
Abstract
Background
Maple syrup urine disease (MSUD) is an autosomal recessive inherited metabolic disease caused by deficient activity of the branched-chain α-keto acid dehydrogenase (BCKD) enzymatic complex. BCKD is a mitochondrial complex encoded by BCKDHA, BCKDHB, DBT, and DLD genes. MSUD is predominantly caused by Variants in BCKDHA, BCKDHB, and DBT genes encoding the E1α, E1β, and E2 subunits of BCKD complex, respectively. The aim of this study was to characterize the genetic basis of MSUD by identifying the point variants in BCKDHA, BCKDHB, and DBT genes in a cohort of Brazilian MSUD patients and to describe their phenotypic heterogeneity. It is a descriptive cross-sectional study with 21 MSUD patients involving molecular genotyping by Sanger sequencing.
Results
Eight new variants predicted as pathogenic were found between 30 variants (damaging and non-damaging) identified in the 21 patients analyzed: one in the BCKDHA gene (p.Tyr120Ter); five in the BCKDHB gene (p.Gly131Val, p.Glu146Glnfs * 13, p.Phe149Cysfs * 9, p.Cys207Phe, and p.Lys211Asn); and two in the DBT gene (p.Glu148Ter and p.Glu417Val). Seventeen pathogenic variants were previously described and five variants showed no pathogenicity according to in silico analysis.
Conclusion
Given that most of the patients received late diagnoses, the study results do not allow us to state that the molecular features of MSUD variant phenotypes are predictive of clinical severity.
Similar content being viewed by others

Background
Maple syrup urine disease (MSUD) (OMIM #24860), also known as leucinosis, is an inborn error of metabolism (IEM) caused by the deficiency of thiamine-dependent branched-chain α-ketoacid dehydrogenase (BCKD), composed of the subunits E1α, E1β, E2, and E3. This particular enzymatic deficiency is found in the several tissues, resulting in elevated blood plasma leves for ketoacids (α-ketoisocaproic, α-ketoisovaleric, α-keto-β-methylvaleric), alloisoleucine, and branch chain aminoacids (valine, leucine and isoleucine). Elevated levels of these amino acids mainly damage the central nervous system (CNS) [1]; in patients with MSUD, branched-chain α-ketoacids cannot be oxidized by the dehydrogenase complex and leucine tolerance reflects unmeasured protein losses and the balance between endogenous protein synthesis and degradation [2]. Some studies suggest that MSUD is characterized by an increase in oxidative stress, even in treated patients [3, 4].
Several mutations causing MSUD have already been described in the catalytic subunits of the BCKD complex encoded by four different genes. Based on altered loci, four genetic subtypes have been proposed: type Ia or E1α (MIM # 608348) for the mutations found in the BCKDHA (E1α subunit) gene; type Ib or E1β (MIM # 248611) for the mutations found in the BCKDHB (E1β subunit) gene, type II or E2 (MIM # 248610) for mutations in the DBT gene (E2 subunit) and type III or E3 (MIM # 238331) for mutations in the DLD gene (E3 subunit) [1, 5,6,7,8,9]. Another two different genes were more recently described how members of the BCKD complex in MSUD: BCKDK gene (MIM # 614923) and PPM1K gene (MIM # 615135); defects of PPM1K may account for a subset of human MSUD; inactivating variants of BCKDK in humans are associated with BCAA deficiency, MSUD, autism, epilepsy, and intellectual disability [10].
Although six genes (BCKDHA, BCKDHB, BCKDK, DBT, DLD and PPM1K) involved in BCAA metabolism have been reported, MSUD is predominantly caused by mutations in the BCKDHA, BCKDHB, and DBT genes encoding the E1α, E1β, and E2 subunits of the BCKD complex, respectively [11]. Mutations in the DLD gene alter the function of four different enzymes, since the E3 component is used by other mitochondrial complexes [9]. Considering the three major genes determining the disease, more than 300 mutations have been described in The Human Gene Mutation Database—HGMD to date. The HGMD currently identifies 104 mutations for the BCKDHA gene, 127 mutations for the BCKDHB gene, and 84 mutations for the DBT gene. In ClinVar has 211 entries only for BCKDHA gene and 278 for BCKDHB gene.
Genomic alterations that impair BCKD activity may occur in any of the three genes, but since the inheritance follows an autosomal recessive Mendelian pattern, both alleles at a single locus of one of the genes must harbor a mutation. The altered or missing protein product renders the BCKD complex inactive. Most of the mutations described affect the BCKDHA gene or the BCKDHB gene [1, 8, 9].
