
Abstract
Background/objective
Arginase 1 Deficiency (ARG1-D) is a rare inherited metabolic disease with progressive, devastating neurological manifestations with early mortality and high unmet need. Information on prevalence is scarce and highly variable due to limited newborn screening (NBS) availability, variability of arginine levels in the first days of life, and high rates of misdiagnosis. US birth prevalence was recently estimated via indirect methods at 1.1 cases per million live births. Due to the autosomal recessive nature of ARG1-D we hypothesize that the global prevalence may be more accurately estimated using genetic population databases.
Methods
MEDLINE and EMBASE were systematically searched for previously reported disease variants. Disease variants in ARG1-D were annotated wherever possible with allele frequencies from gnomAD. Ethnicity-specific prevalence was calculated using the Hardy–Weinberg equation and applied to generate country-specific carrier frequencies for 38 countries. Finally, documented consanguinity rates were applied to establish a birth prevalence for each country.
Results
133 of 228 (58%) known causative alleles were annotated with ethnic-specific frequencies. Global birth prevalence for ARG1-D was estimated at 2.8 cases per million live births (country-specific estimates ranged from 0.92 to 17.5) and population prevalence to be 1.4 cases per million people (approximately 1/726,000 people). Birth prevalence estimates were dependent on population demographics and consanguinity rate.
Conclusion
Birth prevalence of ARG1-D based on genetic database analysis was estimated to be more frequent than previous NBS studies have indicated. There was a higher degree of confidence in North American and European countries due to availability of genetic databases and mutational analysis versus other regions. These findings suggest the need for greater disease education around signs and manifestations of ARG1-D, as well as more widespread testing and standardization of screening for this severe disease in order to appropriately identify patients prior to disease progression.
Similar content being viewed by others


Introduction
Arginase 1 Deficiency (ARG1-D) is a debilitating and progressive metabolic disease characterized by persistent elevation of arginine and its metabolites, ultimately leading to significant morbidity and early mortality [1, 2]. It is one of a group of urea cycle disorders (UCDs) presenting in childhood with manifestations that include lower-limb muscle weakness, spasticity, developmental and motor delays, intellectual disability, and seizures [3]. Spasticity is the most overt sign and differentiates ARG1-D from other UCDs. Preliminary symptoms also include irritability, vomiting and feeding issues, and failure to thrive [1]. Hyperammonemia can occur but is usually less frequent and less severe when compared to other UCDs [4] .
ARG1-D is currently estimated to account for approximately 3.5% of all UCD cases but estimates of disease prevalence are inconsistent [5]. Previous reports of incidence vary by an order of magnitude, from 0.5 to 5.0 cases per million [6, 7]. US birth prevalence of ARG1-D was recently estimated via indirect methods at 1.1 cases per million live births [8]. A US-based newborn screening (NBS) study predicted a similar rate, estimating birth prevalence at 0.9 cases per million live births [9]. However, estimating prevalence of ARG1-D via NBS can be challenging due to the variability of arginine levels in the first days of life and since ARG1-D is not universally included on all NBS panels [10]. Arginine accumulates slowly after birth and as a result the range of levels in affected and unaffected newborns can overlap, making it difficult to determine an appropriate diagnostic threshold to accurately assess arginine deficiency in neonates [9]. For this reason, patients can often be missed at the newborn stage and these studies could underestimate the true prevalence of ARG1-D. There is also published evidence that ARG1-D patients are misdiagnosed as suffering from more recognized disorders such as hereditary spastic paraplegia (HSP) and cerebral palsy (CP) [11].
In the neonate, given the complications of diagnosis via arginine levels, genetic diagnosis of ARG1-D is the most reliable method of determining a positive case of the disease [3]. More than 60 causative ARG1 genetic variants have been identified to date [12]. Due to the autosomal recessive inheritance of ARG1-D, we hypothesize that the global prevalence of the disease may be more accurately estimated via genetic population databases, such as seen in other autosomal recessive diseases [13]. However, birth prevalence based on carrier frequency assumes random mating, an assumption which is violated in the event of consanguineous marriage. Birth defects in consanguineous marriages are estimated to be 2–3% higher compared to the general population, and lead to higher rates of autosomal recessive disorders [14].
Today, the primary treatment of ARG1-D is elimination of protein from the diet to minimize arginine intake, with EAA supplementation to maintain nutritional status; use of nitrogen scavengers is also commonplace [3]. Early intervention is critical to slow disease progression, so understanding the true birth prevalence of the disease is becoming increasingly important, especially as evidence of misdiagnosis builds. This study aims to estimate the global genetic prevalence of ARG1-D via genetic population databases and is the first study to adjust for consanguinity within these methods.
