T, et al. Impact of interventional and non-interventional variables on anthropometric long‐term development in glutaric aciduria type 1: a national prospective multi‐centre study. J Inherit Metab Dis. 2021;44:629–38.
Shibata N, Hasegawa Y, Yamada K, Kobayashi H, Purevsuren J, Yang Y, et al. Diversity in the incidence and spectrum of organic acidemias, fatty acid oxidation disorders, and amino acid disorders in asian countries: selective screening vs. expanded newborn screening. Mol Genet Metab Rep. 2018;16:5–10.
Sitta A, Guerreiro G, de Moura Coelho D, da Rocha VV, dos Reis BG, Sousa C, et al. Clinical, biochemical and molecular findings of 24 brazilian patients with glutaric acidemia type 1: 4 novel mutations in the GCDH gene. Metab Brain Dis. 2021;36:205–12.
Tsai F-C, Lee H-J, Wang A-G, Hsieh S-C, Lu Y-H, Lee M-C, et al. Experiences during newborn screening for glutaric aciduria type 1: diagnosis, treatment, genotype, phenotype, and outcomes. J Chin Med Association. 2017;80:253–61.
Viau K, Ernst SL, Vanzo RJ, Botto LD, Pasquali M, Longo N. Glutaric acidemia type 1: outcomes before and after expanded newborn screening. Mol Genet Metab. 2012;106:430–8.
Lin Y, Zhu X, Zhang C, Yin X, Miao H, Hu Z, et al. Biochemical, molecular, and clinical features of patients with glutaric acidemia type 1 identified through large-scale newborn screening in Zhejiang Province, China. Clin Chim Acta. 2022;530:113–8.
Lin Y, Wang W, Lin C, Zheng Z, Fu Q, Peng W, et al. Biochemical and molecular features of chinese patients with glutaric acidemia type 1 detected through newborn screening. Orphanet J Rare Dis. 2021;16:339.
Yang C, Zhou C, Xu P, Jin X, Liu W, Wang W, et al. Newborn screening and diagnosis of inborn errors of metabolism: a 5-year study in an eastern chinese population. Clin Chim Acta. 2020;502:133–8.
Wang T, Ma J, Zhang Q, Gao A, Wang Q, Li H et al. Expanded newborn screening for inborn errors of metabolism by Tandem Mass Spectrometry in Suzhou, China: Disease Spectrum, Prevalence, genetic characteristics in a Chinese Population. Front Genet. 2019;10.
Zhang R, Qiang R, Song C, Ma X, Zhang Y, Li F, et al. Spectrum analysis of inborn errors of metabolism for expanded newborn screening in a northwestern chinese population. Sci Rep. 2021;11:2699.
Yang Y, Wang L, Wang B, Liu S, Yu B, Wang T. Application of next-generation sequencing following Tandem Mass Spectrometry to Expand Newborn Screening for Inborn errors of metabolism: a Multicenter Study. Front Genet. 2019;10:86.
Al-Dirbashi OY, Kölker S, Ng D, Fisher L, Rupar T, Lepage N, et al. Diagnosis of glutaric aciduria type 1 by measuring 3‐hydroxyglutaric acid in dried urine spots by liquid chromatography tandem mass spectrometry. J Inherit Metab Dis. 2011;34:173–80.
Kölker S, Garbade SF, Boy N, Maier EM, Meissner T, Mühlhausen C, et al. Decline of Acute Encephalopathic crises in children with Glutaryl-CoA dehydrogenase Deficiency identified by Newborn Screening in Germany. Pediatr Res. 2007;62:357–63.
van der Watt G, Owen EP, Berman P, Meldau S, Watermeyer N, Olpin SE, et al. Glutaric aciduria type 1 in South Africa—high incidence of glutaryl-CoA dehydrogenase deficiency in black South Africans. Mol Genet Metab. 2010;101:178–82.