The standard treatment involves dietary restriction of branched-chain amino acids from natural protein and a BCAA-free amino acid specialized formula; however, liver transplantation has been performed in certain patients and may restore some enzymatic activity in MSUD patients [12], allowing diet liberalization and avoiding the occurrence of metabolic decompensation during infectious intercurrences [13]. Decreased leucine concentration has been observed hours after hepatic transplantation in patients with MSUD, using livers from cadaveric donors [12, 14,15,16].
The worldwide incidence of MSUD is estimated at one in 185 000 births. Although it is a rare defect, the estimated incidence of MSUD is one in 200 live births for certain Mennonite communities in Pennsylvania and various cities in the United States [1]. A study carried out in Portugal by Quental et al. [17] that analyzed cases diagnosed by mass spectrometry found an incidence of one in 86 800 live births. There is no data reported in the literature on its prevalence in the Brazilian population [17].
Since there are no common mutations, the molecular approach usually involves direct genetic sequencing. In this study is, we hypothesized that the Brazilian population has a characteristic genotypic profile which provided the basis of our objective to characterize the nucleotide variants identified as point mutations in the BCKDHA, BCKDHB, and DBT in a cohort of Brazilian patients diagnosed clinically and biochemically with MSUD. We also sought to describe the observed phenotypic heterogeneity in MSUD patients.
Methods
Patients
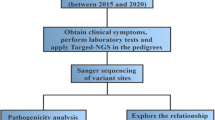
Peripheral blood samples were obtained from 21 patients (all patients who were added in MSUD Network from 2013 to 2015, including one pair of siblings), who belong to the MSUD Network, Brazilian Network for Assistance and Research in Maple Syrup Urine Disease, with various clinical phenotypes. The MSUD Network seeks to structure a national organization of reference for the investigation, diagnosis, management and follow-up of MSUD patients.
Molecular analysis of genes
DNA was extracted from peripheral blood leukocytes for the molecular analysis of the three genes involved in MSUD (BCKDHA, BCKDHB, and DBT).
Primer design
The forward and reverse primers of the 30 exons to be studied were designed manually using the GenBank® Reference with the following access numbers: BCKDHA, NM_000709.3; BCKDHB, NM_000056.3; and DBT, NM_001918.2 (https://www.ncbi.nlm.nih.gov/genbank/). All primer sequences were located in the intron regions and the amplified fragments spanned all the exons. Additional tables files shows primer sequences, PCR conditions and amplified fragment sizes in more detail (see Additional file 1 for BCKDHA gene, Additional file 2 for BCKDHB gene and Additional file 3 for DBT gene).
DNA sequencing
PCR amplified fragments were sequenced on an ABI 3500xL Genetic Analyzer capillary sequencer—24-capillary DNA analysis system (Applied Biosystems, Foster City, CA, USA) using BigDye® Terminator v3.1 cycle sequencing kit (Applied Biosystems).
Variant analysis
The sequencing results were visualized using FinchTV® version 1.4.0 software (Geospiza, Seattle, WA, USA) and compared with the relevant reference sequences from the GenBank® database (NIH, Bethesda, MD, USA). The nomenclature for the description of sequence variants detected was derived from the recommendations of the Human Genome Variation Society (https://www.hgvs.org/mutnomen) [18]. The allelic frequency of each variant was investigated in the Genome Aggregation Database (or gnomAD, data from 123,136 exomes and 15,496 genomes, people around the world) [19] and in Online Archive of Brazilian Mutations (or ABraOM, data from 609 exomes, people from the city of São Paulo, Southeast of Brazil) [20]. In order to verify the pathogenicity potential of the missense mutations, in silico analysis was performed using MutPred® v1.2 [21], Polyphen-2®-Polymorphism Phenotyping v2 software [22] and SIFT® [23]. Sequence variants were evaluated for their disease-causing potential using the Mutation Taster application [24].
Clinical data analysis
Clinical data analysis results of information such as age at diagnosis, family history of the disease, consanguinity, results of the first dosage levels of the branched-chain amino acids (at diagnosis), neurodevelopment delay, and phenotypic classification of the patients were obtained from the registry of clinical records standardized by the Brazilian MSUD Network, which was conducted by the professional responsible for patient care at the Reference Center.