Materials and methods
Search strategy
To evaluate the full genetic spectrum of ARG1-D, a systematic literature review was performed using MEDLINE (PubMed) and EMBASE (Scopus). The following search criteria terms were used: ‘ARG1 AND deficiency’; ‘(arginase 1) AND deficiency’; ‘ARG1 AND mutation’; ‘(arginase 1) AND mutation’. Abstracts of all studies identified via this search were first screened by two independent reviewers (S.A. and C.C.) to identify potentially relevant articles reporting information on ARG1-D cases. Publications that met the inclusion criteria at the abstract stage were flagged for full-text review, and any disagreements were resolved through discussion. Studies were then thoroughly reviewed (S.A. and C.C.) for reports of any ARG1-D cases with verified genetic information. Reference lists of articles selected for full-text review were also searched to identify additional publications that may report on ARG1-D cases. The search was not limited by date or geographical region. All diagnosed ARG1-D cases identified with at least one known causative genetic mutation were recorded into a database. Care was taken to deduplicate cases that were reported more than once in the literature.
Frequency annotation via gnomAD
A list of all known pathogenic ARG1 variants currently identified as causative for ARG1-D, including the frequency of each within ARG1-D cases, was compiled from the case database referenced above. Each reported mutation was standardized and matched to its Reference SNP cluster ID (rsID) number (Additional file 1: Table S1) [15]. Subsequently, each mutation identified was queried via rsID number in the gnomAD database (v2) [16]. Where possible, ARG1 variants were annotated with population frequencies obtained from the gnomAD database for the following ethnic groups: African/African-American (AFR), Latino/Admixed American (AMR), East Asian (EAS), European (non-Finnish) (NFE) and South Asian (SAS). These ethnic groups are the most extensively genotyped and catalogued in the gnomAD database [16]. None of the pathogenic variants were found in the Finnish population so the FIN population were excluded due to a small non-representative sample size. Once the carrier frequency for each variant identified in the case database was obtained, the frequencies across all pathogenic variants were summed within each ethnic sub-group to obtain overall carrier frequencies for ARG1 by ethnicity (Table 1). Ninety-five percent confidence intervals (CIs) for these estimates were calculated using the binomial (Clopper-Pearson) "exact" method based on the beta distribution. Because census data used in this study does not always distinguish between Asian sub-groups, the EAS and SAS populations were then combined into a single Asian population to enable analysis.
Birth prevalence and population prevalence estimates
Using the identified ARG1 carrier frequencies by ethnic sub-group from the case database, country-specific birth and population ARG1-D prevalence estimates were then generated. This analysis focused on 38 countries that together account for 23.3% of the world population (Table 2) [17]. These countries were selected due to the availability of census data needed to estimate the ethnic breakdown, and to provide a representative sampling of the global population. For each of the 38 countries, the population percentage breakdown across the four main ethnic groups referenced above was estimated using census data where available [18,19,20,21,22,23,24,25,26,27,28,29], as well as other sources [30, 31]. For each country, the countrywide percentage of each ethnic group was multiplied by the ARG1 carrier frequency in that group and then summed to obtain baseline country-specific ARG1 mutation carrier frequencies (Table 2). Next, a baseline birth prevalence was calculated for each country assuming complete autosomal recessive inheritance and random mating according to the Hardy–Weinberg equilibrium. Affected individuals need two copies of a mutation to develop disease, thus the expected case frequency was equal to the carrier frequency squared. To adjust for consanguinity, consanguinity estimates for each of the 38 countries were obtained [32, 33]. Consanguinity adjustments were conservative; the percentage of consanguineous marriage that were first cousins or closer was used and more distant relationships were ignored. Consanguinity-adjusted birth prevalence estimates were calculated using the following equation:
where \(f\) is equal to carrier frequency and \(p\) is equal to birth prevalence.
Finally, to estimate overall country-specific prevalence, the adjusted birth prevalence rates were converted to represent population prevalence by assuming an average life expectancy for ARG1-D of 40 years.
Results
Study selection
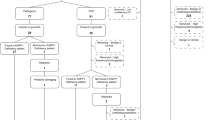
A total of 836 articles were identified in the primary literature search (Fig. 1). Of these, 762 studies were excluded during first-stage screening if the title/abstract clearly indicated that no mutation data on ARG1-D cases were reported. Of the remaining 74 studies, 41 were excluded after full assessment by the reviewers. Finally, a total of 33 unique publications containing genetic information on 114 total ARG1-D cases were retrieved. These 114 cases, along with their complete genetic information (where available), were entered into the database.

