
Abstract
Acid ceramidase (ACDase) deficiency is a spectrum of disorders that includes a rare lysosomal storage disorder called Farber disease (FD) and a rare epileptic disorder called spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME). Both disorders are caused by mutations in the ASAH1 gene that encodes the lysosomal hydrolase that breaks down the bioactive lipid ceramide. To date, there have been fewer than 200 reported cases of FD and SMA-PME in the literature. Typical textbook manifestations of classical FD include the formation of subcutaneous nodules, accumulation of joint contractures, and development of a hoarse voice. In reality, however, the clinical presentation is much broader. Patients may develop severe pathologies leading to death in infancy or may develop attenuated forms of the disorder wherein they are often misdiagnosed or not diagnosed until adulthood. A clinical variability also exists for SMA-PME, in which patients develop progressive muscle weakness and seizures. Currently, there is no known cure for FD or for SMA-PME. The main treatment is symptom management. In rare cases, treatment may include surgery or hematopoietic stem cell transplantation. Research using disease models has provided insights into the pathology as well as the role of ACDase in the development of these conditions. Recent studies have highlighted possible biomarkers for an effective diagnosis of ACDase deficiency. Ongoing work is being conducted to evaluate the use of recombinant human ACDase (rhACDase) for the treatment of FD. Finally, gene therapy strategies for the treatment of ACDase deficiency are actively being pursued. This review highlights the broad clinical definition and outlines key studies that have improved our understanding of inherited ACDase deficiency-related conditions.
Similar content being viewed by others


Background
Dr. Sidney Farber described the first case of “disseminated lipogranulomatosis” in a 14-month-old infant at a Mayo Foundation lecture in 1947. Farber later published a case series of three patients in 1952, as a transaction for the 62nd annual meeting of the American Pediatric Society. He later expanded the descriptions in 1957 [1, 2]. Farber originally hypothesized that the disease shared the lipid storage aspects of Niemann-Pick disease as well as the inflammation observed in Hand-Schüller-Christian disease. Although Farber demonstrated an increase in lipids in his early biochemical studies, the main lipid that accumulates in Farber disease (FD), i.e., ceramide, was not identified until 1967, when it was isolated from a biopsy of a patient’s kidney [3]. Acid ceramidase (ACDase), which was first purified in 1963, catalyzes the synthesis and degradation of ceramide into sphingosine and fatty acid [4]. In 1972, Sugita and colleagues established that ACDase activity was not detectable in post-mortem tissue from a FD patient [5]. In 1996, the ASAH1 gene that encodes ACDase was fully sequenced and characterized [6].
Our literature search spans 70 years and identifies 201 patients described as having ACDase deficiency (Tables 1 and 2). We included cases that were published in English, French, German, Chinese, Russian, and Arabic. While most of the cases we reviewed involved the classical FD phenotype, some were related to the rare motor neuron disease, SMA-PME (Tables 1 and 2). In this review, we will outline the clinical spectrum of ACDase deficiency and summarize key biochemical, genetic, and clinical studies related to this disorder.
Traditional classifications of Farber disease
Farber disease (FD; OMIM #228000), also known as Farber’s lipogranulomatosis, is an ultra-rare lysosomal storage disorder (LSD). It is caused by mutations in ASAH1, which lead to decreased ACDase activity and in turn, to ceramide accumulation and various pathological manifestations (Fig. 1). Moser and colleagues first categorized FD into 5 subtypes in a review in 1989, later adding two other phenotypes [7, 8]. Type 1, also termed the “classical” variant of FD, includes patients with the cardinal symptoms of subcutaneous nodules, joint contractures, and voice hoarseness. These patients may also develop enlarged liver and spleen along with neurological and respiratory complications [8, 9]. Traditionally, Type 1 FD patients exhibit symptoms during infancy and typically do not live past the age of 2–3 years [2, 10]. Types 2 and 3 FD patients have been termed the “intermediate” and “mild” variants, respectively; patients with these phenotypes usually have a longer lifespan due to reduced neurological involvement. However, Types 2 and 3 FD patients suffer from subcutaneous nodules, joint contractures, and aphonia due to inflammation. Types 4 and 5 FD patients have severe disease manifestations. Type 4 is associated with the “Neonatal-Visceral” variant, wherein neonates experience severe organomegaly and visceral histiocytosis [8, 11]. Type 5 is the “Neurological Progressive” variant, which is manifested by progressive neurological deterioration and seizures. Nodules and joint involvement are present in Type 5; however, they are less severe. Type 6 FD is termed “Combined Farber and Sandhoff Disease variant.” In this single co-incidental case, the patient had combined Farber and Sandhoff (OMIM #268800) diseases [12]. The patient presented with clinical signs of FD, and demonstrated a deficiency in both ACDase and hexosaminidases A and B [12]. Finally, Type 7 FD is termed “Prosaposin Deficiency.” This phenotype was identified in one patient and his infant sibling [13]; a mutation was identified in the precursor protein of saposins (i.e., prosaposin, encoded by the PSAP gene) [14]. A total of 4 saposins have been identified, and these proteins, along with the GM2 ganglioside activator protein, collectively belong to a group of sphingolipid activator proteins (SAPs). Only a handful of patients with Type 7 FD have been reported [15]. Similar to Type 6 FD, these patients often have multiple enzyme deficiencies, such as reduced glucocerebrosidase, galactocerebrosidase and ceramidase activities. While patients with prosaposin deficiency may show some biochemical and clinical signs that overlap with FD, it is considered a separate disease (OMIM #176801). Increasingly, many of the more recently reported cases simply identify FD as either the classic childhood or the mild and attenuated form [16,17,18]. Since some of these subtypes are rare and represent separate conditions, an updated classification should be considered to incorporate the existing and emerging phenotypes of ACDase deficiency.

The typical clinical manifestations by organ type that have been reported in cases of Farber Disease (FD) and SMA-PME in the published literature. FD symptoms organized by neurological symptoms, ophthalmic symptoms, cardinal triad symptoms, respiratory symptoms, hematopoietic symptoms, gastrointestinal involvement, dermatological manifestations, liver disease, motor neuron and muscle weakness, and bone disease phenotypes
Biochemistry, genetics and diagnosis
Acid ceramidase and ceramides
Acid ceramidase (ACDase) (E.C. #3.5.1.23) was first identified in 1963 by Gatt in rat brain extracts, where he demonstrated that ACDase was the catalyst for the hydrolysis of the amide bond of ceramides (Fig. 2) [4]. The optimal pH of ACDase is 4.5–5, and this enzyme is responsible for the hydrolysis of ceramide into a sphingosine and a free fatty acid. Due to the low pH, it had been suggested that the enzyme may have a role in the lysosomal system [19]. The first large purification of the enzyme was not performed until 1995 using human urine samples [20]. The purified enzyme was later identified as a heterodimer consisting of an α (13 kDa) and a β (40 kDa) subunits. Studies utilizing the first anti-ACDase polyclonal antibody revealed that ACDase is initially synthesized as a precursor polypeptide and then post-transcriptionally modified and processed into the α and β subunits within the lysosome [21]. These studies also revealed that cleavage into its subunits is essential for enzymatic activity. Later studies using rhACDase showed that cleavage of the precursor polypeptide occurs through an autoproteolytic reaction that is dependent on the cysteine residue 143 [22, 23]. Recently the crystal structure of mammalian ACDase was elucidated for both the proenzyme and the mature form [24]. This study showed that autocleavage of ACDase triggers a conformational change that uncovers the active site for ceramide entry [24]. Additional modeling demonstrated distinct catalytic mechanisms for autocleavage and for hydrolysis of substrate [24]. ACDase, like other enzymes, also exhibits a reverse reaction, in which ACDase can use C12:0 fatty acid and sphingosine to form ceramide at a pH of 6 rather than the lower pH of 4.5 [25]. Similar to other acidic hydrolases, ACDase is tagged with a mannose-6-phosphate residue for transport to the lysosomal compartment.

Reaction schema of the hydrolysis of ceramide by acid ceramidase into sphingosine and free fatty acid
Ceramide lies in one of the central steps in the breakdown and formation of other sphingolipids [26, 27]. Many of these lipids, such as sphingomyelin and complex glycolipids, play important roles in cell membranes [26, 27]. Other sphingolipids, such as glucosylceramides and sulfatides, are essential for the formation and breakdown of myelin [28]. Many species of ceramides exist, and each species is defined by the length, saturation, and hydroxylation of both the fatty acid and sphingoid base moieties [26]. Due to the central role of ceramide in sphingolipid metabolism, any imbalance in ceramide metabolism may lead to significant downstream effects and disease. This topic has been covered in a number of reviews [29,30,31,32,33].
Prevalence of ACDase deficiency
FD is an ultra-rare disease where the prevalence and incidence are not accurately known. According to the epidemiological Orphanet report, FD (ORPHA 333) has a predicted prevalence of < 1/1,000,000 (https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=333). While no formal comprehensive international epidemiological study has been performed for FD, one recent quantitative analysis of 96 case studies found that India and the USA had more than 10 reported cases, followed by Saudi Arabia, Germany, France, and Italy all of which had between 6 to 10 cases [34].
Based on our literature search, we identified 158 reported cases of FD between 1952 and 2018 (Table 1). SMA-PME currently has its own OMIM entry and is usually categorized as a subtype of SMA [35]. We identified 23 cases of SMA-PME associated with mutations in ASAH1 gene since Zhou and colleagues first reported this finding [36]. Due to the rare nature of both disorders, they originally appeared to be two very separate conditions; however, as more cases of each are characterized, the clinical pictures are starting to overlap. For example, a recent case described a patient who presented with muscle weakness typical of SMA-PME, but who also had joint pain synonymous with FD [37]. Additionally, several cases of FD have shown neurological involvement such as delayed mental development, seizures, and muscle weakness as predominant pathologies [9, 38,39,40].