Results
The BCKDHA, BCKDHB, and DBT genes from 21 patients (52.4% female), including one pair of siblings, were studied. Eight new variants predicted as pathogenic were found between 30 variants (damaging and non-damaging) identified in the 21 patients analyzed: one in the BCKDHA gene, five in the BCKDHB gene and two in the DBT gene. Another seventeen pathogenic variants found in Brazilian patients have been reported in the literature: five in the BCKDHA gene, eleven in the BCKDHB gene, and one in the DBT gene. It can be seen that most of the mutations found in the BCKDHA gene were in exons 2, 3, and 4 and in BCKDHB gene, mainly in exons 4 and 5, characterizing hot spots.
Among the 21 patients, six were homozygotes for a pathogenic allele. In 18 of the 21 patients, only one pathogenic allele could be identified. We also identified five non-pathogenic variants among our cohort (Tables 1, 2).
Among pathogenic variants in our patient cohort, the Pro200Ter in the BCKDHB gene was the most prevalent (19.0% of the patients, n = 4, one of whom was homozygous). Among non-pathogenic variants (per in silico analysis), the Pro39His variant in the BCKDHA gene was the most frequent (23.8% of the patients, n = 5) and was always found in the setting of two other pathogenic variants that explains the MSUD phenotype.
Of the 24 pathogenic variants found in the cohort of patients studied, the most prevalent type of pathogenic mutation was missense (n = 13). There were eight variants that exhibited changes in the codon-scoring matrix or frameshift, with five resulting from deletion (four in the BCKDHB gene and one in the BCKDHA gene), one resulting from insertion into the BCKDHA gene, one resulting from deletion and insertion into the BCKDHB gene, and one resulting from a duplication in the BCKDHB gene. The least frequent type was nonsense (n = 3, one variant in each gene studied); the next two least frequent types (frameshift and nonsense mutations) truncate the protein prematurely by the presence of the terminator codon and are considered pathogenic a priori.
Regarding possible genotype–phenotype associations, out of 12 patients exhibiting the classical clinical phenotype, nine presented the genetic subtype EIb, two presented the genetic subtype EII and one presented the genetic subtype EIa. The two patients with intermediate clinical phenotype had different genetic subtypes (EIa and EII) and the EIa genetic subtype was detected in the patient with intermediate or thiamine responsive phenotype. Hence, there is likely no consistent association between clinical phenotypes with the genetic subtypes. However, it is thought that mutations at different locations in the three genes can trigger clinical signs and varied sequelae; these mutations are also classically described in the literature for other diseases characterized by great variability in frequency and severity of clinical manifestations and complications, suggesting, for example, a worse prognosis in the presence of a localized mutation in the gene’s functional domain (Table 1).
Evaluating the genetic subtype of the patients who presented EIa, the phenotype was extremely varied (one patient had a classical phenotype, one intermediate, and one intermediate or thiamine responsive); eight EIb patients presented classical phenotype; EII cases were classical (n = 2) and intermediate (n = 1) phenotypes (Table 1).
Discussion
To date, this is the first study in Brazil to identify variants in the BCKDHA, BCKDHB, and DBT genes in clinically and biochemically diagnosed MSUD patients enrolled in the MSUD Network. We found that out of the 42 alleles, the disease-causing variant was located in the BCKDHA gene in 23.8% (5 patients), in the BCKDHB gene in 57.1% (12 patients), and in the DBT gene in 19.1% (4 patients).
Some of the novel variants have already been detected in database projects involving the search for variants in a large number of individuals but never related to patients. We investigate the variants in “The Exome Aggregation Consortium”—ExAC—composed of 60,706 unrelated individuals, and the Online Archive of Brazilian Mutations—AbraOM—composed of 609 elderly individuals [19, 20]. Three of 11 novel variants were in ExAC, all in very low frequencies (Table 2).
Nellis and Danner [7] studied 63 patients diagnosed clinically with MSUD and found the following frequency of mutations: 33% in the BCKDHA gene, 38% in the BCKDHB gene, and 19% in the DBT gene. In agreement with the results of the present study, most patients (of varied ethnicity) evaluated by Henneke et al. [25] presented mutations of the subtype Ib, followed by Ia, with the least frequent mutation occurring in the DBT gene (subtype II). In a cohort of 32 unrelated Turkish patients, out of 64 alleles, the disease-causing mutation was located in the BCKDHA gene in 37% (12 patients), in the BCKDHB gene in 44% (14 patients), and in the DBT gene in 19% (6 patients) [26]. However, Quental et al. [6], in a study with 30 Portuguese patients, found 17 mutations with no difference in the frequency of these genes, with six in the BCKDHA gene, five in the BCKDHB gene, and six in the DBT gene. A study published by Abiri et al. [27] investigating 20 Iranian families of MSUD patients, six patients demonstrated homozygous haplotype for the BCKDHA gene, nine for the BCKDHB gene, and two for the DBT gene. Gupta et al. [28], studying 24 unrelated Indian patients, found 20 mutations in 22 of the patients, with 11 novel mutations—four in the BCKDHA gene, six in the BCKDHB gene, with a novel mutation identified in the DBT gene.
