
Abstract
Background
Congenital hereditary endothelial dystrophy (CHED) is a rare form of corneal dystrophy caused by SLC4A11 gene variations. This study aims to find the genetic alterations in SLC4A11, in two Indian familial CHED cases with affected members n = 3 and n = 2 respectively and five sporadic CHED cases using direct sequencing, followed by in silico analysis and characterization of the identified variants.
Results
All three affected members of the first CHED family were identified with a novel homozygous c.1514C > G (p.Ser489Trp) variation while second family showed presence of a compound heterozygous variation c.529A > C (p.Arg161Arg) + c.2461insT (p.Val805fs). Among five sporadic cases, two showed novel changes, homozygous c.1487G > T (p.Ser480Ile) and c.620-2A > G, while the other one had previously reported homozygous c.2653C > T (p.Arg869Cys) variation. The remaining two cases did not reveal the presence of SLC4A11-related pathogenic variations. The identified variations were excluded from the Indian control (n = 80). In silico analysis using homology-based protein modeling and pathogenicity prediction tools, which revealed these alterations as pathogenic, changing their protein stability, local flexibility, residue contact clashes, and the hydrogen bond interactions.
Conclusions
This study contributed to the CHED mutational spectrum, adding four novel variations and confirming a previously reported one. It demonstrates different type of variations in CHED cases, including coding, non-coding, homozygous, synonymous, and compound heterozygous variations. The identified variations revealed different degrees of pathogenic effects in silico. Moreover, two sporadic cases could not be identified with pathogenic variation emphasizing the involvement of other genes or genetic mechanisms.
Similar content being viewed by others


Background
Congenital hereditary endothelial dystrophy (CHED) [MIM: 217700] is a rare corneal dystrophy and holds a significant indication of corneal transplantation in India and middle east countries [1]. The disease causes dysfunction in the corneal endothelium, presenile senescence of corneal endothelial cells, a thickened Descemet membrane, and progressive corneal clouding in the early stages of life. Patients may present with varying degrees of amblyopia, nystagmus, and glaucomatous features in the advanced stages of the disease [2]. Genetic causes of CHED have been identified in British [3] and Irish [4] familial cases using linkage analysis, describing CHED1 (autosomal dominant) and CHED2 (autosomal recessive) with a common genetic locus. Later studies distinguished them as two separate loci [5]. A study by Vithana et al. in a consanguineous family from Myanmar refined the CHED2 locus and confirmed the underlying genetic factor, the solute carrier family 4 member 11 (SLC4A11) as the prime candidate gene [6]. Subsequent studies confirmed these findings, and many SLC4A11 mutations were reported (Additional file 1: Table S1) establishing SLC4A11 as the causative gene for CHED2. SLC4A11 knockout (K/O) mice also corroborate with the CHED phenotype, confirming its role in disease pathogenesis [7]. In the current international classification of corneal dystrophy (IC3D), CHED2 is an autosomal recessive form of corneal endothelial dystrophy or CHED. CHED1 is the posterior polymorphous corneal dystrophy (PPCD) or posterior polymorphous dystrophy part of an autosomal dominant form of corneal endothelial dystrophy [8]. The SLC4A11 gene, located on chromosome 20p13, encodes for a transmembrane sodium bicarbonate transporter-like protein-11. On the corneal endothelial surface, it acts as a “pump” to transport ions across the endothelium to the corneal stroma. It helps in water movement from the stroma to the aqueous humour, maintaining the dehydrated state of the corneal stroma [9]. Apart from CHED, a mutation in SLC4A11 also causes a form of autosomal dominant Fuch’s corneal endothelial dystrophy (FECD) and Harboyan syndrome or corneal dystrophy-perceptive deafness [10]. The clinical CHED phenotype overlaps with primary congenital glaucoma and PPCD, whose genetic origins are different. Screening for SLC4A11 variations can help in the differential and confirmative diagnosis of CHED to manage the available corneal transplant options, including Descemet’s membrane endothelial keratoplasty (DMEK) and Descemet’s stripping automated endothelial keratoplasty (DSAEK) [11]. Some SLC4A11 single nucleotide polymorphisms (SNPs) are linked with bisphenol A induced ovarian carcinoma [12]. Specific non-steroidal anti-inflammatory drugs are being investigated for correcting SLC4A11-specific mutant effects [13]. The SLC4A11 mutational spectrum can help to develop medicinal approaches and regenerative medicine, such as Gene Therapy (GT) or Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) based gene editing, [14,15,16]. Also showed in our recent publication on corneal [17]. Screening for familial CHED cases also helps to measure the risk and manage the disease. This study analysed two familial cases of CHED with multiple affected members and five sporadic cases, screening all exons with flanking intronic regions of the SLC4A11 gene using the direct sequencing method. We also characterized the identified variations in silico using homology modelling and pathogenicity prediction tools and looked at the disease phenotype.