Genetics and mutations
The human acid ceramidase gene (ASAH1) is approximately 30 kb in total length. It contains 14 exons that range from 46 to 1200 bp long and is mapped to the short arm of chromosome 8 (8p21.3/22) [41]. The first mutation identified, c.665C > A (p.T222K), was from a patient with a severe form of FD [6]. Based on the literature, we identified 61 pathologic mutations leading to FD or SMA-PME. These mutations are summarized in Tables 3 and 4. Additionally, at the time this review was being written, more than 120 genetic variants had been submitted to the NCBI ClinVar public archive [42]. While a number of these represent published mutations with a pathogenic role, most of the variants were submitted by clinical diagnostic testing facilities and did not include confirmed pathological details. Therefore, our curated list is likely an under-representation of all the sequenced pathologic mutations in FD. Nonetheless, several observations can be extrapolated. Mutations have been identified throughout the ASAH1 gene, but most of the mutations appear to be missense mutations (Fig. 3a-e). Among the recorded mutations that result in the diagnosis of FD, a majority is located within the β-subunit. Eighteen patients were identified to have a mutation in exon 8 and 9 patients had mutations in exon 13. In contrast, a larger number of mutations in SMA-PME have been identified within the α-subunit. One interesting observation is that the T42A and T42M mutations in exon 2 accounted for more than half of the total number of reported cases of SMA-PME. While some of these cases are siblings, they have also occurred within independent families [36, 43,44,45,46,47]. There is currently no definitive genotype-to-phenotype relationship in the noted mutations, which is especially true based on the observation that one patient with SMA-PME and another with FD had the same Y137C mutation [48, 49]. Another patient presented with polyarticular arthritic symptoms synonymous with FD and later developed muscle weakness with no PME [37]. These examples indicate that mutations in ASAH1 can result in a broad range of phenotypes.

Structure of the human ASAH1 gene with the protein and distribution of mutations. a ASAH1 genomic structure. b ASAH1 mature transcript structure. c Schematic of the ACDase protein with annotations for the signal peptide, α-subunit, and β-subunit. d Percentages of the reported 65 ASAH1 mutations by type for FD and SMA-PME. e Frequency of mutations by subunit and reported disease phenotype
In the same year that the relationship between ASAH1 and SMA-PME was established, another report also demonstrated that Han Chinese patients diagnosed with schizophrenia showed a down-regulation of the ASAH1 gene. Furthermore, this study identified two ASAH1 SNPs (rs7830490, and rs3753118) associated with schizophrenia [50]. This observation was also reported in a separate and larger study that analyzed the exomes of 12,332 Swedish individuals, of which 4,877 were affected by schizophrenia [51]. That study found patients with schizophrenia had a higher abundance of ultra-rare variants, of which 7 SNPs loci were in the ASAH1 gene (rs781294134, rs759037498, rs761518207, rs13785393, rs764327759, rs757058563, and rs773025886) [51]. One final example of the broad ACDase deficiency phenotype that can occur is the aforementioned case regarding keloid formation and the L386P mutation in ASAH1 [52].
Clinical diagnosis
FD is inherited in an autosomal recessive manner. Due to its rarity, prenatal screening/neonatal testing is typically not performed unless an older sibling has been previously diagnosed. Consideration of FD is typically based on the manifestation of the cardinal triad symptoms: 1) subcutaneous nodules, 2) joint pain, and 3) voice hoarseness [8]. Diagnosis of the mild and attenuated variants of FD is more troublesome since one or more of the featured symptoms may be absent or missed at the time of diagnosis. One report describes a patient who had no apparent subcutaneous nodule formation until the age of 12 years [53]. As mentioned, other cases have been misdiagnosed as juvenile idiopathic arthritis (JIA) [54, 55]. In fact, one cohort study demonstrated that as many as 71% of FD with mild to intermediate variants of FD were initially misdiagnosed as JIA [55]. Thus, the incidence of FD is likely underestimated. JIA patients who have symptoms consistent with the cardinal triad should also be encouraged to be tested for FD as part of their diagnosis.
In addition to JIA, the differential diagnosis includes rheumatoid arthritis, juvenile hyaline fibromatosis, and multi-centric histiocytosis, due to the similarity in joint and subcutaneous manifestations [8]. In severe cases, misdiagnosis may also occur since the main clinical picture is histiocytosis and hepatosplenomegaly [56]. In these cases, the cardinal symptoms are often masked or have not yet developed since these severe symptoms usually manifest early in infancy.
While the diagnosis of FD often requires further biochemical and genetic analyses, several case reports originating from developing countries have relied on clinical and histological diagnoses due to limited resources and lack of access to specialized diagnostic centers. Morphologic characterization is often achieved through analyses of subcutaneous nodules or other biopsied tissue. Common features reported include the presence of granulomas and large lipid-laden macrophages. A variety of studies have used ultrastructural analyses to demonstrate the presence of semi-curvilinear inclusions, also known as ‘Farber bodies, Banana bodies, and Zebra bodies’, in various tissue types [57,58,59,60].
Biochemical and genetic diagnosis
One method that has been adopted to assist in the diagnosis of FD is a lipid loading test on cultured living cells. In this technique, exogenously labeled sphingolipids are added to patient cells and ceramide turnover is assessed. A variety of precursors have been used, including [14C] stearic acid–labeled cerebroside sulfate in skin fibroblasts, [3H] sphingomyelin in both patient cultured fibroblasts and transformed lymphocytes, and [14C] serine, a precursor substrate in the de novo ceramide synthesis pathway, to demonstrate impaired ceramide degradation in FD [61,62,63].
The most common biochemical method in use for a definitive diagnosis of FD is an enzyme activity assay using cultured patient fibroblasts. Enzyme activity in FD cells is typically < 10% of normal controls, whereas SMA-PME cells have been reported to have as much as 32% of the activity of controls [8, 36]. In addition to fibroblasts, the enzyme activity assay has been tested using leukocytes, plasma, post-mortem tissue, and cultured amniocytes from prenatal testing [64,65,66,67,68]. Conventionally, ACDase activity is determined by the use of either radiolabeled ceramides or fluorescent ceramide analogues. Many of these compounds are not water-soluble and require the use of detergents in addition to specialized technical equipment for analyses [20, 64, 68,69,70,71,72,73]. This drawback means that diagnosis is available in only a very limited number of laboratories. Currently, ACDase activity can be detected with the use of the fluorogenic substrate Rbm14–12 in a 96-well plate in a high-throughput manner [74, 75].
Quantitation of excess ceramides is another method to assist in the diagnosis. The diacylglycerol kinase assay was commonly used in early studies to measure total ceramides, but it was limited because it did not provide information about individual ceramide species [76]. Later, chromatographic methods such as thin-layer chromatography and high-performance liquid chromatography, were also employed to quantify ceramides [77,78,79]. The major drawbacks to these methods were the requirement for radiolabeling or fluorophore incorporation. These methods were found to be difficult to perform and provided limited information on individual ceramide species. Mass spectrometry (MS), in particular electrospray ionization mass spectrometry (ESI/MS), is currently the most sensitive method for the discrimination and detection of sphingolipids [80,81,82,83,84,85]. These methods have been implemented to demonstrate excess ceramide in biopsy samples of subcutaneous nodules, post-mortem liver samples, urine samples, and cultured cells [8, 49, 57, 71, 77, 86,87,88,89].
Genetic testing
The first few mutations in ASAH1 were identified in patient cultured fibroblasts and required amplification of genomic sequences of ASAH1 and a combination of PCR and Sanger sequencing [6, 90]. Exome sequencing is now commonly performed and, in conjunction with biochemical assays, provides a conclusive diagnosis of ACDase deficiency [37, 91]. This is particularly informative in patients with non-classical FD, SMA-PME, and in cases in which the symptoms are suggestive of ACDase deficiency but have atypical presentations [47, 48, 92, 93].
Biomarkers
Increased inflammation and the formation of histiocytes are common in many cases of FD. Recent studies from our laboratories identified monocyte chemoattractant protein 1 (MCP-1) as a potential biomarker [94, 95]. A multiplex cytokine analysis was performed using plasma obtained from FD, JIA, and FD patients who underwent HSCT. This study demonstrated an elevation of MCP-1 in FD samples, but low levels in JIA and normalized levels in FD patients who underwent HSCT [94]. MCP-1 may thus be a beneficial biomarker and could help address the issue of misdiagnosis in mild cases of FD.
Another potential biomarker for the diagnosis of FD is C26:0 ceramide, which was identified by lipid MS quantification of ceramides from lipids extracted from dried blood spots [49]. Two isoforms of C26:0 have been described, with isoform 1 being expressed at a significantly higher level in the newborn (0–6 months) cohort versus the juvenile (0.5–4 years) and adult (> 17 years) cohorts. No details were provided regarding the clinical phenotypes of these patients, but the application of a platform for bloodspot analysis for newborns could be an important step in earlier diagnosis of ACDase deficiency.