Li et al. [29] described eight cases of MSUD (in four females and four males) from unrelated Chinese families who were diagnosed through serum BCAA and genetic analysis between 9 days to 1 year and 8 months of life (six patients presented the neonatal form of the disease); 12 different mutations were found with six in the BCKDHA gene, five in the BCKDHB gene, and one in the DBT gene, of which only one mutation located in the BCKDHA gene had been reported in the literature.
In agreement with the present study, all the Spanish patients studied (belonging to a cohort of 33 MSUD patients) by Rodríguez-Pombo et al. [9] who had a mutation in the BCKDHB gene (subtype Ib) also had the classical phenotype; however, inconsistencies were found between the biochemical parameters of the patients and their clinical phenotype, as in the case of three patients who were homozygotic for the same mutation and exhibited a residual enzymatic activity variable between < 1 and 13%.
Abiri et al. [30] reported six Iranian patients who were homozygous for mutations in the BCKDHA gene, demonstrating that most of the mutations found were in exons 2, 4, and 6. Recently, 20 new mutations were identified in the BCKDHA, BCKDHB, and DBT genes of 52 MSUD patients from Saudi Arabia, with no mutation in the DLD gene identified [31].
The leucine levels at diagnosis were high and are, in the majority of the studied patients, considered critical, and can produce irreversible damage or even death of the patient [32]. Leucine can increase rapidly during catabolic states, altering brain chemistry by competing with nine other amino acids for entry into the brain via the facilitative SLC7A5 transporter [2]. Branched-chain amino acid transaminase (BCAT1) catalyzes the formation of α-ketoisocaproic acid (αKIC) from leucine and α-ketoglutarate (αKG); αKIC enters brain (via the monocarboxylate transporter) and is neurotoxic at high concentrations [2]. Elevated tissue αKIC reverses normal flow through BCAT1, depletes tissues of glutamate (a substrate for glutamine and γ-aminobutyric acid—GABA), and indirectly drives flux through glutamate-pyruvate transaminase to form pyruvate from αKG and alanine [2].
According to Strauss et al. [2], in classic MSUD patients, these interconversions explain inverse relationships of leucine to glutamate, glutamine and alanine, and likely underlie the depletion of glutamate and elevation of lactate observed in brain tissue during metabolic encephalopathy. However, the results of a Brazilian study [33] on the MSUD panorama over the last 2 decades in the country did not indicate a significant association between the severity of developmental delay and leucine levels at diagnosis; this can be attributed to the fact that early diagnosis and long-term metabolic control are considered more decisive factors for psychomotor and cognitive development than leucine levels at the time of diagnosis. Therefore, regarding the characteristics and clinical situation of the patients, it is thought that the association of phenotype and genotype is not ideal in that neurological deterioration is directly associated with delayed diagnosis and thus the absence of appropriate nutritional support, which is essential for the control of BCAA serum levels as well as those of their metabolites (metabolic control).
Although some patients were diagnosed in the first month of life, as can be seen in Table 1, diagnosis was not made at the beginning of the presentation of symptoms in the majority of patients (only patients 6, 11, and 15 were diagnosed at the time at which symptoms appeared and patient 18 was diagnosed when still asymptomatic). In Brazil, the time between diagnosis and receipt of the metabolic formula is long and variable [33], which can also be observed in the registry of the clinical files of patients in our study. Only four patients (patients 4, 6, 11, and 18) received the metabolic formula for treatment of MSUD immediately after diagnosis; receipt varied for the other patients between 10 days and 2 months after diagnosis, delaying appropriate treatment of the disease. When patients are diagnosed in the acute phase of the disease during a hospital stay due to metabolic decompensation, treatment with a metabolic formula is started at the time of diagnosis only if the formula is available at the admitting hospital. In contrast, patients diagnosed in the non-acute phase of the disease and treated on an outpatient basis are prone to receive the formula late, with the guarantee of access to the metabolic formula occurring only through judicial measures [33]. These facts reinforce the need to include neonatal screening for MSUD in the Brazilian Newborn Screening Program.