Naughten ER, Mayne PD, Monavari AA, Goodman SI, Sulaiman G, Croke DT. Glutaric aciduria type I: outcome in the Republic of Ireland. J Inherit Metab Dis. 2004;27:917–20.
Therrell BL, Lloyd-Puryear MA, Camp KM, Mann MY. Inborn errors of metabolism identified via newborn screening: ten-year incidence data and costs of nutritional interventions for research agenda planning. Mol Genet Metab. 2014;113:14–26.
Boy N, Mengler K, Thimm E, Schiergens KA, Marquardt T, Weinhold N, et al. Newborn screening: a disease-changing intervention for glutaric aciduria type 1. Ann Neurol. 2018;83:970–9.
Deng K, Zhu J, Yu E, Xiang L, Yuan X, Yao Y, et al. Incidence of inborn errors of metabolism detected by tandem mass spectrometry in China: a census of over seven million newborns between 2016 and 2017. J Med Screen. 2021;28:223–9.
Liang EH, Zhang L, Qiu H, Ye W, Xu J. Evaluation of the clinical, biochemical, neurological, and genetic presentations of glutaric aciduria type 1 in patients from China. Front Genet. 2021;12:702374.
Sitta A, Guerreiro G, de Moura Coelho D, da Rocha VV, dos Reis BG, Sousa C, et al. Clinical, biochemical and molecular findings of 24 brazilian patients with glutaric acidemia type 1: 4 novel mutations in the GCDH gene. Metab Brain Dis. 2021;36:205–12.
Gupta N, Singh PK, Kumar M, Shastri S, Gulati S, Kumar A, et al. Glutaric acidemia type 1-Clinico-molecular Profile and Novel mutations in GCDH Gene in indian patients. JIMD Rep. 2015;21:45–55.
Boy N, Mühlhausen C, Maier EM, Heringer J, Assmann B, Burgard P, et al. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision. J Inherit Metab Dis. 2017;40:75–101.
Kurkina MV, Mihaylova SV, Baydakova GV, Saifullina EV, Korostelev SA, Pyankov DV, et al. Molecular and biochemical study of glutaric aciduria type 1 in 49 russian families: nine novel mutations in the GCDH gene. Metab Brain Dis. 2020;35:1009–16.
Mushimoto Y, Fukuda S, Hasegawa Y, Kobayashi H, Purevsuren J, Li H, et al. Clinical and molecular investigation of 19 japanese cases of glutaric acidemia type 1. Mol Genet Metab. 2011;102:343–8.
Zhou J, Zeng Y, Qiu X, Lin Q, Chen W, Luo J, et al. Characterization of phenylalanine hydroxylase gene variants and analysis of genotype–phenotype correlation in patients with phenylalanine hydroxylase deficiency from Fujian Province, Southeastern China. Mol Biol Rep. 2022;49:10409–19.
Acknowledgements
We thank the patients and their families for their contribution to this study.
Funding
This work was supported by the Key Project on Science and Technology Program of the Fujian Health Commission (Grant No. 2021ZD01002), the Natural Science Foundation of Fujian Province (Grant No. 2020J01327), the Fujian Provincial Health Technology Project (Grant No. 2020GGB017), and the Joint Funds for the Innovation of Science and Technology, Fujian Province (Grant No. 2020Y9143).
Author information
Authors and Affiliations
Contributions
JZ conceptualized the study and drafted the manuscript. GL curated and analyzed the data. LD designed the study. PZ performed the experiments. YZ supervised the study. XQ performed the experiments. JL performed project administration and reviewed and edited the manuscript. LX conceptualized the study and reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Ethics Review Committee of Fujian Provincial Maternity and Child Hospital (permission no. 2017-037). Written informed consent was obtained from the guardians of all participants after providing a detailed description of the purpose of the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, J., Li, G., Deng, L. et al. Biochemical and molecular features of chinese patients with glutaric acidemia type 1 from Fujian Province, southeastern China. Orphanet J Rare Dis 18, 215 (2023). https://doi.org/10.1186/s13023-023-02833-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-023-02833-z