The diverse signs and symptoms in ACDase deficiency
Cardinal triad symptoms of FD
The classical triad of symptoms that manifest in FD is the formation of subcutaneous nodules, painful and swollen joints, and the development of a hoarse voice and aphonia [9]. Subcutaneous nodules are palpable and may cause hyperesthesia; this is often evident within the first few weeks of nodule development in severe cases [2, 10, 12]. However, nodule formation may present later in life in attenuated forms of the disease [55, 96, 97]. Nodules typically appear on joints and over pressure points. With time, the nodules may thicken and increase in size and number, causing significant swelling. Joint contractures can manifest in a number of locations, ranging from the interphalangeal, metacarpal, wrist, elbow, knee, ankle, and facet joints of the spine [98,99,100,101]. Joint contractures are progressive, and the resulting lack of movement can severely limit mobility for some patients [99, 102]. The development of a hoarse voice also occurs as a result of nodule formation in the larynx. Infants are often reported to have a weak cry, which progresses to dysphonia and eventually an inability to speak [98]. The formation of the nodules in the upper airway may also expand to the epiglottis and cause swelling, which results in feeding and respiratory difficulties [10, 89, 103]. If the nodule formation is extreme, tracheostomy may be required [10, 86].
While a definitive diagnosis of FD ideally incorporates the measurement of ACDase enzyme activity, accessibility to the assay and/or a reference diagnostic center is an issue in certain developing countries [100, 104, 105]. In these circumstances, diagnosis of FD is made by relying on the triad symptoms and histological analysis.
Hematologic findings
Nodule formation and inflammation are ubiquitous within the spectrum of FD. This feature highlights the role that the hematopoietic system may play in the disease. The nodules are composed of foamy histiocytes and macrophages. This distinctive foamy phenotype is caused by the accumulation of storage material [98,99,100,101, 106, 107]. Ultrastructural analysis of nodules has revealed the presence of Zebra bodies and curved semi-linear tubular bodies (Farber bodies) [108,109,110]. Bloodwork samples from patients have also revealed an increased leukocyte count and erythrocyte sedimentation rate and moderately elevated plasma chitotriosidase and C-reactive protein (CRP) in severe cases [54, 111,112,113,114,115]. The formation of nodules and histiocytic infiltration may extend beyond the extremities and joints, and it has also been observed within the reticuloendothelial system, including the bone marrow, liver, lung, lymph node, and spleen, as well as the thymus and heart, in a number of patients [106, 116, 117]. In one case, solely the presence of invading histiocytes in a patient’s bone marrow aspirate led to the appropriate clinical identification of FD [118].
Several other hematologic findings have been reported. Enlarged lymph nodes have been noted in autopsy reports [2, 77, 116, 119]. Lymphadenopathy and calcification of the axillary lymph nodes have been detected on X-rays [100]. Finally, anemia, thrombocytopenia, and the presence of nucleated red blood cells have also been reported in FD patients [99, 100, 116].
Neurological findings
Neurological manifestations are usually only seen in patients with Type 5 or classical FD [8]; the epileptic picture that is characteristic of SMA-PME is described in a separate paragraph below. Neurological involvement in FD is broad and can affect the central or peripheral nervous systems. Within the brain, hydrocephalus and cortical brain atrophy have been detected by magnetic resonance imaging [120, 121]. Storage pathology has been reported in a variety of neural tissues, including the anterior horns of the spinal cord, the brain stem, the cerebral cortex, and the cerebellum [17, 120, 122,123,124]. Storage pathology has also been reported in cells of the peripheral nervous system (PNS), where both myelinating and non-myelinating Schwann cells have large membrane-bound inclusions [60, 124, 125]. Pathology descriptions suggest that compression of the axonal body may affect proper nerve conduction [98, 124, 125]. A number of case reports have documented the occurrence of seizures and developmental delay leading to intellectual disability [38, 120, 122, 124]. Due to the pathology in the anterior horn cells and peripheral neuropathy, patients may also present with hypotonia, muscle weakness, and atrophy, leading to them requiring wheelchairs [38, 120, 122,123,124].
Pulmonary findings
Beyond the development of the cardinal phenotypes, pulmonary complications are one of the more common occurrences in both classic and attenuated variants of FD [9]. Clinical signs may include sternal retraction, expiratory stridor, aphonia, and labored breathing [1, 38, 77, 97, 102]. As mentioned above, when nodule formation in the larynx and upper airway is extreme, tracheostomy may be required [56, 102, 126]. X-rays have shown presence of consolidation, nodular opacities, and lung atelectasis [86, 97, 102, 125]. Bronchial alveolar lavage and post-mortem analyses of patients have revealed significant inflammation with large lipid-laden macrophages and cellular infiltration throughout the bronchioles and alveoli [1, 67]. The lung tissue of one patient was described as poorly expanded with excessive connective tissue, and its ultrastructural analysis revealed lung histiocytes containing curvilinear storage bodies [123]. Pulmonary distress, infection, and pneumonia are the main causes of mortality [2, 8, 97, 101, 123, 127].
Ophthalmic findings
Ocular manifestations have mostly been associated with the classic form of FD and those with neurological involvement [8]. In Farber’s original description of the disorder, he reported that his second patient was blind; however, limited analysis was performed [2]. A variety of ophthalmic findings have been documented in the literature; the most common sign is a cherry red spot [77, 115, 125, 126, 128, 129]. Additional ocular manifestations include retinal opacification, corneal opacities, and macular degeneration [10, 59, 128, 130]. Other findings related to the eyes have included the presence of xanthoma-like growths in the conjunctiva, poor visual fixation, and nystagmus [102, 120, 127]. Post-mortem analyses of the eyes showed no abnormalities in the anterior segment, but the posterior segment contained birefringent lipids within the ganglion cell layer and displayed significant storage pathology in other cell types in the eye [128, 131].
Gastrointestinal findings
There are several cases in the literature describing gastrointestinal manifestations of FD. Persistent diarrhea has occasionally been seen in infants [99, 110]. One patient also exhibited extensive gastrointestinal lesions with widespread erosion of the gastrointestinal mucosa [110]. Another study that biopsied colonic tissue in a patient with severe disease demonstrated an increased level of apoptosis of cells within the crypt of the colon. This study also demonstrated that the caspase-3 positive cells co-localized with cells that were positive for GD3 gangliosides, concluding that colonocyte apoptosis may be triggered by the synthesis of GD3 as a consequence of ceramide accumulation [132].
Hepatic findings
A palpable liver and hepatomegaly are commonly reported in patients with the classic variant of FD [1, 59, 67, 86, 96]. Zebra bodies and Farber bodies have been observed in hepatocytes, endothelial cells, and Kupffer cells [133, 134]. The most significant liver pathology seen is in patients with severe type 4 FD [8]. Infants have presented with cholestatic jaundice, ascites, liver fibrosis, and elevated liver enzymes [11, 56, 135]. In a unique case, a 6-month-old infant showed significant liver failure and was misdiagnosed with neonatal hepatitis; he/she underwent liver transplantation, which subsequently normalized the liver function [56]. FD was properly diagnosed in that instance after the appearance of nodules and histiocytic infiltrates. In these few severe cases, the enlargement of visceral organs and histiocyte formation may mask or precede the appearance of nodules [56].
Bone findings
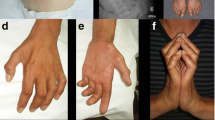
When joint involvement is present in FD patients, there may also be juxta-articular bone erosion and demineralization [86, 87, 96, 103]. In addition to the joints, bone erosion has been observed in long bones, metacarpals, metatarsals, and phalanges [111, 116, 136,137,138]. Osteoporosis is often progressive during the course of disease [97, 99, 102]. One patient, a 9-year-old girl, grew a tumorous osseous lesion in her spine, resulting in destruction of the odontoid by inflammatory cells. She underwent two HSCTs, which improved her mobility, but episodes of myoclonic epilepsy were still persistent [139]. In the milder spectrum, Bonafé et al. presented a case series of three siblings who displayed peripheral osteolysis between the ages of 40–60 years [93]. The patients all had shortened fingers and toes, as well as redundant skin. One of the siblings had limited movement of his knees and toes [73]. An unrelated 29-year-old patient also displayed deformities of the hands, demonstrating shortened fingers and redundant skin [97]. These patients had longer than average lifespans and were not formally diagnosed with FD until well into adulthood, which indicates that such milder cases may be underrepresented.
Dermatological findings
In addition to the formation of subcutaneous nodules, skin lesions and plaques have been reported in some FD patients [99, 140, 141]. Analyses of dermal biopsies have revealed hyalinized collagen in the dermis, hyperkeratosis, and the presence of large foamy histiocytes [99, 131, 134]. The storage pathology in dermal tissue and histiocytes revealed the presence of Farber bodies [134, 141, 142]. A rare presentation featured an infant with clinical signs that overlapped with stiff skin syndrome [113]. The infant displayed thick indurated skin since birth, a stiff neck, and scleroderma-like areas; he/she died at around 2 years of age [113]. Recently a study has demonstrated that heterozygous ASAH1 mutations may increase the susceptibility for keloid formation. This report performed genetic analyses on a Yoruba family in Nigeria, and of 24 members, 9 had keloids and 2 others had hypertrophic or stretched scars [52]. The L386P mutation (clinVar ID SCV000538196) was identified through a combination of linkage analyses and exome sequencing [52]. The appearance of keloids in this family ranged from 2 to 57 years of age. Additionally, the locations of keloid formation varied. Unfortunately, no lipid analysis or enzyme activity was reported. However, this variant nonetheless expands the clinical picture of ACDase deficiency [52].
Hydrops Fetalis
In the literature to date, there have been two FD patients presenting with hydrops fetalis [34]. One report is of a 29-week-old stillborn fetus with mild internal hydrops, a well-preserved spleen, and the presence of foamy cells [117]. The second report is of a 3-day-old neonate with an extreme phenotype of hydrops [106, 143]. The latter infant presented with an enlarged abdomen filled with hemorrhagic ascites, hepatosplenomegaly, and many white nodules on the peritoneal surfaces of the liver, spleen, and other organs. These two cases of fetal hydrops represent the shortest-lived patients recorded in the Farber literature.
Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME)
A new variant of ACDase deficiency has emerged that shares no classical signs and symptoms of FD. These patients have a separate disease called spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME) (OMIM #159950). SMA-PME was first described in 1978 by Jankovic and colleagues. He described patients from a family in Louisiana and Texas who first developed muscle weakness and wasting, which gradually progressed to jerking of the limbs and myoclonus [144]. Most patients who suffer from SMA typically have a mutation in SMA1 or SMD2 [36]. However, some patients who have SMA-PME have now been identified to carry mutations in ASAH1 [36, 44, 46, 91, 145, 146]. To the best of our knowledge, there have been 23 confirmed cases of SMA-PME with ASAH1 mutations reported in the literature to date (Table 1). Additionally, from 1978 to 2009, 20 cases were reported to have a SMA-PME-like clinical presentation, which include the original case described by Jankovic [144, 147,148,149,150,151].
Symptoms of SMA-PME may appear as early as 2 years of age [44] and include increasing difficulty in walking, sporadic falls, muscle weakness, and tremors [35, 36, 151]. Development of lower motor neuron disease in the form of muscle weakness is often the first manifestation of SMA-PME in patients [152]. Muscle weakness has been reported in young children between 3 and 7 years of age up to adolescents at 15 years of age [44, 145]. Lower motor neuron disease also affects the respiratory muscles. Death is usually attributed to respiratory failure and has been recorded as early as the teenage years [36, 44, 91, 145]. Epilepsy usually develops after the onset of neuronal disease during late childhood, though exceptions have occurred such as in the case report by Filosto and colleagues where two sister-patients both developed an adult SMA phenotype with no myoclonic epilepsy [35, 47]. The most common form of epilepsy is myoclonic seizures which appear as a series of shock-like upper limb proximal jerks [152]. Action myoclonus and myoclonic status have also been documented in some patients [44]. Lastly, other manifestations include development of generalized tremors, scoliosis, and sensorineural hearing loss [44, 91, 145, 152]. As the disease progresses, patients experience increasing seizure activity [151]. Impaired mobility, cognitive decline, and difficulty swallowing occur near the end of life [152].
Generalizations of the clinical picture should be made with caution since the number of identified SMA-PME patients is limited, and most reported cases share the same T42M mutation (Table 4). However, several cases encourage a broader understanding of SMA-PME. For example, the first description of an adult SMA patient with a mutation in ASAH1 did not present myoclonic epilepsy [47]. Another patient presented with eyelid myoclonic status epilepticus, in addition to muscle weakness, which has not been previously observed in SMA-PME [153].
Phenotypic variability in ACDase deficiency
ASAH1 mutations seem to result in two separate disorders, demonstrating the broad importance of ACDase for the proper maintenance of health. We have highlighted the diverse clinical spectrum that can be seen in various forms of ACDase deficiency. Interestingly, phenotypic variability is also seen in reports involving siblings. In one such case, one sibling demonstrated a classic Farber phenotype and died at 6 months of age, whereas the other sibling survived to 12 weeks of age and had extreme histiocytic infiltration throughout the body [67]. Surprisingly, post-mortem analyses of liver tissue from both patients revealed a similar level of enzyme activity [67]. Another case showed hepatosplenomegaly in a 3-month-old male [119]. While no nodules were noted in this patient, histiocytosis was the dominant phenotype. His sister, who was 5 and a half months old, displayed a classical phenotype of FD [119]. Fiumara et al. featured two sisters and one female cousin with a mild variant of FD and significant symptom variability [97]. Clinically, all three patients displayed nodule formation, joint involvement, and the presence of erosions [97]. However, variability in symptom onset and longevity was observed. One sister developed symptoms in her second year of life and lived to 30 years of age, whereas the other sister was symptomatic at 20 months of age and died when she was 18 years old. While ACDase enzyme activity was not reported for the shorter-lived sister, the assay was performed on cells cultured from the cousin who developed symptoms even earlier and died the earliest (at 11 years of age), the long-lived sister, and an established FD control cell line (FD patient who died at 1.8 years of age). In this enzyme activity assay, both the long-lived sister, the cousin, and the FD control showed an enzyme activity between 4 and 6% of normal controls [97]. Presumably, the three mild FD patients shared a similar mutation, yet there was obvious variability in symptom onset and patient longevity [97]. Similarly, enzyme activity for classical patients, who have shorter lifespans, may be comparable to those of patients who are long-lived. Therefore, while enzyme activity is important for diagnosis of FD, there is not a complete correlation between in vitro enzyme activity levels and patient outcomes.
Research, treatment and future therapy
Animal models
An ACDase knock-out mouse model was previously generated through insertional mutagenesis into the Asah1 gene. Heterozygous mice (Asah1+/−) did not show any overt changes in phenotype and had a normal life span of at least 1.5 years [154]. However, analyses of the organs of heterozygous mice 6 months of age and older revealed lipid accumulation and inclusions in the liver, lung, skin, and bone [154]. The heterozygous liver was the most affected; it became fibrous and pale. While most hepatic cell types were filled with lipids, the most significant effect was observed in Kupffer cells. By 9 months of age, some ceramides were also elevated in the heterozygous animals, in which the greatest accumulation was detected in the liver, with a 1.5–2-fold increase compared with wild-type animals. Homozygous mice (Asah1−/−) were embryonic lethal; none were detected at day E8.5 or later [154]. A second knock-out mouse was generated via a targeted ES cell clone [155]. Analyses of this model demonstrated that homozygous embryos did not survive beyond the 2-cell to the 4-cell stage and underwent apoptotic cell death, highlighting the importance of ACDase as a vital enzyme for early embryonic development.
A tamoxifen-induced conditional Asah1 knock-out mouse has also been developed [156]. Intraperitoneal delivery of tamoxifen in 5-week-old female mice resulted in impaired fertility due to lack of mature follicles in the ovaries. The follicles were not able to fully develop, and apoptosis occurred between the transition from the secondary to the antral stage. This observation supports the essential role of ACDase in ovary maturation and its importance in fertility [156]. Tamoxifen injection showed variable penetrance, where 100% Asah1 ablation was reported in skin and 70% in ovaries [156]. Reports using this conditional knockout have focused exclusively on the ovary phenotype. While classical and severe cases of FD do not survive to sexual maturity, data derived from these studies may prove relevant with regard to mild and attenuated FD patients. While no overt FD phenotypes have been reported, this model nonetheless may serve as an important tool for fertility studies.
Finally, a knock-in model has also been developed, in which an ASAH1 patient mutation (P362R) was introduced into the analogous murine locus (P361R), resulting in a mouse that recapitulates many of the phenotypes observed in classical cases of FD [157]. The P362R mutation has been identified in two patients with FD. One patient, who died at 1.5 years of age, had a classical form of FD and was homoallelic for the mutation [41]. The other patient, who died at 8 years of age, was heteroallelic for P362R and E138V [41]. Furthermore, this mutation site was selected because it represents the most conserved region of the gene between the species [157]. Homozygous (Asah1P361R/P361R) mice have a decreased lifespan and reduced weight. These mice develop a significant inflammatory phenotype and the accumulation of large foamy macrophages in many tissues. Recent studies have also shown that these mice have impaired hematopoiesis, central nervous pathology, abnormal skin development, and impaired lungs [94, 158,159,160]. The Asah1P361R/P361R model does not develop nodules, but it does exhibit many features that are seen in patients, such as inflammation; enlarged organs, including hepatosplenomegaly; respiratory distress; and neurological and behavioral impairment [94, 158,159,160]. Animal studies have thus provided key insights into ACDase biology and Asah1 mutant pathology. In addition, they have and will continue to serve as important models that will ultimately guide and inform the use of future therapies in patients.
Current treatment
There is currently no cure for ACDase deficiency. Current treatment strategies focus on symptom management. Anti-inflammatory medications and physical therapy can help address pain and mobility issues [18, 161, 162]. Surgical intervention may occasionally be applied for the removal of nodules in the hands and oral cavity [138, 163]. In one severe case in which a patient was misdiagnosed with hemangioendothelioma, a series of five surgeries to remove sacrococcygeal masses and three surgeries for scalp masses were performed over the course of a patient’s life. He/she eventually expired at 5 years of age [164]. HSCT is another therapeutic option and has been demonstrated to substantially improve mobility and pain in a number of FD patients lacking CNS involvement [165, 166]. Early studies in which HSCT was performed in two patients with classical FD with CNS complications were promising because they showed an elevation in ACDase activity and resolution of voice hoarseness, subcutaneous nodules, and painful joints [167, 168]. However, in both cases, HSCT did not reverse the neurological phenotypes, and the patients deteriorated over time. A recent article has provided long-term follow-up data on 10 FD patients who underwent HSCT within the last 15 years [169]. Eight of the 10 transplanted FD patients in that study are still alive with a mean survival time to date of 10.4 years [169]. Inflammatory joint disease was resolved in all the surviving patients, respiratory findings were variable, and the neurological deficits persisted (and even progressed in some of the patients) [169]. Despite the scarcity of patient data, HSCT appears to be a promising treatment for mild and attenuated FD.
For SMA-PME, most patients are prescribed anti-epileptic drugs to assist with seizure control, with mixed efficacy [146, 153]. Since respiratory complications are progressive, some patients may also require mechanical ventilation and gastric feeding [44, 153].