Molecular genetics analysis is critical for the accurate diagnosis of MSUD patients, can aid in identifying variant phenotypes and offer guidance on prognosis and treatment. In addition, the molecular screening of affected patients provides epidemiological data for implementation of the test in populations, facilitating prenatal diagnosis in families at risk [28, 30].
Flaschker et al. [34], who described mutations in 15 individuals from Germany, Austria, and Switzerland with different phenotypic variants of MSUD, concluded that in the cohort of patients analyzed, the most severe enzymatic and clinical phenotypes of the disease forms (MSUD variants) were associated mainly with specific genotypes in the BCKDHA gene, whereas milder enzymatic and clinical phenotypes were associated with specific genotypes in the BCKDHB and DBT genes; the results of this study led to the conclusion that genotyping may be predictive of metabolic and clinical phenotype and may have a prognostic value, particularly in individuals with a variant of MSUD identified in neonatal screening such that early treatment will effectively delay the natural course of the disease.
However, in a study by Gupta et al. [28], it was not possible to establish any genotype and phenotype correlation in Indian MSUD patients; most of the cases (66.6%) had the classic neonatal type of MSUD, most of the classic neonatal patients (10 of 14 cases) having mutations in the BCKDHB gene.
Patients with the intermittent form of the disease, evaluated by Tsuruta et al. [35], presented mutations exclusively in the DBT gene, giving rise to low but significant residual activity of the BCKD complex. However, in the present study, no patient with the genetic subtype EII presented this phenotypic form of the disease.
Wang et al. [36] suggested that DNA sequencing associated with in silico analysis is a simple and rapid method to predict the severity of MSUD and that neonatal screening has been demonstrated to be an effective approach for screening MSUD patients in a diverse ethnic population [34], allowing early intervention with dietary therapy [36]. The identification of different missense-type mutations provides valuable evidence for the accuracy of the analysis, as well as the determination of its effect on the catalytic subunit. DNA sequencing analysis, together with in silico analysis, has been suggested to predict the clinical manifestations of MSUD, since it is a simple and rapid method [36].
According to Feier et al. [14], liver transplantation provides metabolic control, allowing the patient to resume a normal diet and avoid further neurological damage, helping the individual's neurodevelopment as well as eliminating the chance of cerebral edema and death. In this case report, Feier et al. [14] described the situation of a 2-year-old child with MSUD who underwent liver transplantation (domino transplantation); the donor was the patient’s mother. The use of living relative donors has been controversial because parents are mandatory heterozygotes, but in this case, the data suggest that the use of a donor associated with MSUD was effective. Although the traditional treatment during the maintenance phase is basically the dietary restriction of BCAA and supplementation with thiamine and a food formula without BCAA, liver transplantation is indicated in the literature as a good treatment option [14, 15, 27].
No somatic cell complementation studies were performed to assign enzymatic subtypes Ia, Ib or II, which can be a limitation. Another limitation of this study is about the ability or lack thereof of performing deletion/duplication analysis at the exon or whole gene level, and this can be related to not being able to make a molecular diagnosis in some of the patients studied. In addition, other molecular characteristics (type and location of mutation) were not considered in this analysis of this study.
Conclusions
The point variants identified in the BCKDHA, BCKDHB, and DBT genes of the cohort of Brazilian patients diagnosed clinically and biochemically with the MSUD did not present a similar pattern or frequency among the genes. Eight new variants predicted as pathogenic were identified: one in the BCKDHA gene, five in the BCKDHB gene and two in the DBT gene. We detected a broad spectrum of clinical presentation in the cohort of Brazilian patients with MSUD, and there was no apparent association between phenotypic characteristics and the genetic subtype. Given that most Brazilian patients were diagnosed late and did not receive immediate treatment, the study results indicate no consistent relationship between molecular and clinical MSUD phenotype; however, we speculate that genetic screening may have prognostic value particularly in individuals with MSUD variants identified in expanded newborn screening, in which early treatment can likely alter the natural course of the disease.