Search strategy and flow chart of study selection. Inclusion criteria are listed in methods
Full genetic spectrum for ARG1-D
A total of 62 unique mutations responsible for ARG1-D were identified in the 114 cases found in the literature. GnomAD allele frequency data were available for 28/62 (45.2%) of these variants (Additional file 1: Table S1). 28 identified mutations accounted for 133 of the 228 (58.3%) total alleles found in the 114 ARG1-D cases (Table 1). Each mutation identified in the gnomAD database was annotated with carrier frequencies for each of the five ethnic groups (NFE, AFR, AMR, EAS, SAS). After summing the 28 mutation frequencies across the ethnic groups, the results were adjusted (divided by 58.3%) to account for the unknown alleles. This resulted in birth prevalence frequencies for the differing ethnic groups as follows—NFE: 0.75 cases per million live births; AFR: 1.30 cases per million live births; AMR: 1.97 cases per million live births; EAS/SAS: 1.51 cases per million live births. Thus, ARG1-D is more likely to occur in Latino populations, followed by Asian and African populations, and least likely in European populations.
Country-specific birth and population prevalence estimates
Country-specific birth prevalence estimates accounting for ethnic-specific carrier frequencies are presented in Table 2. Prior to adjusting for consanguinity, the Latin countries had the highest birth prevalence estimates owing to the increased carrier frequency in the Latino/Admixed American (AMR) ethnic group. Unadjusted for consanguinity, the country with the highest ARG1-D birth prevalence is Argentina with 2.9 cases per million live births, closely followed by Colombia, Chile and Mexico, all with a birth prevalence of 2.6 per million live births.
After adjusting for consanguinity, it became apparent that interfamily marriage is the main driver behind the four countries with the highest birth prevalence of this disease (Table 2). The most prevalent countries are all from the Middle East as follows: Qatar (17.5 per million live births), Saudi Arabia (14.6 per million live births), Kuwait (13.0 per million live births), and United Arab Emirates (12.8 per million live births). Countries with homogeneous white European populations and very low consanguinity such as Norway, Finland, and the Netherlands, have the lowest estimates, each with an adjusted ARG1-D birth prevalence of 0.5 per million live births. The overall global birth prevalence of ARG1-D across all 38 countries was calculated at 2.8 cases per million live births.
A total of 2511 total cases of ARG1-D are estimated across the 38 countries investigated, resulting in an overall prevalence of 1.38 cases per million population (Table 3). Although this analysis only considered 38 countries, a racially and geographically diverse selection of countries was used, and thus these results are expected to reflect the true global prevalence. Similar to birth prevalence, the highest population prevalence estimates are seen in countries with high consanguinity (Qatar, Kuwait, UAE and Saudi Arabia) where estimates ranged from 6.4 to 8.7 cases per million population. Birth prevalence also has a high range in countries with a higher proportion of Latin population, such as Argentina, Brazil, Chile, and Colombia, with estimates within the range of 2.9 to 3.8 cases per million population. Similarly to birth prevalence, countries with predominantly homogeneous white European populations and very low consanguinity have the lowest population prevalence of ARG1-D.
Discussion
This report presents the findings of a genetic analysis that estimated global birth prevalence for ARG1-D to be 2.8 cases per million live births (1/357,000 live births) and population prevalence to be 1.4 cases per million people (approximately 1/726,000 people). Prevalence estimates from this genetic analysis are higher than reported in most previous studies.
Screening for ARG1-D is conducted by measuring the elevation of arginine in newborn screening blood spots via tandem mass spectrometry, and is currently included as a secondary target on the US Recommended Uniform Screening Panel [9]. A US newborn screening study published in 2017 reported a birth prevalence of approximately 0.9 per million live births [9]. A separate study published in 2013 using both newborn screening data and data from the Urea Cycle Disorders Consortium (UCDC) found a similar ARG1-D birth prevalence at 1.1 per million live births (1/950,000 live births) [8]. The estimate for birth prevalence presented in this study indicate the US birth prevalence of ARG1-D at 1.5 cases per million live births, approximately 50% higher than these studies report. Newborn screening studies are often considered the gold standard of birth prevalence estimates; however, with ARG1-D in particular, screening can be challenging as there is variability in arginine levels in the first days of life [10]. Moreover, the appropriate arginine level to diagnose a case at birth has been difficult to determine due to the overlap between arginine levels in affected and unaffected newborns. The cutoff for a positive screening result also varies across states, further complicating the calculation of birth prevalence estimates from newborn screening studies [9]. The higher genetic analysis estimates presented in this study suggest that the newborn screenings are currently under-capturing potential cases.