Gene therapy
ACDase deficiency is an attractive target for gene therapy because it is caused by a single gene defect. In fact, several gene therapies for monogenic lysosomal storage disorders are currently being investigated in clinical trials [170,171,172]. In the context of ACDase deficiency, one early study demonstrated that FD patient cells recovered ACDase activity when the cells were infected with an onco-retroviral vector that engineered expression of human ACDase [173]. This study confirmed that the transduced cells had increased ACDase activity and normalized ceramide levels [173]. Additionally, the treated cells could also cross-correct untreated cells when supplemented with medium from transduced cells that secreted human ACDase [173]. Through the mannose-6-phosphate receptor pathway, the non-infected cells acquired functional enzyme, demonstrating the effect of metabolic co-cooperativity. A later study reproduced this same effect using lentiviral vectors as the delivery vehicle and showed successful gene correction in hematopoietic stem cells [174]. That same study also showed that direct injection of vector into murine neonates could provide long-term expression of ACDase for up to 13 weeks [174]. This same approach was applied to the P361R FD mouse model and demonstrated an increased lifespan from 9 to 10 weeks to 16.5 weeks of age [157].
Ex vivo gene therapy is a treatment strategy that may deliver a longer-lasting therapeutic benefit than traditional HSCT. In this approach, stem, progenitor, or differentiated cells are isolated from a patient or donor, modified by genetic correction, and subsequently transplanted back into the patient [175, 176]. HSCs are a promising cell type for such gene therapy strategies since they are readily accessible and easily separated from a patient’s blood and can expand/differentiate into long-lived cell types [177, 178]. Ex vivo gene therapy followed by transplant represents an improvement over HSCT alone because the transduced cells express enzyme derived from the therapeutic vector in addition to their endogenous gene expression, which, in theory, allows for increased enzyme production, lysosomal activity, and potential cross-correction.
Many active gene therapy protocols are investigating such ex vivo HSCT transductions/transplantations to treat genetic disorders [179]. Ex vivo gene therapy followed by transplant may circumvent the limitations of HSCT alone to improve neurological symptoms, as in the case of metachromatic leukodystrophy [180]. In the case of ACDase deficiency, ex vivo transduction followed by HSCT is also a promising option. A series of proof-of-concept studies have demonstrated the successful transduction of the huACDase cDNA into murine CD34+ stem/progenitor cells and later into analogous cells from non-human primates [174, 181]. In the latter study, higher than normal ACDase enzyme activity was detectable in peripheral blood cells, in the bone marrow, the spleen and liver for more than a year [181]. Additionally, the animals had decreased ceramide levels [181].
At the time that this manuscript was written, a gene therapy trial was initiated for the treatment of SMA type I (clinicaltrials.gov ID NCT02122952). This trial involves the use of adeno-associated virus serotype 9 (AAV9), a non-integrating virus that encodes the SMA1 cDNA, infused through a peripheral vein. While the trial is still ongoing, preliminary data demonstrate a reduced need for pulmonary support, and patients could feed themselves, indicating a potential improvement in swallowing function [182, 183]. Although these results are for a different type of SMA, it is possible that a similar gene therapy approach may also be promising for patients with the SMA-PME phenotypes.
Enzyme replacement therapy
Enzyme replacement therapy (ERT) is currently the standard of care for several LSDs. Since early studies demonstrating the efficacy of ERT in Gaucher disease, this treatment strategy has been developed for a wide assortment of LSDs. It has been implemented to treat Pompe disease, Fabry disease, MPSI, II, VI, neuronal ceroid lipofuscinosis type 2 (CLN2), and Niemann-Pick B [184,185,186,187,188,189,190]. ERT with rhACDase is currently under development and represents a promising therapy for ACDase deficiency and several other conditions in which ceramide accumulation is pathologic, such as cystic fibrosis [191, 192]. Currently, large volume production of rhACDase is achieved by the amplification and transfection of Chinese hamster ovary (CHO) cells [193]. Overexpression of ACDase in CHO cells results in the secretion of enzyme into the medium, which is then purified by a series of chromatography steps [193].
A recent proof-of-concept study using the CHO-derived rhACDase as treatment in the P361R FD mouse model has shown promise [193]. Treatment with the recombinant enzyme resulted in decreased ceramide accumulation, less macrophage infiltration, lower MCP-1 expression, and a normalized spleen weight in FD mice [193]. This initial study holds promise for future FD treatments, but further investigations are required to better delineate the dose response in this model and to determine how this effect can be better translated to the human variant of FD or SMA-PME. One limitation of ERT is a reduced ability to cross the blood-brain barrier, which represents an issue for those LSDs that manifest with neurologic components, such as severe cases of FD. However, targeted CNS administration of enzyme has been observed to circumvent this limitation, and the use of fusion proteins with CNS-targeting moieties is currently being evaluated as a promising method for enzyme delivery to the CNS [194].
Conclusion
Over 70 years have passed since Farber’s Mayo Foundation lecture. Included in this historic transition is a brief transcript where Farber states: “The clinical picture I describe may be found to be typical for these 3 cases and may not be encountered in the next 20 or 30. We should, with a disease of this kind, expect to see a number of unrelated clinical pictures in the future” [1]. Farber’s comment and insight are highly relevant to this day. ACDase deficiency is a spectrum disorder that includes FD, SMA-PME, and potentially keloid formation or susceptibility to schizophrenia. Even amongst the individual conditions, there is a wide clinical spectrum. In mild cases, a misdiagnosis or a delay in diagnosis could impact the treatment plan and adversely affect the ability to properly manage symptoms [34]. A natural history study is currently underway on clinicaltrials.gov (ID NCT03233841), which aims to gain greater insight into the natural history of ACDase deficiency through retrospective and prospective patient data. It also aims to establish clinical information, biomarkers and other functional data to access the efficacy of future therapies, such as rhACDase ERT. The establishment of a complete natural history will greatly improve and potentially fill in gaps in the current definition of ACDase deficiency. Finally, due to the wide spectrum of clinical presentations, the precise number of patients is likely to be underrepresented. An improved understanding of the disease and increasingly effective knowledge translation will allow more patients to be identified, efficiently diagnosed, and effectively managed.
Abbreviations
- AAV9:
-
Adeno-associated virus serotype 9
- ACDase:
-
Acid ceramidase
- CHO:
-
Chinese hamster ovary
- CLN2:
-
Neuronal ceroid lipofuscinosis type 2
- CRP:
-
C-reactive protein
- CNS:
-
Central nervous system
- EEG:
-
Electroencephalogram
- EMG:
-
Electromyogram
- ERT:
-
Enzyme replacement therapy
- ESI/MS:
-
Electrospray ionization mass spectrometry
- FD:
-
Farber disease
- JIA:
-
Juvenile idiopathic arthritis
- LSD:
-
Lysosomal storage disorder
- MCP-1:
-
Monocyte chemoattractant protein 1
- MS:
-
Mass spectrometry
- PNS:
-
Peripheral nervous system
- rhACDase:
-
Recombinant human ACDase
- SAPs:
-
Sphingolipid activator proteins
- SMA-PME:
-
Spinal muscular atrophy with progressive myoclonic epilepsy
References
Farber S. A lipid metabolic disorder: disseminated lipogranulomatosis; a syndrome with similarity to, and important difference from, Niemann-pick and hand-Schuller-Christian disease. AMA Am J Dis Child. 1952;84(4):499–500.
Farber S, Cohen J, Uzman LL. Lipogranulomatosis; a new lipo-glycoprotein storage disease. J Mt Sinai Hosp NY. 1957;24(6):816–37.
Prensky AL, Ferreira G, Carr S, Moser HW. Ceramide and ganglioside accumulation in Farber's lipogranulomatosis.∗. Proc Soc Exp Biol Med . 1967;126(3):725–8.
Gatt S. Enzymic hydrolysis and synthesis of ceramides. J Biol Chem. 1963;238:3131–3.
Sugita M, Dulaney JT, Moser HW. Ceramidase deficiency in Farber's disease (lipogranulomatosis). Science. 1972;178(4065):1100–2.
Koch J, Gärtner S, Li C, Quintern LE, Bernardo K, Levran O, Schnabel D, Desnick RJ, Schuchman EH, Sandhoff K. Molecular cloning and characterization of a full-length complementary DNA encoding human acid ceramidase identification of the first molecular lesion causing Farber disease. J Biol Chem. 1996;271(51):33110–5.
Moser HW. Ceramidase deficiency: Farber lipogranulomatosis. The Metabolic Basis of Inherited Disease1989.
Levade T, Sandhoff K, Schulze H, Medin JA. Acid ceramidase deficiency: Farber lipogranulomatosis. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, editors. Scriver's OMMBID (Online Metabolic and Molecular Bses of Inherited Diseases). New York: McGraw-Hill; 2014.
Beck M, Moser H, Sandhoff K. Acid Ceramidase Deficiency: Farber Lipogranulomatosis and Spinal Muscular Atrophy Associated with Progressive Myoclonic Epilepsy. In: Rosenberg RNPJ, editor. Rosenberg's molecular and genetic basis of neurological and psychiatric disease. London: Elsevier; 2014. p. 395–402.
Zetterström R. Disseminated lipogranulomatosis (Farber's disease). Acta Paediatr. 1958;47(5):501–10.
Willis A, VanHuse C, Newton KP, Wasserstein M, Morotti RA. Farber's disease type IV presenting with cholestasis and neonatal liver failure: report of two cases. Pediatr Dev Pathol. 2008;11(4):305–8.
Fusch C, Huenges R, Moser HW, Sewell AC, Roggendorf W, Kustermann-Kuhn B, Poulos A, Carey WF, Harzer K. A case of combined Farber and Sandhoff disease. Eur J Pediatr. 1989;148(6):558–62.