Availability of data and materials
All data generated or analysed during this study are included in this published article. For more information, the datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BCAA:
-
Branched-chain amino acids
- BCAT1:
-
Branched-chain amino acid transaminase
- BCKD:
-
Branched-chain α-keto acid dehydrogenase
- BCKDHA:
-
Branched chain keto acid dehydrogenase E1, alpha polypeptide
- BCKDHB:
-
Branched chain keto acid dehydrogenase E1, beta polypeptide
- CNS:
-
Central nervous system
- DBT:
-
Dihydrolipoamide branched chain transacylase E2
- DLD:
-
Dihydrolipoamide dehydrogenase
- DNA:
-
Desoxirribonucleic acid
- IEM:
-
Inborn error of metabolism
- MSUD:
-
Maple syrup urine disease
- PCR:
-
Polimerase chain reaction
- αKG:
-
α-Ketoglutarate
- αKIC:
-
α-Ketoisocaproic acid
References
Chuang DT, Shih VE, et al. Maple syrup urine disease. In: Scriver CR, et al., editors. The metabolic and molecular bases of inherited diseases. 8th ed. New York: McGraw-Hill; 2001. p. 1971–2005.
Strauss KA, Carsona VJ, Soltys K, Young ME, Bowser LE, Puffenberger EG, Brigatti KW, Williams KB, Robinson DL, Hendrickson C, Beiler K, Morton DH. Branched-chain α-ketoacid dehydrogenase deficiency (maple syrup urine disease): Treatment, biomarkers, and outcomes. Mol Genet Metab. 2020;129:193–206.
Barschak AG, et al. Oxidative stress in plasma from maple syrup urine disease patients during treatment. Metab Brain Dis. 2008a;23(1):711–80.
Barschak AG, et al. Maple syrup urine disease in treated patients: biochemical and oxidative stress profiles. Clin Biochem. 2008b;41(4–5):317–24.
Fisher CR, Fisher CW, Chuang DT, Cox RP. Occurrence of a Tyr393Asn (Y393N) mutation in the E1 alpha gene of the branched-chain alpha-keto acid dehydrogenase complex in maple syrup urine disease patients from a Mennonite population. Am J Hum Genet. 1991;49(2):429–34.
Quental S, Macedo-Ribeiro S, Matos R, Vilarinho L, Martins E, Teles EL, Rodrigues E, Diogo L, Garcia P, Eusébio F, Gaspar A, Prata MJ. Molecular and structural analyses of maple syrup urine disease and identification of a founder mutation in a Portuguese Gypsy community. Mol Genet Metab. 2008;94:148–56.
Nellis MM, Danner DJ. Gene preference in maple syrup urine disease. Am J Hum Genet. 2001;68:232–7.
Online Mendelian Inheritance in Man, OMIM (TM). National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD, USA) (2011). https://www.ncbi.nlm.nih.gov/omim/
Rodríguez-Pombo P, Navarrete R, Merinero B, Gómez-Puertas P, Ugarte M. Mutational spectrum of maple syrup urine disease in Spain. Hum Mutat. 2006;27:715–27.
Strauss KA, Puffenberger EG, Carson VJ. Maple syrup urine disease. 2006 Jan 30 [Updated 2020 Apr 23]. In: Adam MP, Ardinger HH, Pagon RA et al (eds) GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020.
Yang N, Han L, Gu X, Ye J, Qiu W, Zhang H, Gong Z, Zhang Y. Analysis of gene mutations in Chinese patients with maple syrup urine disease. Mol Genet Metab. 2012;106(4):412–8.
Strauss KA, Mazariegos GV, Sindhi R, Squires R, Finegold DN, Vockley G, et al. Elective liver transplantation for the treatment of classical maple syrup urine disease. Am J Transplant. 2006;6:557–64.
Wendel U, Baulny, & H. O. . Branched-chain organic acidurias/acidemias. In: Fernandes J, Saudubray JM, Berghe GVD, Walter JH, editors. In born metabolic diseases. 4th ed. Germany: Würzburg; 2006. p. 247–56.
Feier FH, Miura IK, Fonseca EA, Porta G, Pugliese R, Porta A, Schwartz IVD, Margutti AVB, Camelo JS Jr, Yamaguchi SN, Taveira AT, Seda Neto J. Successful domino liver transplantation in maple syrup urine disease with a related living donor. Braz J Med Biol Res. 2014;47(6):522–6.
Feier FH, Schwartz IVD, Benkert AR, Seda Neto J, Miura I, Chapchap P, da Fonseca EA, Vieira S, Zanotelli ML, e Vairo, F.P. and Camelo Jr, J.S., Strauss, K. A. . Living related versus deceased donor liver transplantation for maple syrup urine disease. Mol Genet Metab. 2016;117(3):336–43.