Methods
Ethics statement
This study was approved by the institutional review board of the LV Prasad Eye Institute (Ethics Ref. No. LEC-BHR-01–20-381) and followed the tenets of the declaration of Helsinki.
Subjects and clinical examination
Patients were recruited at the LV Prasad Eye Institute, India, between 2007 and 2021. Every participant, or the guardian of a minor, signed the informed consent. The demographic and clinical details of the participants are shown in Table 1. The mean age of the affected individuals at presentation was 9.6 y with a range of 7–15 y. The majority were females (n = 6) and males (n = 4). The condition was diagnosed based on ground glass corneal clouding in the presence of normal anterior segment architecture. All the patients had a detailed ophthalmic examination, including a slit lamp, optical coherence tomography, and specular microscopic imaging. The corneal opacity was classified as mild with visible iris, moderate obscuring iris details, and severe obscuring the intraocular structure details.
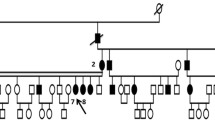
The increase in corneal thickness, caused by edema, was measured using Fourier domain optical coherence tomography (FD-OCT) RTVue-100 (Optovue, Fremont, California, USA) (Fig. 1). Pedigrees were constructed for each affected individuals based on the information provided by the guardians (Fig. 2). Eighty healthy individuals from the Indian population, (47.5% males, 52.5% females) (mean age- 31.75y) without any clinical history of ocular dystrophy were recruited as control.

Left to right: Slit lamp and AS-OCT images of the familial (F1 and F2) and sporadic (N1-N5) CHED cases. A cloudy and hazy cornea is observed using the slit lamp. The OCT images indicating edematous haze in the individuals. OD: Right eye; OS: Left Eye
Clinical slit lamp and anterior segment-optical coherence tomography (AS-OCT) images of the eyes of the recruited participants.

Panels I-V show the representative pedigrees and their sequenced chromatograms for all the CHED cases. The members underwent sequencing analysis are marked with asterisked*.The affected members sequence chromatogram were compared with the wild-type (WT) chromatogram (bottom). I and II represent familial (F1 and F2) cases identified with homozygous c.1514C>G (p.Ser489Trp) and compound heterozygous c.529A>C (p.Arg161Arg) + c.2461insT (p.Val805fs) variations, respectively. II, III, and IV represent sporadic cases (N1, N2, and N3) identified with homozygous c.1487G>T (p.Ser480Ile), c.2653C>T (p.Arg869Cys), and c.620-2A>G variations, respectively
Pedigree and chromatograms of familial and sporadic CHED cases identified with SLC4A11 variations.
Genetic analysis
For each participant, 2–3 mL of blood was drawn from a radial vein. The parents were also genetically screened along with the affected individuals of the family. The genomic DNA was extracted from blood samples using a DNA extraction kit (JetFlex™ Genomic DNA Purification Kit, A30700, Invitrogen USA). Nineteen coding exons and flanking intronic regions of the corneal specific gene transcript SLC4A11-201 (NCBI reference sequence: NM_032034.3) (Transcript ID: ENST00000380056.7) were targeted using 13 sets of published [18] and newly designed primers. PCR amplification of each amplicon was performed using EmeraldAmp MAX HS PCR Master Mix (RR330, Takara, Japan) as described in Additional file 1: Table S2. The diluted (1/10 to 1/15) amplified PCR product was subjected to Sanger sequencing using a BigDye Terminator ready reaction mix on an ABI-3130XL sequence analyser (Applied Biosystems, USA). The sequence chromatograms were analysed using CHROMAS/FINCH TV [19], and the manual DNA sequence annotation was conducted using ApE [20]. The location and position of the identified variations were mapped against the ENSEMBL genome browser (https://www.ensembl.org/).