Estimating prevalence via genetic databases is reliant on the amount and precision of currently available information. In this study, ethnic-specific frequencies were annotated from 133 of 228 (58%) causative ARG1-D alleles. While the most common mutations are captured here, uncertainty remains regarding the remaining 42% of alleles. Over time it is expected that it will be possible to refine and improve these estimates as more sample genomes and information on more genetic variants are added to databases such as gnomAD. However, adjusting the currently annotated mutations by the percentage of unknown variants currently existing in cases should result in a very good approximation of the true prevalence. When estimating the global prevalence and birth prevalence of ARG1-D, 38 countries that had the most available information on racial breakdown were included, predominantly using census data. While these countries make up nearly 25% of the global population, we have not yet estimated prevalence in every country globally. However, since the countries represented here are geographically varied and racially diverse, the reported estimates in this study should be an appropriate representation of a global estimate. To the best of the authors’ knowledge, this is the first study to adjust prevalence estimates based on carrier frequency for consanguinity in each country. It has been established that high consanguinity (such as seen in many Arab countries) leads to a sharp increase in the birth rate of autosomal recessive disease [32, 34]. Thus, adjusting for this factor provides a more accurate reflection of the current state of ARG1-D birth prevalence globally. This can be seen reflected in the study case database; for example, within European ARG1-D cases, patients with a Turkish origin where consanguinity is higher than the rest of Europe are much more represented than we would expect based on genetic carrier frequency alone.
A potential limitation of this study is that assumptions of consanguinity and life expectancy may not be generalizable to all populations. Consanguinity in the current study was limited to marriage of first cousins or closer, and rates of consanguinity, even using this conservative definition, are likely higher in reality than estimated here. Further, although an estimated median life expectancy of 40 years was selected based on the literature and clinical experience, actual life expectancy is likely lower in developing countries and regions where disease awareness, access to diagnostic procedures, and implementation of management strategies remain a challenge.
The findings from this present study suggest that ARG1-D may be more prevalent than previously thought, indicating a lack of diagnosis or misdiagnosis. Awareness of ARG1-D and the burden it brings to patients, caregivers and healthcare systems, may minimize delays in diagnosis which are otherwise associated with poor outcomes. Diagnosis of ARG1-D is possible through simple amino acid testing (since elevated levels of arginine are seen in the majority of patients) and can be confirmed with genetic testing or analysis of red blood cell arginase levels. The utility of this analysis is to highlight that ARG1-D disease prevalence may be currently underestimated, because of poor disease awareness. Greater awareness to better recognize the signs and symptoms of ARG1-D, as well as more widespread and standardized screening, is needed to ensure proper and timely diagnosis of this severe disease.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are available in GnomAD, https://gnomad.broadinstitute.org/
References
Sin YY, Baron G, Schulze A, Funk CD. Arginase-1 deficiency. J Mol Med (Berl). 2015;93(12):1287–96. https://doi.org/10.1007/s00109-015-1354-3.
Adam MP, Ardinger HH, Pagon RA, et al. GeneReviews. Seattle, WA: University of Washington; 1993.
Häberle J, Boddaert N, Burlina A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. https://doi.org/10.1186/1750-1172-7-32.
Cui D, Liu Y, Jin L, Hu L, Cao L. A novel compound heterozygous mutation in the arginase-1 gene identified in a Chinese patient with argininemia: a case report. Medicine (Baltimore). 2020;99(32): e21634. https://doi.org/10.1097/MD.0000000000021634.
Burrage LC, Jain M, Gandolfo L, Lee BH, Nagamani SC, Urea Cycle Disorders Consortium. Sodium phenylbutyrate decreases plasma branched-chain amino acids in patients with urea cycle disorders. Mol Genet Metab. 2014;113(1–2):131–5. https://doi.org/10.1016/j.ymgme.2014.06.005.
Nagata N, Matsuda I, Oyanagi K. Estimated frequency of urea cycle enzymopathies in Japan. Am J Med Genet. 1991;39(2):228–9. https://doi.org/10.1002/ajmg.1320390226.
Marsden D. Expanded newborn screening by tandem mass spectrometry: the Massachusetts and New England experience, Southeast Asian. J Trop Med Public Health. 2003;34(Suppl. 3):111–4.
Summar ML, Koelker S, Freedenberg D, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110(1–2):179–80. https://doi.org/10.1016/j.ymgme.2013.07.008.
