
Abstract
Background
Schaaf-Yang syndrome (SYS) is a newly recognized imprinting related syndrome, which is caused by a truncating variant in maternally imprinted MAGEL2 located in 15q11-q13. Yet, precise pathomechanism remains to be solved. We sequenced MAGEL2 in patients suspected Prader-Willi syndrome (PWS) to delineate clinical presentation of SYS. We examined 105 patients with clinically suspected PWS but without a specific PWS genetic alteration. Sanger sequencing of the entire MAGEL2 gene and methylation-specific restriction enzyme treatment to detect the parent of origin were performed. Clinical presentation was retrospectively assessed in detail.
Results
Truncating variants in MAGEL2 were detected in six patients (5.7%), including a pair of siblings. All truncating variants in affected patients were on the paternally derived chromosome, while the healthy father of the affected siblings inherited the variant from his mother. Patients with MAGEL2 variants shared several features with PWS, such as neonatal hypotonia, poor suck, and obesity; however, there were also unique features, including arthrogryposis and a failure to acquire meaningful words. Additionally, an episode of neurological deterioration following febrile illness was confirmed in four of the six patients, which caused severe neurological sequalae.
Conclusions
SYS can be present in infants suspected with PWS but some unique features, such as arthrogryposis, can help discriminate between the two syndromes. An episode of neurological deterioration following febrile illness should be recognized as an important complication.
Similar content being viewed by others

Background
Chromosome 15q11-q13 contains a cluster of imprinted genes essential for normal mammalian neurodevelopment [1]. Paternal deletions of this region result in Prader-Willi syndrome (PWS, OMIM #176270) [1, 2]. It was recently reported that truncating variants in MAGEL2, one of the paternally expressed genes located in this region, caused Schaaf-Yang syndrome (SYS, OMIM #615547) [3,4,5,6,7,8,9,10]. SYS patients present with several symptoms typical of PWS, such as developmental delay, neonatal hypotonia, poor suck that requires special feeding techniques, and excessive weight gain. However, they also experience symptoms not typically seen in PWS, including arthrogryposis and autism spectrum disorder (ASD) [3,4,5,6,7,8,9,10]. Truncating variants in MAGEL2 cause severe arthrogryposis with reduced fetal movement resulting in perinatal death [11]. Recently, pathogenic variants in MAGEL2 were reported as causes of Chitayat-Hall syndrome, which is characterized by distal arthrogryposis, intellectual disability, dysmorphic features and hypopituitarism, and in particular, growth hormone deficiency (OMIM #208080) [12,13,14]. These reports demonstrated that SYS and Chitayat-Hall syndromes are allelic disorders.
The pathological mechanism underlying SYS is thought to be the loss of functional MAGEL2 [3, 5], yet a gain-of-function mechanism has also been suggested [9, 15]. MAGEL2 is located in the PWS critical region and is deleted in deletion-positive PWS patients. Therefore, a loss-of-function phenotype in MAGEL2 should also be associated with PWS. However, the paradox is that patients with SYS show more severe phenotypes than deletion-positive PWS patients. Interestingly, deletions of the entire paternal copy of MAGEL2 can cause different phenotypes like other conditions with a mild phenotype caused by a segmental deletion including other genes [16, 17].
In this study, we sequenced 105 patients with clinically suspected PWS but without a genetic alteration specific for PWS, and examined their clinical features.
Methods
Study subjects
The subjects were 105 patients in whom PWS was clinically suspected but not genetically diagnosed. We initially performed the SNURF-SNRPN and MEG3 DNA methylation tests. The MEG3 methylation test was performed to detect Temple syndrome, which demonstrates a PWS-like phenotype [18]. Subjects who tested negative in both tests were enrolled in the study. Experimental protocols were approved by the Ethical Committee for the Study of Human Gene Analysis at Nagoya City University Graduate School of Medical Sciences, and were carried out in accordance with the approved guidelines. Written informed consent was obtained from the parents of patients.
Genetic analyses
The SNURF-SNRPN and MEG3 DNA methylation tests were performed as described previously [18, 19]. Long-range PCR of the MAGEL2 coding region was performed as previously reported [3], followed by Sanger sequencing using a 3130xl DNA Analyzer (Thermo Fisher Scientific, Waltham, MA, USA) with six sequencing primer pairs (primer sequences are available on request). If a variant was detected, the parental origin of the variant was determined using the methylation-specific restriction enzyme, SmaI (New England Biolabs, Beverly, MA, USA), as previously reported [3].