Harzer K, Paton BC, Poulos A, Kustermann-Kuhn B, Roggendorf W, Grisar T, Popp M. Sphingolipid activator protein deficiency in a 16-week-old atypical Gaucher disease patient and his fetal sibling: biochemical signs of combined sphingolipidoses. Eur J Pediatr. 1989;149(1):31–9.
Schnabel D, Schroder M, Furst W, Klein A, Hurwitz R, Zenk T, Weber J, Harzer K, Paton BC, Poulos A. Simultaneous deficiency of sphingolipid activator proteins 1 and 2 is caused by a mutation in Initiation codon of their common gene. J Biol Chem. 1992;267(5):3312–5.
Hulkova H, Cervenkova M, Ledvinova J, Tochackova M, Hrebicek M, Poupetova H, Befekadu A, Berna L, Paton BC, Harzer K. A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation. Hum Mol Genet. 2001;10(9):927–40.
Solyom A, Simonaro CM, He X, Schuchman EH. Acid ceramidase deficiency: clinical implications of an emerging phenotypic spectrum and potential therapies. Mol Genet Metab. 2015;114(2):S109.
Bao XH, Tian JM, Ji TY, Chang XZ. A case report of childhood Farber's disease and literature review. Zhonghua Er Ke Za Zhi. 2017;55(1):54–8.
Schuchman EH, Mitchell J, Solyom A. Morbidity and mortality associated with Farber disease and prospects for therapy. Expert Opin Orphan Drugs. 2017;5(9):717–26.
De Duve C. The function of intracellular hydrolases. Exp Cell Res. 1959;7:169–82.
Bernardo K, Hurwitz R, Zenk T, Desnick RJ, Ferlinz K, Schuchman EH, Sandhoff K. Purification, characterization, and biosynthesis of human acid ceramidase. J Biol Chem. 1995;270(19):11098–102.
Ferlinz K, Kopal G, Bernardo K, Linke T, Bar J, Breiden B, Neumann U, Lang F, Schuchman EH, Sandhoff K. Human acid ceramidase: processing, glycosylation, and lysosomal targeting. J Biol Chem. 2001;276(38):35352–60.
He X, Okino N, Dhami R, Dagan A, Gatt S, Schulze H, Sandhoff K, Schuchman EH. Purification and characterization of recombinant, human acid ceramidase. Catalytic reactions and interactions with acid sphingomyelinase. J Biol Chem. 2003;278(35):32978–86.
Shtraizent N, Eliyahu E, Park JH, He X, Shalgi R, Schuchman EH. Autoproteolytic cleavage and activation of human acid ceramidase. J Biol Chem. 2008;283(17):11253–9.
Gebai A, Gorelik A, Li Z, Illes K, Nagar B. Structural basis for the activation of acid ceramidase. Nat Commun. 2018;9(1):1621.
Okino N, He X, Gatt S, Sandhoff K, Ito M, Schuchman EH. The reverse activity of human acid ceramidase. J Biol Chem. 2003;278(32):29948–53.
Futerman AH, Hannun YA. The complex life of simple sphingolipids. EMBO Rep. 2004;5(8):777–82.
Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9(2):139–50.
Yu RK, Nakatani Y, Yanagisawa M. The role of glycosphingolipid metabolism in the developing brain. J Lipid Res. 2009;50(Suppl):440.
Arana L, Gangoiti P, Ouro A, Trueba M, Gómez-Muñoz A. Ceramide and ceramide 1-phosphate in health and disease. Lipids Health Dis. 2010;9(1):15.
Soreghan B, Thomas SN, Yang AJ. Aberrant sphingomyelin/ceramide metabolic-induced neuronal endosomal/lysosomal dysfunction: potential pathological consequences in age-related neurodegeneration. Adv Drug Deliv Rev. 2003;55(11):1515–24.
Gangoiti P, Camacho L, Arana L, Ouro A, Granado MH, Brizuela L, Casas J, Fabriás G, Abad JL, Delgado A. Control of metabolism and signaling of simple bioactive sphingolipids: implications in disease. Prog Lipid Res. 2010;49(4):316–34.
Petrache I, Berdyshev EV. Ceramide signaling and metabolism in pathophysiological states of the lung. Annu Rev Physiol. 2016;78:463–80.
Gómez-Muñoz A, Ouro A, Arana L, Rivera I, Ordoñez M, Gangoiti P, Trueba M. Inhibition of ceramide metabolism key enzymes and its implication in cell physiology and pathology. Curr Enzym Inhib. 2011;7(4):191–204.
Zielonka M, Garbade SF, Kölker S, Hoffmann GF, Ries M. A cross-sectional quantitative analysis of the natural history of Farber disease: an ultra-orphan condition with rheumatologic and neurological cardinal disease features. Genet Med. 2017;20(5):524–30.
Topaloglu H, Melki J. Spinal muscular atrophy associated with progressive myoclonus epilepsy. Epileptic Disord. 2016;18(s2):128–34.
Zhou J, Tawk M, Tiziano FD, Veillet J, Bayes M, Nolent F, Garcia V, Servidei S, Bertini E, Castro-Giner F. Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1. Am J Hum Genet. 2012;91(1):5–14.
Teoh HL, Solyom A, Schuchman EH, Mowat D, Roscioli T, Farrar M, Sampaio H. Polyarticular arthritis and spinal muscular atrophy in acid ceramidase deficiency. Pediatrics. 2016;138(4):e20161068. Epub 2016 Sep 20
Eviatar L, Sklower SL, Wisniewski K, Feldman RS, Gochoco A. Farber lipogranulomatosis: an unusual presentation in a black child. Pediatr Neurol. 1986;2(6):371–4.
Colamaria V, Giardina L, Simeone M, Salviati A, Fensom AH, Dalla Bernardina B: Neurologic progressive form (type 5) of Farber's lipogranulomatosis (ceramidase deficiency) in monozygotic twins [abstract]. 1992
Ahmad A, Mazhar AU, Anwar M. Farber disease: a rare neurodegenerative disorder. J Coll Physicians Surg Pak. 2009;19(1):67–8.
Li C, Park J, He X, Levy B, Chen F, Arai K, Adler DA, Disteche CM, Koch J, Sandhoff K. The human acid ceramidase gene (ASAH): structure, chromosomal location, mutation analysis, and expression. Genomics. 1999;62(2):223–31.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2015;44(D1):D868.
Sathe S, Pearson T. Phenotypic characterization of the spinal muscular atrophy with progressive myoclonus epilepsy syndrome caused by ASAH1 mutations. Mol Genet Metab. 2014;111(2):S93.
Rubboli G, Veggiotti P, Pini A, Berardinelli A, Cantalupo G, Bertini E, Tiziano FD, D'amico A, Piazza E, Abiusi E. Spinal muscular atrophy associated with progressive myoclonic epilepsy: a rare condition caused by mutations in ASAH1. Epilepsia. 2015;56(5):692–8.
Johannsen J, Heinemeyer J, Denecke J. ASAH1 mutation in a boy with non-5q SMA and progressive myoclonic epilepsy. Neuropediatrics. 2015;46((S 01):25.
Giráldez BG, Guerrero-López R, Ortega-Moreno L, Verdú A, Carrascosa-Romero MC, García-Campos Ó, García-Muñozguren S, Pardal-Fernández JM, Serratosa JM. Uniparental disomy as a cause of spinal muscular atrophy and progressive myoclonic epilepsy: phenotypic homogeneity due to the homozygous c. 125C> T mutation in ASAH1. Neuromuscul Disord. 2015;25(3):222–4.
Filosto M, Aureli M, Castellotti B, Rinaldi F, Schiumarini D, Valsecchi M, Lualdi S, Mazzotti R, Pensato V, Rota S. ASAH1 variant causing a mild SMA phenotype with no myoclonic epilepsy: a clinical, biochemical and molecular study. Eur J Hum Genet. 2016;24(11):1578–83.
Kernohan KD, Frésard L, Zappala Z, Hartley T, Smith KS, Wagner J, Xu H, McBride A, Bourque PR, Consortium CC. Whole-transcriptome sequencing in blood provides a diagnosis of spinal muscular atrophy with progressive myoclonic epilepsy. Hum Mutat. 2017;38(6):611–4.
Cozma C, Iurascu MI, Eichler S, Hovakimyan M, Brandau O, Zielke S, Bottcher T, Giese AK, Lukas J, Rolfs A. C26-ceramide as highly sensitive biomarker for the diagnosis of Farber disease. Sci Rep. 2017;7(1):2.
Zhang H, Li D, Su Y, Jiang S, Xu Y, Jiang K, Cui D. Identification of the N-acylsphingosine amidohydrolase 1 gene (ASAH1) for susceptibility to schizophrenia in a Han Chinese population. World J Biol Psychiatry. 2012;13(2):106–13.
Genovese G, Fromer M, Stahl EA, Ruderfer DM, Chambert K, Landen M, Moran JL, Purcell SM, Sklar P, Sullivan PF, Hultman CM, McCarroll SA. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat Neurosci. 2016;19(11):1433–41.
Santos-Cortez RLP, Hu Y, Sun F, Benahmed-Miniuk F, Tao J, Kanaujiya JK, Ademola S, Fadiora S, Odesina V, Nickerson DA, Bamshad MJ, Olaitan PB, Oluwatosin OM, Leal SM, Reichenberger EJ. Identification of ASAH1 as a susceptibility gene for familial keloids. Eur J Hum Genet. 2017;25(10):1155–61.
Al Jasmi F. A novel mutation in an atypical presentation of the rare infantile Farber disease. Brain and Development. 2012;34(6):533–5.