Therrell BL, Currier R, Lapidus D, Grimm M, Cederbaum SD. Newborn screening for hyperargininemia due to arginase 1 deficiency. Mol Genet Metab. 2017;121(4):308–13. https://doi.org/10.1016/j.ymgme.2017.06.003.
Jay A, Seeterlin M, Stanley E, Grier R. Case report of argininemia: the utility of the arginine/ornithine ratio for newborn screening (NBS). JIMD Rep. 2013;9:121–4. https://doi.org/10.1007/8904_2012_190.
Carvalho DR, Brum JM, Speck-Martins CE, et al. Clinical features and neurologic progression of hyperargininemia. Pediatr Neurol. 2012;46(6):369–74. https://doi.org/10.1016/j.pediatrneurol.2012.03.016.
Diez-Fernandez C, Rüfenacht V, Gemperle C, Fingerhut R, Häberle J. Mutations and common variants in the human arginase 1 (ARG1) gene: impact on patients, diagnostics, and protein structure considerations. Hum Mutat. 2018;39(8):1029–50. https://doi.org/10.1002/humu.23545.
Gao J, Brackley S, Mann JP. The global prevalence of Wilson disease from next-generation sequencing data. Genet Med. 2019;21(5):1155–63. https://doi.org/10.1038/s41436-018-0309-9.
Hamamy H. Consanguineous marriages: preconception consultation in primary health care settings. J Community Genet. 2012;3(3):185–92. https://doi.org/10.1007/s12687-011-0072-y.
Sherry ST, Ward M, Sirotkin K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999;9(8):677–9.
Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43. https://doi.org/10.1038/s41586-020-2308-7.
United Nations, Department of Economic and Social Affairs, Population Division.
Instituto Nacional de Estadistica y Censos, Republica de Argentina; Censo 2010.
Australian Bureau of Statistics. Australia Population and Housing Census 2001. Canberra, Australia: Australian Bureau of Statistics.
Statistics Austria; Census 2013.
Statistics Canada; 2016 Census.
Instituto Nacional de Estadisticas de Chile; Census 2017.
Official Statistics of Finland (OSF): population structure [e-publication]. ISSN=1797-5395. Helsinki: Statistics Finland [referred: 6 May 2019]. Access method: http://www.stat.fi/til/vaerak/tau_en.html.
Istituto Nazionale di Statistica, Italia 2013.
Statistics Korea (South Korea). South Korea Population and Housing Census 2017.
New Zealand Population and Housing Census 2013. Statistics New Zealand.
Embassy of the Russian Federation; Federal State Statistics Service (Russia). Russia Census 2010.
Instituto Nacional de Estadistica; Spain Population Estimates January 2019.
U.S. Census Bureau, 2013–2017 American Community Survey 5-Year Estimates.
Central Intelligence Agency. In the world factbook. 2019. Retrieved from https://www.cia.gov/library/publications/the-world-factbook/geos/br.html.
Population of Qatar by nationality—2019 report. https://priyadsouza.com/population-of-qatar-by-nationality-in-2017/.
Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health. 2009;6:17. https://doi.org/10.1186/1742-4755-6-17.
Bittles AH. Empirical estimates of the global prevalence of consanguineous marriage in contemporary societies. 1998.
Tadmouri GO, Sastry KS, Chouchane L. Arab gene geography: From population diversities to personalized medical genomics. Glob Cardiol Sci Pract. 2014;2014(4):394–408. https://doi.org/10.5339/gcsp.2014.54.
Acknowledgements
Not applicable.
Funding
Funding was provided by Aeglea BioTherapeutics, Inc. MB And NU are employees of Aeglea and contributed in writing and editing the manuscript.
Author information
Authors and Affiliations
Contributions
CC analyzed and interpreted the data regarding ARG1-D. CC, MB, and SA contributed to writing the manuscript. All authors edited, read, and approved final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was based on cases identified from a search of published literature and cross-referenced in a publicly available genetics research database. There were no human subjects in this study; ethics approval/consent to participate were not required.
Consent for publication
Not applicable.
competing interests
CC and SA performed as consultants and received funding from Aeglea BioTherapeutics, Inc. MB and NU are employees of Aeglea BioTherapeutics, Inc.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Each ARG1 variant reported among the 114 cases in the literature was standardized and matched to its Reference SNP cluster ID (rsID) and queried in the gnomAD database. GnomAD allele frequency data were available for 28 of 68 of these variants (45%).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Catsburg, C., Anderson, S., Upadhyaya, N. et al. Arginase 1 Deficiency: using genetic databases as a tool to establish global prevalence. Orphanet J Rare Dis 17, 94 (2022). https://doi.org/10.1186/s13023-022-02226-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02226-8