In silico analysis
The pathogenicity score of the identified variations was analysed using the Predict SNP server (https://loschmidt.chemi.muni.cz/predictsnp) [21] with several tools, including SIFT [22] and PolyPhen [23]. The mutated residue conservation was analysed using the ConSurf server (https://consurf.tau.ac.il) [24] (Table 3). As the crystal protein structure of SLC4A11 is not yet solved, a homology-based modelling approach was executed for structural analysis. Using Modeller-10.1, a basic wild type (WT) three-dimensional homology model of the human SLC4A11 trans-membrane domain was created (Fig. 3). It was constructed using the crystal structure template of the anion exchanger domain of the human erythrocyte band-3 domain at 3.5 Å resolution (PDB 4YZF), with ~ 30% sequence identity. The structure was validated using SAVES-v6. The wild-type (WT) modelled structure was used as the template to build the mutated protein structure. The structure was visualised using CHIMERA [25]. The altered residues were analysed for any contact or clash with amino acid in the surrounding region caused due to variation (Fig. 3A1–C2). The number of hydrogen bond interactions around the 5 Å region of the variation was compared between the WT and mutated protein structures (Fig. 4). The impact on protein stability was predicted using DUET (http://marid.bioc.cam.ac.uk/sdm2/prediction, DUET (unimelb.edu.au) [26]. Protein conformation and flexibility was performed using DynaMut (http://biosig.unimelb.edu.au/dynamut/prediction) [27]. For identified intronic changes, the potential pathogenic effect for splice-site prediction was determined by the varSEAK online tool (https://varseak.bio) [28].

3A, 3B, 3C shows modelled WT SLC4A11 structures in which zone around site of alteration is analysed. 3A1, 3B1,3C1 shows focused zone around WD residue and 3A2,3B2,3C2 shows corresponding focused zone around altered residue for S489W, R869C and S480I variations respectively (encircled). Number of amino acid residue clashes in surrounding zone was found two in S489W, zero in R869C and two in S480I
Homology-based protein modelling of SLC4A11 protein and structural analysis.

SLC4A11 mutational effects on the number of hydrogen bond interactions around the 5 Å region of the mutation. A1, B1, and C1 shows the number of H bonds of WT SLC4A11 at S489, R869, and S480 positions, respectively. A2, B2, and C2 shows the corresponding number of H bonds after it mutates to W489, C869, and I480, respectively
Results
Clinical and sequencing analysis
The variations identified in the two CHED familial and three sporadic cases are listed in Table 2.
CHED familial case 1
Three siblings presented with a CHED clinical phenotype in this consanguineous family (F1). They showed varying degrees of corneal clouding. All three cases underwent DSAEK surgery to manage their corneal dystrophy. The parents and the fourth sibling were unaffected. The SLC4A11 sequencing identified a homozygous C to G DNA-nucleotide substitution at the 1514 position in the cDNA in all three affected cases. This caused, codon change from TCG to TGG (c.1514TCG > TGG), changing the amino acid from serine to tryptophan at the 489th amino acid position (p.Ser489Trp). The parental analysis found both the parents as heterozygous for this variant, thus acting as carriers (Fig. 2I). This change was not observed in the 80 healthy Indian controls.
CHED familial case 2
Two siblings were affected in this non-consanguineous family (F2). The younger girl presented with significant corneal clouding, while the older boy showed a milder phenotype. The genetic analysis identified compound heterozygous changes in both siblings, comprising the substitution of A > C at the 529th position of the cDNA and insertion of a T at the 2653rd position of the cDNA (Fig. 2II). A > C at the 529th position is a synonymous change (p.Arg161Arg). Insertion of a T at the 2653rd position likely creates a frameshift at Valine in the 805th position and generates a premature stop codon at the 877th position, 15 amino acids before the natural termination codon. This change was not observed in the 80 healthy Indian controls.