Results
Molecular genetics
Among 105 patients with clinically suspected PWS but no PWS-specific genetic alteration, we identified truncating variants in MAGEL2 in six patients (five nonsense variants, one frameshift variant), including a pair of siblings (Fig. 1a). The variants were all centrally located within the single exon gene in amino acids 587 to 666. All truncating MAGEL2 variants were on the paternal allele. Additionally, 25 nonsynonymous single nucleotide variants were also identified (Table 1). The latter are likely to have no pathologic significance as they are rare SNPs registered in public databases, including the Human Genetic Variation Database (HGVD; http://www.hgvd.genome.med.kyoto-u.ac.jp) [20] and Exome Aggregation Consortium (EXaC; http://exac.broadinstitute.org), or they are maternally inherited.

a MAGEL2 variants and protein domain structures. Truncating MAGEL2 variants found in our cohort and previously reported in literature (RefSeq NM_019066.4). The six variants identified in our patients with Sanger sequencing confirmation are shown above the gene. The proline-rich region (Proline Rich; residues 13–700), USP7 binding site (U7BS; residues 949–1004), and MAGE homology domain (MAGE; residues 1020–1219) are indicated by their positions in the coding sequence. b Pedigree of a familial case and phasing of the MAGEL2 variant. The c.1762C > T variant identified in patients 5 and 6, who were siblings, was found to be inherited from the unaffected father and unaffected paternal grandmother. Sanger sequencing following SmaI digestion detected only the methylated allele (maternal allele). Red arrows indicate the MAGEL2 c.1762C > T variant site. Black arrows indicate the variant site after SmaI digestion
The c.1762C > T; p (Q588*) variant identified in the siblings (patients 5 and 6) was found to be inherited from the unaffected father, where the variant of the father was on his maternal allele (Fig. 1b). The remaining three variants in patients for whom both parent’s DNA were available for analysis were confirmed to be de novo. Parent samples from Patient 1 were not available.
Clinical features
Table 2 summarizes the clinical features of the six patients harboring truncating variants in MAGEL2. Neonatal hypotonia, poor suck, and developmental delay, all major symptoms of PWS, were confirmed in all subjects. Typical clinical features are illustrated in Fig. 2. Characteristic facial appearance, and hypogonadism, also major symptoms of PWS, were confirmed in the majority of patients. Short stature and small hands, minor criteria of PWS, were also confirmed in 5/6 and 6/6 patients, respectively. All patients showed arthrogryposis, which is not typically seen in PWS. In addition, growth hormone (GH) deficiency, seen in Chitayat-Hall syndrome, was observed in all three patients in whom GH was evaluated. The developmental quotient, measured in only three patients, was 13–21, indicating severe intellectual disability. Interestingly, an episode of neurological deterioration following febrile illness was confirmed in four of the six patients. Patient 2 developed a seizure and impaired consciousness on the second day after the onset of fever. Brain MRI revealed high signal intensity areas in the right parietal white matter on diffusion-weighted imaging. He became unable to walk because of neurological sequelae. Patient 6 developed vomiting and a cluster of seizures on the third day after the onset of fever and was placed on a mechanical ventilator. Brain MRI revealed high signal intensity areas in bilateral putamen and globus pallidus on T2-weighted imaging. He became bedridden after an episode of acute encephalopathy.