Kostik MM, Chikova IA, Avramenko VV, Vasyakina LI, Le Trionnaire E, Chasnyk VG, Levade T. Farber lipogranulomatosis with predominant joint involvement mimicking juvenile idiopathic arthritis. J Inherit Metab Dis. 2013;36(6):1079–80.
Solyom A, Mitchell J, Beck M, Hügle B, Schuchman EH. Farber disease: design of the first observational and cross-sectional cohort study capturing retrospective and prospective data on the natural history and phenotypic spectrum of patients, including novel methodologies for assessment of disease-specific symptoms. Mol Genet Metab. 2017;120(1):S125.
Salo MK, Karikoski R, Hällström M, Jalanko H, Holmberg C. Farber disease diagnosed after liver transplantation. J Pediatr Gastroenterol Nutr. 2003;36(2):274–7.
Dustin P, Tondeur M, Jonniaux G, Vamos-Hurwitz E, Pelc S. La maladie de Farber. Etude anatomo-clinique et ultrastructurale. Bull Acad R Med Belg. 1973;128:733–62.
Rivel J, Vital C, Battin J, Heheunstre JP, Leger H. La lipogranulomatose disséminée de Farber études anatomoclinique et ultrastructurale de deux observations familiales. Arch Anat Cytol Pathol. 1977;25:37–42.
Tanaka T, Takahashi K, Hakozaki H, Kimoto H, Suzuki Y. Farber's disease (disseminated lipogranulomatosis) A pathological, histochemical and ultrastructural study. Pathol Int. 1979;29(1):135–55.
Schmoeckel C, Hohlfed M. A specific ultrastructural marker for disseminated lipogranulomatosis (Farber). Arch Dermatol Res. 1979;266(2):187–96.
Kudoh T, Wenger DA. Diagnosis of metachromatic leukodystrophy, Krabbe disease, and Farber disease after uptake of fatty acid-labeled cerebroside sulfate into cultured skin fibroblasts. J Clin Invest. 1982;70(1):89–97.
Levade T, Enders H, Schliephacke M, Harzer K. A family with combined Farber and Sandhoff, isolated Sandhoff and isolated fetal Farber disease: postnatal exclusion and prenatal diagnosis of Farber disease using lipid loading tests on intact cultured cells. Eur J Pediatr. 1995;154(8):643–8.
van Echten-Deckert G, Klein A, Linke T, Heinemann T, Weisgerber J, Sandhoff K. Turnover of endogenous ceramide in cultured normal and Farber fibroblasts. J Lipid Res. 1997;38(12):2569–79.
Sugita M, Iwamori M, Evans J, McCluer RH, Dulaney JT, Moser HW. High performance liquid chromatography of ceramides: application to analysis in human tissues and demonstration of ceramide excess in Farber's disease. J Lipid Res. 1974;15(3):223–6.
Dulaney JT, Milunsky A, Sidbury JB, Hobolth N, Moser HW. Diagnosis of lipogranulomatosis (Farber disease) by use of cultured fibroblasts. J Pediatr. 1976;89(1):59–61.
Fensom AH, Neville B, Moser A, Benson PF, Moser HW, Dulaney JT. Prenatal diagnosis of Farber's disease. Lancet. 1979;314(8150):990–2.
Antonarakis S, Valle D, Moser HW, Moser A, Qualman SJ, Zinkham WH. Phenotypic variability in siblings with Farber disease. J Pediatr. 1984;104(3):406–9.
Ben-Yoseph Y, Gagne R, Parvathy MR, Mitchell DA, Momoi T. Leukocyte and plasma N-laurylsphingosine deacylase (ceramidase) in Farber disease. Clin Genet. 1989;36(1):38–42.
Momoi T, Ben-Yoseph Y, Nadler HL. Substrate-specificities of acid and alkaline ceramidases in fibroblasts from patients with Farber disease and controls. Biochem J. 1982;205(2):419–25.
Azuma N, Obrien JS, Moser HW, Kishimoto Y. Stimulation of acid ceramidase activity by saposin D. Arch Biochem Biophys. 1994;311(2):354–7.
Chatelut M, Feunteun J, Harzer K, Fensom AH, Basile J, Salvayre R, Levade T. A simple method for screening for Farber disease on cultured skin fibroblasts. Clin Chim Acta. 1996;245(1):61–71.
Tani M, Okino N, Mitsutake S, Ito M. Specific and sensitive assay for alkaline and neutral ceramidases involving C12-NBD-ceramide. J Biochem. 1999;125(4):746–9.
Dagan A, Agmon V, Gatt S, Dinur T. Section II. Methods for analyzing aspects of sphingolipid metabolism in intact Cells-23 synthesis of fluorescent substrates and their application to study of sphingolipid metabolism in vitro and in. Meth Enzymol. 2000;312:293–303.
Bedia C, Casas J, Garcia V, Levade T, Fabriàs G. Synthesis of a novel ceramide analogue and its use in a high-throughput Fluorogenic assay for ceramidases. Chembiochem. 2007;8(6):642–8.
Bedia C, Camacho L, Abad JL, Fabrias G, Levade T. A simple fluorogenic method for determination of acid ceramidase activity and diagnosis of Farber disease. J Lipid Res. 2010;51(12):3542–7.
Van Veldhoven PP, Bishop WR, Yurivich DA, Bell RM. Ceramide quantitation: evaluation of a mixed micellar assay using E. coli diacylglycerol kinase. Biochem Mol Biol Int. 1995;36(1):21–30.
Moser HW, Prensky AL, Wolfe HJ, Rosman NP. Farber's lipogranulomatosis: Report of a case and demonstration of an excess of free ceramide and ganglioside. Am J Med. 1969;47(6):869–90.
Iwamori M, Costello C, Moser HW. Analysis and quantitation of free ceramide containing nonhydroxy and 2-hydroxy fatty acids, and phytosphingosine by high-performance liquid chromatography. J Lipid Res. 1979;20(1):86–96.
Cremesti AE, Fischl AS. Current methods for the identification and quantitation of ceramides: an overview. Lipids. 2000;35(9):937–45.
Gu M, Kerwin JL, Watts JD, Aebersold R. Ceramide profiling of complex lipid mixtures by electrospray ionization mass spectrometry. Anal Biochem. 1997;244(2):347–56.
Raith K, Wolf R, Wagner J, Neubert RH. Separation of phospholipids by nonaqueous capillary electrophoresis with electrospray ionisation mass spectrometry. J Chromatogr A. 1998;802(1):185–8.
Raith K, Neubert RH. Liquid chromatography–electrospray mass spectrometry and tandem mass spectrometry of ceramides. Anal Chim Acta. 2000;403(1):295–303.
Sullards MC. Analysis of sphingomyelin, glucosylceramide, ceramide, sphingosine, and sphingosine 1-phosphate by tandem mass spectrometry. Methods Enzymol. 2000;312:32–45.
Han X. Characterization and direct quantitation of ceramide molecular species from lipid extracts of biological samples by electrospray ionization tandem mass spectrometry. Anal Biochem. 2002;302(2):199–212.
Kasumov T, Huang H, Chung Y, Zhang R, McCullough AJ, Kirwan JP. Quantification of ceramide species in biological samples by liquid chromatography electrospray ionization tandem mass spectrometry. Anal Biochem. 2010;401(1):154–61.
Samuelsson K, Zetterström R. Ceramides in a patient with lipogranulomatosis (Farber's disease) with chronic course. Scand J Clin Lab Invest. 1971;27(4):393–405.
Amirhakimi GH, Haghighi P, Ghalambor MA, Honari S. Familial lipogranulomatosis (Farber's disease). Clin Genet. 1976;9(6):625–30.
Toppet M, Vamos-Hurwitz E, Jonniaux G, Cremer N, Tondeur M, Pelc S. Farber's disease as a ceramidosis: clinical, radiological and biochemical aspects. Acta Paediatrica Scand. 1978;67(1):113–9.
Ozaki H, Mizutani M, Hayashi H, Oka E, Ohtahara S, Kimoto H, Tanaka T, Hakozaki H, Takahashi K, Suzuki Y. Farber's disease (disseminated lipogranulomatosis): the first case reported in Japan. Acta Med Okayama. 1978;32(1):69–79.
Li C, Hong S, Kopal G, He X, Linke T, Hou W, Koch J, Gatt S, Sandhoff K, Schuchman EH. Cloning and characterization of the full-length cDNA and genomic sequences encoding murine acid ceramidase. Genomics. 1998;50(2):267–74.
Gan JJ, Garcia V, Tian J, Tagliati M, Parisi JE, Chung JM, Lewis R, Baloh R, Levade T, Pierson TM. Acid ceramidase deficiency associated with spinal muscular atrophy with progressive myoclonic epilepsy. Neuromuscul Disord. 2015;25(12):959–63.
Kim SY, Choi SA, Lee S, Lee JS, Hong CR, Lim BC, Kang HJ, Kim KJ, Park S, Choi M. Atypical presentation of infantile-onset farber disease with novel ASAH1 mutations. Am J Med Genet. 2016;170(11):3023–7.
Bonafé L, Kariminejad A, Li J, Royer-Bertrand B, Garcia V, Mahdavi S, Bozorgmehr B, Lachman RL, Mittaz-Crettol L, Campos-Xavier B. Brief report: peripheral Osteolysis in adults linked to ASAH1 (acid ceramidase) mutations: a new presentation of Farber's disease. Arthritis Rheum. 2016;68(9):2323–7.