CHED sporadic cases
Five cases (N1 to N5) with no familial history of CHED were sequenced to identify SLC4A11 variations. Case-N1 presented with congenital bilateral decreased vision. It was identified with the c.1487 G > T variation, which was previously unreported and was not observed in the 80 Indian controls (Fig. 2III). Case-N2 showed congenital white opacity along with glaucoma. It was identified as the substitution variation, c.2653 C > T (p.Arg869Cys), reported in Indian CHED patients in two previous studies [6, 18] (Fig. 2IV). Case-N3 presented with a progressive decrease in vision along with glaucoma. It was identified as the homozygous substitution of A to G. Two bases upstream of the 620 cDNA position in the flanking intronic region at the beginning of exon 5 (Fig. 2V). This change was previously unreported and excluded in the 80 Indian controls. CHED cases N4 and N5 did not show the presence of any SLC4A11 pathogenic changes.
Mutational characterization—in silico analysis
c.1514TCG > TGG (p.Ser489Trp)
This novel variation found in family-F1 had high penetrance and persistence of occurrence as it affected three children in successive births. The wild-type Ser489 region was conserved in 147 of the 150 species protein sequences accessed. The SIFT and PolyPhen composite scores, derived from Predicts SNP, showed that the mutation has deleterious effects. The structural analysis of the mutated Trp489 residue showed two clashes in surrounding region (Fig. 3A1 and A2)). The DUET protein stability score of the mutated protein showed a destabilizing effect, and the DynaMut score indicated restricted local flexibility (Table 3). The number of hydrogen bond interactions around the 5 Å region of the mutated residues decreased from 136 to 116 (Fig. 4 A1-A2).
c.2653C > T (p.Arg869Cys)
This variation was identified in the sporadic case-N2. It was previously reported to affect the interaction of the transmembrane helix with the cytoplasmic domain of the protein [18]. The wild-type Arg869 region is conserved across all 150 species accessed. The SIFT and PolyPhen scores showed deleterious effects. The mutated Cys-869 residue showed no clashes in surrounding region (Fig. 3B1and B2). The DUET protein stability score showed the destabilizing effect of the mutation. The DynaMut score indicated increased local flexibility (Table 3). The number of hydrogen bond interactions around the 5 Å region of the mutated residue decreased from 15 to 13 (Fig. 4B1–B2).
c.1487G > T (p.Ser480Ile)
This novel variation was found in case-N1 in a well-conserved Ser480 position in 132 of the 150 species accessed. The Predict SNP score was deleterious. The mutated Ile 480 residue showed two clashes in surrounding region (Fig. 3C1 and C2). The mutated protein showed decreased stability and local flexibility (Table 3). The number of hydrogen bond interactions around the 5 Å region of the mutated residue decreased from 15 to 13 (Fig. 4C1–C2).
c.529A > C + c.2461insT (p.Arg161Arg + p.Val805fs)
Two siblings in the familial case-F2 shared this compound heterozygous change. Arg161Arg synonymous variation was previously described in both FECD cases and a few controls. It is listed as rs3827075 in the SNP database [29]. However, it was not observed in the 80 Indian controls we analysed. In this case, one heterozygous change is a synonymous change, and the other is a novel change. The prediction suggests that a combination of both changes leads to the disease phenotype.
c.620-2A > G
It was identified as a novel splice site variant. Two bases upstream of the exon 5 in sporadic CHED case-N3. The varSEAK online tool predicted a loss of function for the authentic splice site at this position (Table 3).