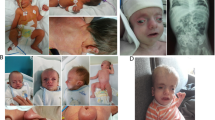
Clinical photographs of patient 2 (a, b, c), patient 4 (d, e, f), patient 5 (h, i, j) and patient 6 (g). Most patients show a shallow nasolabial fold, large ears, and contractures of the proximal and distal interphalangeal joints, small hands, and tapering of the fingers
Discussion
In this study, we identified MAGEL2 truncating variants in six of 105 (5.7%) patients initially suspected to have PWS, but excluded following specific genetic testing. These variants, including two novel variants, were centrally localized to a previously reported hotspot at amino acids 587–666 [5, 9]. To clarify how SYS and PWS share clinical features, we examined the clinical presentation of our patients with SYS. The major symptoms overlapping of PWS were hypotonia, poor sucking and developmental delay confirmed in all patients. We also found that four of the six (66.7%) patients in our cohort showed hypopigmentation, which is commonly seen in PWS but not previously reported in SYS. Hypopigmentation is thought to be caused by a dysfunction of OCA2 or GABRB3 [21], not by MAGEL2, and thus it was not expected. Yet, since our cohort was initially suspected of having PWS, an inclusion bias may be present. It was recently reported that Chitayat-Hall syndrome was also caused by pathogenic variants in MAGEL2 [12, 14]. GH deficiency, which is a characteristic of this syndrome, was found in all three cases in whom GH was evaluated. Therefore, GH deficiency should also be assessed in SYS. Interestingly, in a recent paper, Patak et al. produced a review of their cases and a systematic review and found no clinical or genetic differences between SYS and Chitayat–Hall syndrome [14]. In addition, arthrogryposis, which is not found in PWS, was confirmed in all patients as previously reported [3, 5,6,7,8,9,10]. Many of the intellectual disabilities confirmed in this cohort were severe. No patients acquired meaningful words. The mean intelligence quotient (IQ) of PWS patients is 60–70 [22], therefore intellectual disability in SYS patients is significantly more severe than PWS patients. However, Patients 1, 2, 3 and 6 may have been affected by neurological deterioration due to encephalopathy-like episodes. Interestingly, four of the six patients had an episode of neurological deterioration following febrile illness. Although McCarthy et al. reported that 67% of patients with SYS showed temperature instability [9], no episodes of neurological deterioration have been reported. In Japan, the prevalence of febrile seizures and acute encephalopathy is high [23, 24], which may be associated with the high prevalence of encephalopathy-like episodes in this population. In addition, because post-febrile regression is difficult to identify, particularly in patients with severe intellectual disabilities, some patients may have been overlooked. Therefore, careful observation should be performed for patients with SYS during febrile illness. Although it was reported that SYS patients have a higher prevalence of ASD [3, 5, 9], this could not be evaluated due to the severe intellectual disability in most of our patients. Previous studies report that among adolescent patients with SYS, the proportion of those who presented with hyperphagia and obesity was low, unlike that of patients with PWS, and many of those presented with ASD [9, 14]. In our study, it was difficult to evaluate patients in these respects because we could only observe two patients until puberty; however, we identified some patients without hyperphagia, obesity, or the personality characteristic of PWS. Overall, our study finds that symptoms of SYS are distinct from, and more severe than, those of PWS.
Next, a genotype–phenotype correlation is discussed. Previous reports have mentioned that c.1996dupC is the most common variant, which is more severe than other variants (e.g., frequencies of arthrogryposis, tube feeding, and respiratory dysfunction) [9, 14]. In our study, however, Patient 4 (c.1996dupC) had a history of tube feeding during infancy, but did not show respiratory dysfunction, indicating that the patient’s condition was comparable to that of other patients at least by the age of 6 years.
Buiting et al. [17] reported a 3-year-old boy with a paternally inherited approximately 3.9 Mb deletion that spanned MAGEL2 but not the SNORD116 cluster. The patient showed mild motor delay, which appears to be different to the SYS phenotype. Therefore, the truncated variants of MAGEL2 identified in SYS may cause different phenotypes to a complete deletion of MAGEL2, which is likely to represent loss of function. However, it should be noted that the 3.9 Mb deletion may not represent loss of function of MAGEL2 because it could disturb proper expression of some other genes. This can only be established however, once more cases with a complete deletion of MAGEL2 are reported. It should also be noted that almost only truncated variants but only one missense variant have been identified in MAGEL2 [14]. If loss of function is the major pathomechanism, a missense variant, especially at a functional domain, would be expected. As MAGEL2 is a single exon gene, nonsense-mediated mRNA decay is not induced. Therefore, truncating variants in MAGEL2 may result in abnormal truncated protein products through a gain-of-function mechanism. Although several articles have raised the possibility of a dominant negative mechanism [5, 15], it seems that a gain-of-function mechanism is the most likely pathological mechanism because there is no proof that MAGEL2 forms multimers.
MAGEL2 belongs to the MAGE family proteins that were initially identified as tumor-specific antigens [25]. Proteins encoded by the MAGE gene family, with approximately 40 unique members in humans, share the MAGE homology domain that mediates protein-protein interaction [25, 26]. MAGEL2 is known to bind and to enhance the activity of the TRIM27 E3 RING ubiquitin ligase. The MAGEL2-USP7-TRIM27 (MUST) complex plays an important role in a cellular process that recycles membrane proteins from endosomes through the retromer sorting pathway [15, 27, 28]. Thus, dysregulation of this pathway may be associated with the pathogenesis of SYS. Indeed, retromer has been implicated in several neurodegenerative disorders in humans, including Alzheimer’s and Parkinson’s disease [29, 30].
Conclusions
We identified truncating variants in MAGEL2 in six of 105 patients with suspected PWS. SYS can be present in infants suspected of PWS, but some unique features, such as arthrogryposis, can help discriminate between the two syndromes. An episode of neurological deterioration following febrile illness should be recognized as an important complication.