Mazariegos GV, Morton DH, Sindhi R, Soltys K, Nayyar N, Bond G, Shellmer D, Shneider B, Vockley J, Strauss KA. Liver transplantation for classical maple syrup urine disease: long-term follow-up in 37 patients and comparative united network for organ sharing experience. J Pediatr. 2012;160(1):116–21.
Quental S, Vilarinho L, Martins E, Teles EL, Rodrigues E, Diogo L, Garcia P, Eusébio F, Gaspar A, Sequeira S, Amorim A, Prata MJ. Incidence of maple syrup urine disease in Portugal. Mol Genet Metab. 2010;100:385–7.
Den Dunnen JT, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat. 2000;15(1):7–12.
Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, MacArthur D. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Naslavsky MS, Yamamoto GL, de Almeida TF, Ezquina SA, Sunaga DY, Pho N, Bozoklian D, Sandberg TOM, Brito LA, Lazar M, Bernardo DV, Zatz M. Exomic variants of an elderly cohort of Brazilians in the ABraOM database. Hum Mutat. 2017;38:751–63.
Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, Mooney SD, Radivojac P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25(21):2744–50.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–74.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
Henneke M, Flaschker N, Helbling C, Müller M, Schadewaldt P, Gärtner J, Wendel U. Identification of twelve novel mutations in patients with classic and variant forms of maple syrup urine disease. Hum Mutat. 2003;22(5):417.
Gorzelany K, Dursun A, Coşkun T, Kalkanoğlu-Sivri SH, Gökçay GF, Demirkol M, Feyen O, Wendel U. Molecular genetics of maple syrup urine disease in the Turkish population. Turk J Pediatr. 2009;51(2):97–102.
Abiri M, Karamzadeh R, Mojbafan M, Alaei MR, Jodaki A, Safi M, Kianfar S, Sarhaddi AB, Noori-Daloii MR, Karimipoor M, Zeinali S. In silico analysis of novel mutations in maple syrup urine disease patients from Iran. Metab Brain Dis. 2017;32(1):105–13.
Gupta D, Bijarnia-Mahay S, Saxena R, Kohli S, Dua-Puri R, Verma J, Thomas E, Shigematsu Y, Yamaguchi S, Deb R, Verma IC. Identification of mutations, genotype-phenotype correlation and prenatal diagnosis of maple syrup urine disease in Indian patients. Eur J Med Genet. 2015;58(9):471–8.
Li X, Ding Y, Liu Y, Ma Y, Song J, Wang Q, Li M, Qin Y, Yang Y. Eleven novel mutations of the BCKDHA, BCKDHB and DBT genes associated with maple syrup urine disease in the Chinese population: Report on eight cases. Eur J Med Genet. 2015;58(11):617–23.
Abiri M, Karamzadeh R, Karimipoor M, Ghadami S, Alaei MR, Bagheri SD, Bagherian H, Setoodeh A, Noori-Daloii MR, Zeinali S. Identification of six novel mutations in Iranian patients with maple syrup urine disease and their in silico analysis. Mutat Res. 2016;786:34–40.
Imtiaz F, Al-Mostafa A, Allam R, Ramzan K, Al-Tassan N, Tahir AI, Al-Numair NS, Al-Hamed MH, Al-Hassnan Z, Al-Owain M, Al-Zaidan H, Al-Sayed M. Twenty novel mutations in BCKDHA, BCKDHB and DBT genes in a cohort of 52 Saudi Arabian patients with maple syrup urine disease. Mol Genet Metab Rep. 2017;11:17–23.
Simon E, Fingerhut R, Baumkotter J, Konstantopoulou V, Ratschi-Mann R, Wendel U. Maple syrup urine disease: favourable effect of early diagnosis by newborn screening on the neonatal course of the disease. J Inherit Metab Dis. 2006;29:532–7.
Herber S, Schwartz IV, Nalin T, Netto CB, Camelo JS Jr, Santos ML, Ribeiro EM, Schüler-Faccini L, de Souza CFM. Maple syrup urine disease in Brazil: a panorama of the last two decades. J Pediatr. 2015;91(3):292–8.
Flaschker N, Feyen O, Fend S, Simon E, Schadewaldt P, Wendel U. Description of the mutations in 15 subjects with variant forms of maple syrup urine disease. J Inherit Metab Dis. 2007;30(6):903–9.