Discussions
Ever since the SLC4A11 gene was established as a causative gene for CHED-related phenotype, many pathogenic changes have been identified for its cause. Most of these variations (~ 75%) are single base homozygous changes in coding regions. Changes like compound heterozygous and splice site variants represent ~ 5% of the reported variations (Additional file 1 Table S1). In this study, we have identified a spectrum of changes, including substitution, compound heterozygous changes, and splice site variants in a small set of CHED cases. Among them, 80% were novel, signifying a wide range of genetic variations involved in the CHED pathogenesis. The novel variation p.Ser489Trp, identified in F1, showed different grades of severity of the disease among the three affected members. Previously, Ser489Leu alterations have been reported at the same amino acid position in CHED cases from Pakistan. An in vitro study on the Ser489Leu variant showed diffuse distribution with increased intracellular localization of the bicarbonate transporter-related protein-1 (BTR1) compared to WT within the endoplasmic reticulum of HEK293 cells. Though, WT is highly localized on the cell membrane of the HEK293 cell lines [6].
In this study, F2 carried a compound heterozygous change with a synonymous (p.Arg161Arg) and a novel frameshift (p.Val805fs) variation. The synonymous variant was reported in CHED and FECD patients [29, 30]. This variant is listed as rs3827075 in the SNP database with a global minor allele frequency of G = 0.39 (https://www.ncbi.nlm.nih.gov/snp/rs3827075). Previously, there was no evidence for CHED pathogenicity by a synonymous variant in the SLC4A11 gene. Synonymous variations appear silent and could become pathogenic by altering codon usage, mRNA stability, or inducing translational pausing [33]. For p.Arg161Arg synonymous variation, the pathogenic or non-pathogenic effect depends on the presence of other variants. However, further studies are required to elucidate the mechanisms involved. The compound heterozygous variant severely affected one sibling and mildly affected another, showing varied expressions. The homology-based protein modelling provides a better platform to analyse the physio-chemical changes caused by the variations at various levels of structural analysis. It has been applied in this study as an alternative model to analyse mutational effects in the absence of a known crystal structure for SLC4A11 [34]. In silico analysis showed that the identified novel p.Ser489Trp variation significantly decreased H-bond interactions around the mutational coordinate. The other identified substitution variations, S480I and R869C, showed a marginal decrease in H-bond interactions. Hydrogen bonds in a protein determine most of the directional interactions. Localised change in the number of interactions can affect protein folding, flexibility, conformation, and molecular recognition. A localised decrease in hydrogen bond interactions might affect overall protein conformation or structure. During structural analysis, the mutations compromised the proteins’ flexibility and stability. The identified variations in this study were positioned in conserved regions; therefore, they could potentially have deleterious effects. In this study, two cases were negative for SLC4A11 variations. Few studies have shown similar results. In a study by Hemadevi B et al., 9/20 families of Indian origin did not show mutations in the coding and promoter regions of the SLC4A11 gene [35]. All nine members of a large multi-consanguineous marriage of a CHED family from Saudi showed p.Thr271Met mutation. An affected twin family was the exception, which did not show any pathogenic mutation [36]. In another study, 9/25 CHED families from north India were characterized using SLC4A11 mutations [37]. The remaining 11 families were negative for the SLC4A11 mutations. These studies, including ours, emphasize the involvement of other genes or genetic mechanisms in CHED pathogenesis. Recently, exome sequencing identified a single patient with a variation in MPDZ (Multiple PDZ domain Crumbs cell polarity complex component) gene that might be involved in CHED pathogenesis. The patient did not have the SLC4A11 mutation. The mother carrying the MPDZ gene variation had a mild FECD phenotype, a phenomenon seen in patients with the SLC4A11 gene [38]. However, evidence for MPDZ in SLC4A11 negative cases is scarce and requires further studies. The absence of SLC4A11 mutations in CHED cases could be because of the lack of accurate differential diagnosis. The limitations of Sanger sequencing in identifying large heterozygous deletions might also affect negative results [39]. Next generation sequencing (NGS)-based analysis of SLC4A11 negative cases from different ethnicities could provide insight into more unknown genetic factors. Further studies are needed to establish candidate genes or genetic mechanisms for CHED patients not carrying the SLC4A11 variations.
Conclusions
This study contributed to the CHED mutational spectrum by adding four novel variations and confirming a previously reported variation in two familial cases and three sporadic cases. Our analysis emphasizes the involvement of other genes, genetic mechanisms, or clinical diagnosis for SLC4A11 negative CHED cases. These findings might help in planning gene editing corrections to reverse these variations and improve the management of CHED.