Availability of data and materials
The datasets supporting the conclusions of this article are available in the ClinVar repository (http://www.ncbi.nlm.nih.gov/clinvar/).
Abbreviations
- ASD:
-
Autism spectrum disorder
- GH:
-
Growth hormone
- IQ:
-
Intelligence quotient
- PCR:
-
Polymerase chain reaction
- PWS:
-
Prader-Willi syndrome
- SNP:
-
Single nucleotide polymorphism
- SYS:
-
Schaaf-Yang syndrome
References
Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10–26.
Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR, Whitman BY, Greenberg F. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91(2):398–402.
Schaaf CP, Gonzalez-Garay ML, Xia F, Potocki L, Gripp KW, Zhang B, Peters BA, McElwain MA, Drmanac R, Beaudet AL, et al. Truncating mutations of MAGEL2 cause Prader-Willi phenotypes and autism. Nat Genet. 2013;45:1405.
Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, LePichon JB, Miller NA, Thiffault I, Dinwiddie DL, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6(265):265ra168.
Fountain MD, Aten E, Cho MT, Juusola J, Walkiewicz MA, Ray JW, Xia F, Yang Y, Graham BH, Bacino CA, et al. The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families. Genet Med. 2017;19(1):45–52.
Hidalgo-Santos AD, del Carmen DeMingo-Alemany M, Moreno-Macian F, Rosello M, Orellana C, Martinez F, Caro-Llopis A, Leon-Carinena S, Tomas-Vila M. A novel mutation of MAGEL2 in a patient with Schaaf-Yang syndrome and hypopituitarism. Int J Endocrinol Metab. 2018;16(3):e67329.
Enya T, Okamoto N, Iba Y, Miyazawa T, Okada M, Ida S, Naruto T, Imoto I, Fujita A, Miyake N, et al. Three patients with Schaaf-Yang syndrome exhibiting arthrogryposis and endocrinological abnormalities. Am J Med Genet A. 2018;176(3):707–11.
Matuszewska KE, Badura-Stronka M, Smigiel R, Cabala M, Biernacka A, Kosinska J, Rydzanicz M, Winczewska-Wiktor A, Sasiadek M, Latos-Bielenska A, et al. Phenotype of two polish patients with Schaaf-Yang syndrome confirmed by identifying mutation in MAGEL2 gene. Clin Dysmorphol. 2018;27:49–52.
McCarthy J, Lupo PJ, Kovar E, Rech M, Bostwick B, Scott D, Kraft K, Roscioli T, Charrow J, Schrier Vergano SA, et al. Schaaf-Yang syndrome overview: report of 78 individuals. Am J Med Genet A. 2018;176:2564–74.
McCarthy JM, McCann-Crosby BM, Rech ME, Yin J, Chen CA, Ali MA, Nguyen HN, Miller JL, Schaaf CP. Hormonal, metabolic and skeletal phenotype of Schaaf-Yang syndrome: a comparison to Prader-Willi syndrome. J Med Genet. 2018;55(5):307–15.
Mejlachowicz D, Nolent F, Maluenda J, Ranjatoelina-Randrianaivo H, Giuliano F, Gut I, Sternberg D, Laquerriere A, Melki J. Truncating mutations of MAGEL2, a gene within the Prader-Willi locus, are responsible for severe Arthrogryposis. Am J Hum Genet. 2015;97(4):616–20.
Jobling R, Stavropoulos DJ, Marshall CR, Cytrynbaum C, Axford MM, Londero V, Moalem S, Orr J, Rossignol F, Lopes FD, et al. Chitayat-Hall and Schaaf-Yang syndromes:a common aetiology: expanding the phenotype of MAGEL2-related disorders. J Med Genet. 2018;55(5):316–21.
Chitayat D, Hall JG, Couch RM, Phang MS, Baldwin VJ. Syndrome of mental retardation, facial anomalies, hypopituitarism, and distal arthrogryposis in sibs. Am J Med Genet. 1990;37(1):65–70.
Patak J, Gilfert J, Byler M, Neerukonda V, Thiffault I, Cross L, Amudhavalli S, Pacio-Miguez M, Palomares-Bralo M, Garcia-Minaur S, et al. MAGEL2-related disorders: a study and case series. Clin Genet. https://doi.org/10.1111/cge.13620. Epub 2019 Aug 9.
Tacer KF, Potts PR. Cellular and disease functions of the Prader-Willi syndrome gene MAGEL2. Biochem J. 2017;474(13):2177–90.