Tsuruta M, Mitsubuchi H, Mardy S, Miura Y, Hayashida Y, Kinugasa A, Ishitsu T, Matsuda I, Indo Y. Molecular basis of intermittent maple syrup urine disease: novel mutations in the E2 gene of the branched-chain alpha-keto acid dehydrogenase complex. J Hum Genet. 1998;43(2):91–100.
Wang Y, Qi M, Li T, Zhao Y. Two novel mutations in the BCKDHB gene (R170H, Q346R) cause the classic form of maple syrup urine disease (MSUD). Gene. 2012;498:112–5.
Chinsky J, Appel M, Almashanu S, Costeas P, Ambulos N Jr, Carmi R. A nonsense mutation (R242X) in the branched-chain alpha-keto acid dehydrogenase E1alpha subunit gene (BCKDHA) as a cause of maple syrup urine disease. Mutations in brief no. 160. Online. Hum Mutat. 1997;12(2):136.
Scaini G, Tonon T, Moura de Souza CF, Schuck PF, Ferreira GC, Quevedo J, Neto JS, Amorim T, Camelo JS, Margutti AVB, Tresbach RH, Streck EL. Evaluation of plasma biomarkers of inflammation in patients with maple syrup urine disease. J Inherit Metab Dis. 2018;41:631–40.
Dursun A, Henneke M, Ozgül K, Gartner J, Coşkun T, Tokatli A, Kalkanoğlu HS, Demirkol M, Wendel U, Ozalp I. Maple syrup urine disease: mutation analysis in Turkish patients. J Inherit Metab Dis. 2002;25(2):89–97.
Acknowledgements
Data contributions by the professional responsible for patients care at the Reference Center belonging to the Brazilian MSUD Network. Financial support by CNPq—National Council for Scientific and Technological Development, NAP—Nucleus of Research Support and CISBi USP—Center for Integrated Systems Biology of the University of São Paulo at Ribeirão Preto Medical School.
Funding
This research was funded by the Nucleus of Research Support (NAP), Center for Integrative Systems Biology of University of São Paulo- Brazil (CISBi-USP) and the Brazilian Maple Syrup Urine Disorder Network (Grant MCT/CNPq/CT-SAUDE #57/2010).
Author information
Authors and Affiliations
Contributions
AVBM was the Ph.D. student who performed the participants biomolecular study and analyzed the phenotype and genotype, WASJ helped with the mutations analysis, DFG helped with the methodology standardization and helped with the mutations analysis, GAM helped the biomolecular study, AAM did the sequencing reaction, TA, VMGP, RTBS, IKM, JSN, ESS, MLSFS, CML, TT and CFMS were responsible for collecting the blood samples and all the participants clinical data, FS-L helped with the mutations analysis, IVDS was the researcher responsible for the Brazilian Maple Syrup Urine Disorder Network and helped with the mutations analysis, and JSCJ was DRRC advisor and helped with the biomolecular analysis and the clinical data interpretation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The research was approved by the Scientific Committee and Research Ethics Committee of the Clinics Hospital of Porto Alegre, the main center and headquarter of the MSUD Network, Brazilian Network for Assistance and Research in Maple Syrup Urine Disease, under reference number CAEE 19061113.9.0000.5327 (20 May 2015), according to the Declaration of Helsinki. All other centers gave their ethical approval as participant centers. All patients completed an informed consent form (ICF) signed by the person in charge and their clinical data was recorded in a standardized form, filled out by the doctor responsible for the patient in his/her reference clinical center; these clinics were located in several regions of Brazil.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Primers sequences, PCR conditions and amplified fragment sizes of the BCKDHA gene. Description of data: Primer location, primer sequences, Annealing Temperature and amplified fragment sizes (amplicon size) of the BCKDHA gene.
Additional file 2.
Primers sequences, PCR conditions and amplified fragment sizes of the BCKDHB gene. Description of data: Primer location, primer sequences, Annealing Temperature and amplified fragment sizes (amplicon size) of the BCKDHB gene.
Additional file 3.
Primers sequences, PCR conditions and amplified fragment sizes of the DBT gene. Description of data: Primer location, primer sequences, Annealing Temperature and amplified fragment sizes (amplicon size) of the DBT gene.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Margutti, A.V.B., Silva, W.A., Garcia, D.F. et al. Maple syrup urine disease in Brazilian patients: variants and clinical phenotype heterogeneity. Orphanet J Rare Dis 15, 309 (2020). https://doi.org/10.1186/s13023-020-01590-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01590-7