Availability of data and materials
All patient data has been anonymized, and any further information may be obtained from the corresponding author.
Change history
29 June 2023
A Correction to this paper has been published: https://doi.org/10.1186/s13023-023-02791-6
Abbreviations
- CHED:
-
Congenital hereditary endothelial dystrophy
- SLC4A11 :
-
Sodium bicarbonate transporter-like protein-11
- FECD:
-
Fuchs endothelial corneal dystrophy
- PPCD:
-
Posterior polymorphous corneal dystrophy
- CDPD:
-
Corneal dystrophy and perceptive deafness
- AS-OCT:
-
Anterior segment optical coherence tomography
- IC3D:
-
International classification of corneal dystrophy
- NSAID:
-
Non‐steroidal anti‐inflammatory drugs
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- BPA:
-
Bisphenol A
References
Moshirfar M, Drake TM, Ronquillo Y: Congenital Hereditary Endothelial Dystrophy. In: StatPearls. Treasure Island (FL); 2022.
Jeang LJ, Margo CE, Espana EM. Diseases of the corneal endothelium. Exp Eye Res. 2021;205: 108495.
Toma NM, Ebenezer ND, Inglehearn CF, Plant C, Ficker LA, Bhattacharya SS. Linkage of congenital hereditary endothelial dystrophy to chromosome 20. Hum Mol Genet. 1995;4(12):2395–8.
Hand CK, Harmon DL, Kennedy SM, FitzSimon JS, Collum LM, Parfrey NA. Localization of the gene for autosomal recessive congenital hereditary endothelial dystrophy (CHED2) to chromosome 20 by homozygosity mapping. Genomics. 1999;61(1):1–4.
Callaghan M, Hand CK, Kennedy SM, FitzSimon JS, Collum LM, Parfrey NA. Homozygosity mapping and linkage analysis demonstrate that autosomal recessive congenital hereditary endothelial dystrophy (CHED) and autosomal dominant CHED are genetically distinct. Br J Ophthalmol. 1999;83(1):115–9.
Vithana EN, Morgan P, Sundaresan P, Ebenezer ND, Tan DT, Mohamed MD, Anand S, Khine KO, Venkataraman D, Yong VH, et al. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2). Nat Genet. 2006;38(7):755–7.
Han SB, Ang HP, Poh R, Chaurasia SS, Peh G, Liu J, Tan DT, Vithana EN, Mehta JS. Mice with a targeted disruption of Slc4a11 model the progressive corneal changes of congenital hereditary endothelial dystrophy. Invest Ophthalmol Vis Sci. 2013;54(9):6179–89.
Seitz B, Lisch W, Weiss J. The revised newest IC(3)D classification of corneal dystrophies. Klin Monbl Augenheilkd. 2015;232(3):283–94.
Patel SP, Parker MD. SLC4A11 and the Pathophysiology of congenital hereditary endothelial dystrophy. Biomed Res Int. 2015;2015: 475392.
Desir J, Moya G, Reish O, Van Regemorter N, Deconinck H, David KL, Meire FM, Abramowicz MJ. Borate transporter SLC4A11 mutations cause both Harboyan syndrome and non-syndromic corneal endothelial dystrophy. J Med Genet. 2007;44(5):322–6.
AlArrayedh H, Collum L, Murphy CC. Outcomes of penetrating keratoplasty in congenital hereditary endothelial dystrophy. Br J Ophthalmol. 2018;102(1):19–25.
Zahra A, Dong Q, Hall M, Jeyaneethi J, Silva E, Karteris E, Sisu C. Identification of potential bisphenol A (BPA) exposure biomarkers in ovarian cancer. J Clin Med. 2021;10(9):1979. https://doi.org/10.3390/jcm10091979.
Alka K, Casey JR. Ophthalmic nonsteroidal anti-inflammatory drugs as a therapy for corneal dystrophies caused by SLC4A11 mutation. Invest Ophthalmol Vis Sci. 2018;59(10):4258–67.