Kanber D, Giltay J, Wieczorek D, Zogel C, Hochstenbach R, Caliebe A, Kuechler A, Horsthemke B, Buiting K. A paternal deletion of MKRN3, MAGEL2 and NDN does not result in Prader-Willi syndrome. Eur J Hum Genet. 2009;17(5):582–90.
Buiting K, Di Donato N, Beygo J, Bens S, von der Hagen M, Hackmann K, Horsthemke B. Clinical phenotypes of MAGEL2 mutations and deletions. Orphanet J Rare Dis. 2014;9:40.
Hosoki K, Kagami M, Tanaka T, Kubota M, Kurosawa K, Kato M, Uetake K, Tohyama J, Ogata T, Saitoh S. Maternal uniparental disomy 14 syndrome demonstrates prader-willi syndrome-like phenotype. J Pediatr. 2009;155(6):900–903 e901.
Kubota T, Das S, Christian SL, Baylin SB, Herman JG, Ledbetter DH. Methylation-specific PCR simplifies imprinting analysis. Nat Genet. 1997;16(1):16–7.
Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, Doi K, Shimizu M, Nakabayashi K, Aoki Y, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61(6):547–53.
Delahanty RJ, Zhang Y, Bichell TJ, Shen W, Verdier K, Macdonald RL, Xu L, Boyd K, Williams J, Kang JQ. Beyond epilepsy and autism: disruption of GABRB3 causes ocular hypopigmentation. Cell Rep. 2016;17(12):3115–24.
Roof E, Stone W, MacLean W, Feurer ID, Thompson T, Butler MG. Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. J Intellect Disabil Res. 2000;44(Pt 1):25–30.
Tsuboi T. Epidemiology of febrile and afebrile convulsions in children in Japan. Neurology. 1984;34(2):175–81.
Hoshino A, Saitoh M, Oka A, Okumura A, Kubota M, Saito Y, Takanashi J, Hirose S, Yamagata T, Yamanouchi H, et al. Epidemiology of acute encephalopathy in Japan, with emphasis on the association of viruses and syndromes. Brain Dev. 2012;34(5):337–43.
Chomez P, De Backer O, Bertrand M, De Plaen E, Boon T, Lucas S. An overview of the MAGE gene family with the identification of all human members of the family. Cancer Res. 2001;61(14):5544–51.
Barker PA, Salehi A. The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J Neurosci Res. 2002;67(6):705–12.
Hao YH, Doyle JM, Ramanathan S, Gomez TS, Jia D, Xu M, Chen ZJ, Billadeau DD, Rosen MK, Potts PR. Regulation of WASH-dependent actin polymerization and protein trafficking by ubiquitination. Cell. 2013;152(5):1051–64.
Seaman MN. The retromer complex - endosomal protein recycling and beyond. J Cell Sci. 2012;125(Pt 20):4693–702.
Bonifacino JS, Rojas R. Retrograde transport from endosomes to the trans-Golgi network. Nat Rev Mol Cell Biol. 2006;7(8):568–79.
Small SA, Petsko GA. Retromer in Alzheimer disease, Parkinson disease and other neurological disorders. Nat Rev Neurosci. 2015;16(3):126–32.
Acknowledgements
We are extremely grateful to all the patients and their parents who took part in this study. We wish to acknowledge Core Laboratory, Nagoya City University Graduate School of Medical Sciences. We wish to thank all members in the laboratories of Department of Pediatrics and Neonatology, Nagoya City University Graduate School of Medical Sciences, for their helpful assistance.
Funding
This study was funded by a grant from the Grant-in-Aid for Early-Career Scientists (18 K15682) to Y.N., by the Program for an Integrated Database of Clinical and Genomic Information from the Japanese Agency for Medical Research and Development, AMED (JP17kk0205002) to S.S. and by Grant-in-Aid for Research in Nagoya City University to S.S.
Author information
Authors and Affiliations
Contributions
YN performed the majority of the experimental work, analyzed and interpreted data, and wrote the manuscript. DI and IH performed experiments. YN, TY, HK, JT, KN, and HT provided samples and clinical data. SS initiated the project, analyzed the data and wrote the manuscript. All authors commented on and edited the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Experimental protocols were approved by the Ethical Committee for the Study of Human Gene Analysis at Nagoya City University Graduate School of Medical Sciences (approval number 112).
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this report and any accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Negishi, Y., Ieda, D., Hori, I. et al. Schaaf-Yang syndrome shows a Prader-Willi syndrome-like phenotype during infancy. Orphanet J Rare Dis 14, 277 (2019). https://doi.org/10.1186/s13023-019-1249-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-019-1249-4