Shyam R, Ogando DG, Kim ET, Murugan S, Choi M, Bonanno JA. Rescue of the congenital hereditary endothelial dystrophy mouse model by adeno-associated virus-mediated Slc4a11 replacement. Ophthalmol Sci. 2022;2(1): 100084.
Uehara H, Zhang X, Pereira F, Narendran S, Choi S, Bhuvanagiri S, Liu J, Kumar SR, Bohner A, Carroll L, et al. Start codon disruption with CRISPR/Cas9 prevents murine Fuchs’ endothelial corneal dystrophy. eLife. 2021. https://doi.org/10.7554/eLife.55637.
Lohia A, Sahel DK, Salman M, Singh V, Mariappan I, Mittal A, Chitkara D. Delivery Strategies for CRISPR/Cas Genome editing tool for Retinal Dystrophies: challenges and opportunities. Asian J Pharmaceut Sci. 2022;17(2):153–76. https://doi.org/10.1016/j.ajps.2022.02.001.
Salman M, Verma A, Singh VK, Jaffet J, Chaurasia S, Sahel DK, Ramappa M, Singh V. New frontier in the management of corneal dystrophies: basics, development, and challenges in corneal gene therapy and gene editing. Asia Pac J Ophthalmol (Phila). 2022;11(4):346–59.
Jiao X, Sultana A, Garg P, Ramamurthy B, Vemuganti GK, Gangopadhyay N, Hejtmancik JF, Kannabiran C. Autosomal recessive corneal endothelial dystrophy (CHED2) is associated with mutations in SLC4A11. J Med Genet. 2007;44(1):64–8.
Treves DS. Review of three DNA analysis applications for use in the microbiology or genetics classroom. J Microbiol Biol Educ. 2010;11(2):186–7.
Davis MW, Jorgensen EM. ApE, a plasmid editor: a freely available DNA manipulation and visualization program. Front Bioinf. 2022;2: 818619.
Bendl J, Stourac J, Salanda O, Pavelka A, Wieben ED, Zendulka J, Brezovsky J, Damborsky J. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 2014;10(1): e1003440.
Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc. 2016;11(1):1–9.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Ashkenazy H, Erez E, Martz E, Pupko T, Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38(Web Server):W529–33. https://doi.org/10.1093/nar/gkq399.
Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinf. 2006;7:339.
Pires DEV, Ascher DB, Blundell TL. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014;42(W1):W314–9. https://doi.org/10.1093/nar/gku411.
Rodrigues CH, Pires DE, Ascher DB. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018;46(W1):W350–5.
Yari A, Ali-Nejad RM, Saleh-Gohari N. A novel homozygous splice-site mutation in SCARB2 is associated with progressive myoclonic epilepsy with renal failure. Neurol Sci. 2021;42(12):5077–85.
Vithana EN, Morgan PE, Ramprasad V, Tan DT, Yong VH, Venkataraman D, Venkatraman A, Yam GH, Nagasamy S, Law RW, et al. SLC4A11 mutations in Fuchs endothelial corneal dystrophy. Hum Mol Genet. 2008;17(5):656–66.
Minear MA, Li YJ, Rimmler J, Balajonda E, Watson S, Allingham RR, Hauser MA, Klintworth GK, Afshari NA, Gregory SG. Genetic screen of African Americans with Fuchs endothelial corneal dystrophy. Mol Vis. 2013;19:2508–16.
Ramprasad VL, Ebenezer ND, Aung T, Rajagopal R, Yong VHK, Tuft SJ, Viswanathan D, El-Ashry MF, Liskova P, Tan DTH, et al. Novel SLC4A11 mutations in patients with recessive congenital hereditary endothelial dystrophy (CHED2). Human Mutation. 2007;28(5):522–3. https://doi.org/10.1002/humu.9487.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–24.
Sauna ZE, Kimchi-Sarfaty C. Synonymous mutations as a cause of human genetic disease. Wiley; 2013.
Zahra A, Hall M, Chatterjee J, Sisu C, Karteris E. In Silico study to predict the structural and functional consequences of SNPs on biomarkers of ovarian cancer (OC) and BPA exposure-associated OC. Int J Mol Sci. 2022;23(3):1725. https://doi.org/10.3390/ijms23031725.
Hemadevi B, Veitia RA, Srinivasan M, Arunkumar J, Prajna NV, Lesaffre C, Sundaresan P. Identification of mutations in the SLC4A11 gene in patients with recessive congenital hereditary endothelial dystrophy. Arch Ophthalmol. 2008;126(5):700–8.
Shah SS, Al-Rajhi A, Brandt JD, Mannis MJ, Roos B, Sheffield VC, Syed NA, Stone EM, Fingert JH. Mutation in the SLC4A11 gene associated with autosomal recessive congenital hereditary endothelial dystrophy in a large Saudi family. Ophthalmic Genet. 2008;29(1):41–5.
Paliwal P, Sharma A, Tandon R, Sharma N, Titiyal JS, Sen S, Nag TC, Vajpayee RB. Congenital hereditary endothelial dystrophy - mutation analysis of SLC4A11 and genotype-phenotype correlation in a North Indian patient cohort. Mol Vis. 2010;16:2955–63.
Moazzeni H, Javadi MA, Asgari D, Khani M, Emami M, Moghadam A, Panahi-Bazaz MR, Hosseini Tehrani M, Karimian F, Hosseini B, et al. Observation of nine previously reported and 10 non-reported SLC4A11 mutations among 20 Iranian CHED probands and identification of an MPDZ mutation as possible cause of CHED and FECD in one family. Br J Ophthalmol. 2020;104(11):1621–8.
Puangsricharern V, Yeetong P, Charumalai C, Suphapeetiporn K, Shotelersuk V. Two novel mutations including a large deletion of the SLC4A11 gene causing autosomal recessive hereditary endothelial dystrophy. Br J Ophthalmol. 2014;98(10):1460–2.
Acknowledgements
We would like to thank all our patients and control individuals who gave consent and participated in this study. We would also like to thank Abhinav Sekar and Tejah Balantrapu, Department of science, health data and storytelling, LVPEI for improving the manuscript.
Funding
The authors wish to thank the Indian Council of Medical Research (ICMR) for junior research fellowship to MS [Ref. No. 3/1/3/JRF-2019/ HRD (LS)/25389], senior research fellowship to DP (2021–12785), and Hyderabad Eye Research Foundation (HERF) for financial and infrastructure support.
Author information
Authors and Affiliations
Contributions
MS and AV contributed equally to the design and implementation of the study, including manuscript preparation. SC helped with patient recruitment and clinical analysis. MR contributed to the clinical data analysis, supervised the workflow and helped write the manuscript. Dr VS helped with study design, coordination, and supervision. DP contributed to running the experiments and CK contributed to the genetic analysis and manuscript writing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the institutional review board of the L V Prasad Eye Institute (Ethics Ref. No. LEC-BHR-01–20-381).Written informed consent from all adult participants and legal guardians/parents for minors were obtained for the research study and publications.
Consent for publication
Consent for publications were included in consent form.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of this article was revised: Ethics declaration had to be corrected in the backmatter of the article. It was: “The study was approved by the “Institutional Review committee for protection of research risk to humans, Indian Statistical Institute, 2015”. Written informed consent from all adult participants and legal guardians/parents for minors were obtained for the research study and publications.
It should be: “The study was approved by the institutional review board of the L V Prasad Eye Institute (Ethics Ref. No. LEC-BHR-01–20-381).Written informed consent from all adult participants and legal guardians/parents for minors were obtained for the research study and publications”.
Supplementary Information
Additional file 1.
Table S1. List of identified variants of SLC4A11 reported in CHED/FECD phenotype. Table S2. List of primers and PCR annealing temperature for each amplicon of SLC4A11.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Salman, M., Verma, A., Chaurasia, S. et al. Identification and in silico analysis of a spectrum of SLC4A11 variations in Indian familial and sporadic cases of congenital hereditary endothelial dystrophy. Orphanet J Rare Dis 17, 361 (2022). https://doi.org/10.1186/s13023-022-02521-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02521-4