Dworski S, Lu P, Khan A, Maranda B, Mitchell JJ, Parini R, Di Rocco M, Hugle B, Yoshimitsu M, Magnusson B, Makay B, Arslan N, Guelbert N, Ehlert K, Jarisch A, Gardner-Medwin J, Dagher R, Terreri MT, Marques Lorenco C, Barillas-Arias L, Tanpaiboon P, Solyom A, Norris JS, He X, Schuchman EH, Levade T, Medin JA. Acid ceramidase deficiency is characterized by a unique plasma cytokine and ceramide profile that is altered by therapy. BBA Molecular Basis of Disease. 2017;1863(2):386–94.
Yu FPS, Dworski S, Medin JA. Deletion of MCP-1 impedes pathogenesis of acid ceramidase deficiency. Sci Rep. 2018;8(1):6.
Abul-Haj SK, Martz DG, Douglas WF, Geppert LJ. Farber's disease: Report of a case with observations on its histogenesis and notes on the nature of the stored material. J Pediatr. 1962;61(2):221–32.
Fiumara A, Nigro F, Pavone L, Moser HW. Farber disease with prolonged survival. J Inherit Metab Dis. 1993;16(5):915–6.
Burck U, Moser HW, Goebel HH, Grüttner R, Held KR. A case of lipogranulomatosis Farber: some clinical and ultrastructural aspects. Eur J Pediatr. 1985;143(3):203–8.
Fujiwaki T, Hamanaka S, Koga M, Ishihara T, Nishikomori R, Kinoshita E, Furusho K. A case of Farber disease. Pediatr Int. 1992;34(1):72–9.
Mondal RK, Nandi M, Datta S, Hira M. Disseminated lipogranulomatosis. Indian Pediatr. 2009;46(2):175–77.
Ekici B, Kürkçü D, Çaliskan M. Farber disease: A clinical diagnosis. J Pediatr Neurosci. 2012;7(2):154–55.
Kim YJ, Park SJ, Park CK, Kim SH, Lee CW. A case of Farber lipogranulomatosis. J Korean Med Sci. 1998;13(1):95–8.
Schultze G, Lang EK. Disseminated lipogranulomatosis: report of a case. Radiology. 1960;74(3):428–31.
Erfan M, Haque AU, Ahmed SA. Farber's disease: a case report. IntJPathol. 2015;13:115–9.
Nasreen A, Sunil AK, Pulak MR, Sujit BK. Clinical diagnosis of Farber’s disease-a rare case report. Int J Health Sci Res. 2017;7(7):388–90.
Kattner E, Schäfer A, Harzer K. Hydrops fetalis: manifestation in lysosomal storage diseases including Farber disease. Eur J Pediatr. 1997;156(4):292–5.
Devi ARR, Gopikrishna M, Ratheesh R, Savithri G, Swarnalata G, Bashyam M. Farber lipogranulomatosis: clinical and molecular genetic analysis reveals a novel mutation in an Indian family. J Hum Genet. 2006;51(9):811–4.
Rauch HJ, Auböck L. " Banana bodies" in disseminated lipogranulomatosis (Farber's disease). Am J Dermatopathol. 1983;5(3):263–6.
Cartigny B, Libert J, Fensom AH, Martin JJ, Dhondt JL, Wyart D, Fontaine G, Farriaux JP. Clinical diagnosis of a new case of ceramidase deficiency (Farber's disease). J Inherit Metab Dis. 1985;8(1):8.
Koga M, Ishihara T, Uchino F, Fujiwaki T. An autopsy case of Farber's lipogranulomatosis in a Japanese boy with gastrointestinal involvement. Pathol Int. 1992;42(1):42–8.
Schanche AF, Bierman SM, Sopher RL, O'loughlin BJ. Disseminated lipogranulomatosis: early roentgenographic changes. Radiology. 1964;82(4):675–8.
Klingkowski U, Beck M, Zépp F. An 18-month-old girl with hoarseness, stiff joints and subcutaneous nodules. Eur J Pediatr. 1998;157(6):515–6.
El-Kamah GY, El-darouti MA, Kotoury AI, Mostafa MI. Farber disease overlapping with stiff skin syndrome: Expanding the Spectrum. EJMHG. 2009;10(1):97–104.
Torcoletti M, Petaccia A, Pinto RM, Hladnik U, Locatelli F, Agostoni C, Corona F. Farber disease in infancy resembling juvenile idiopathic arthritis: identification of two new mutations and a good early response to allogeneic haematopoietic stem cell transplantation. Rheumatology. 2014;53(8):1533–4.
Saygi S, Haytoglu Z, Savas T, Alkan O, Erol I. P78 – 2554: Farber disease in a child with a novel homozygous c.92G>T mutation [abstract]. Eur J Paediatr Neurol. 2015;19:S116.
Antonarakis S, Valle D, Moser H, Zinkham W, Qualman S. Farber’s lipogranulomatosis: variability of expression and clinical overlap with histiocytosis. Pediatr Res. 1983;17:206A.
van Lijnschoten G, Groener JE, Maas SM, Ben-Yoseph Y, Dingemans KP, Offerhaus GJA. Intrauterine fetal death due to Farber disease: case report. Pediatr Dev Pathol. 2000;3(6):597–602.
Nivaggioni V, Cano A, Arnoux I, Michel G, Loosveld M. Early morphological diagnosis of Farber disease. Br J Haematol. 2016;175(2):189.
Qualman SJ, Moser HW, Valle D, Moser AE, Antonarakis SE, Boitnott JK, Zinkham WH, Opitz JM, Bernstein J. Farber disease: pathologic diagnosis in sibs with phenotypic variability. Am J Med Genet A. 1987;28(S3):233–41.
Chedrawi AK, Al-Hassnan ZN, Al-Muhaizea M, Colak D, Al-Younes B, Albakheet A, Tulba S, Kaya N. Novel V97G ASAH1 mutation found in Farber disease patients: unique appearance of the disease with an intermediate severity, and marked early involvement of central and peripheral nervous system. Brain and Development. 2012;34(5):400–4.
Muranjan M, Agarwal S, Lahiri K, Bashyam M. Novel biochemical abnormalities and genotype in Farber disease. Indian Pediatr. 2012;49(4):320–2.
Molz G. Farbersche Krankheit. Virchows Arch. 1968;344(1):86–99.
Bierman SM, Edgington T, Newcomer VD, Pearson CM. A disorder of mucopolysaccharide metabolism with articular, respiratory, and neurologic manifestations. Arthritis Rheum. 1966;9(4):620–30.
Zappatini-Tommasi L, Dumontel C, Guibaud P, Girod C. Farber disease: an ultrastructural study. Virchows Archiv A. 1992;420(3):281–90.
Pellissier JF, Berard-Badier M, Pinsard N. Farber's disease in two siblings, sural nerve and subcutaneous biopsies by light and electron microscopy. Acta Neuropathol. 1986;72(2):178–88.
Cvitanovic-Sojat L, Juraski RG, Sabourdy F, Fensom AH, Fumic K, Paschke E, Levade T. Farber lipogranulomatosis type 1–late presentation and early death in a Croatian boy with a novel homozygous ASAH1 mutation. Eur J Paediatr Neurol. 2011;15(2):171–3.
Ohfu M, Abe K, Hirano H, Ogata H, Mitsudome A, Okada S, Inui K, Nishimoto J. Farber Lipogranulomatosis-A report of a case with nystagmus, myoclonus and convulsions. Brain Dev. 1987;9(2):227.
Cogan DG, Kuwabara T, Moser H, Hazard GW. Retinopathy in a case of Farber's lipogranulomatosis. Arch Ophthalmol. 1966;75(6):752–7.
Zarbin MA, Green WR, Moser HW, Morton SJ. Farber's disease: light and electron microscopic study of the eye. Arch Ophthalmol. 1985;103(1):73–80.
Chandwani R, Kuwar AS. Farber's disease. Indian Pediatr. 2002;39(5):502.
Bashyam MD, Chaudhary AK, Kiran M, Reddy V, Nagarajaram HA, Dalal A, Bashyam L, Suri D, Gupta A, Gupta N. Molecular analyses of novel ASAH1 mutations causing Farber lipogranulomatosis: analyses of exonic splicing enhancer inactivating mutation. Clin Genet. 2014;86(6):530–8.
Farina F, Cappello F, Todaro M, Bucchieri F, Peri G, Zummo G, Stassi G. Involvement of caspase-3 and GD3 ganglioside in ceramide-induced apoptosis in Farber disease. J Histochem Cytochem. 2000;48(1):57–62.
Hoof F, Hers HG. The abnormalities of lysosomal enzymes in mucopolysaccharidoses. FEBS J. 1968;7(1):34–44.
Abenoza P, Sibley RK. Farber's disease: a fine structural study. Ultrastruct Pathol. 1987;11(4):397–403.
Nowaczyk M, Feigenbaum A, Silver MM, Callahan J, Levin A, Jay V. Bone marrow involvement and obstructive jaundice in Farber lipogranulomatosis: clinical and autopsy report of a new case. J Inherit Metab Dis. 1996;19(5):655–60.
Crocker AC, Cohen J, Farber S. The “lipogranulomatosis” syndrome; Review, with report of patient showing milder involvement. Inborn disorders of sphingolipid metabolism. New York: Pergamon Press; 1967. p. 485–503.
Pavone L, Moser HW, Mollica F, Reitano C, Durand P. Farber's lipogranulomatosis: ceramidase deficiency and prolonged survival in three relatives. Johns Hopkins Med J. 1980;147(5):193–6.
Moritomo H, Nakase T, Maeda K, Murase T, Yoshikawa H. Surgical treatment of hand disorders in Farber's disease: A case report. J Hand Surg. 2002;27(3):503–7.
Jarisch A, Steward CG, Sörensen J, Porto L, Kieslich M, Klingebiel T, Bader P. Odontoid infiltration and spinal compression in Farber disease: reversal by haematopoietic stem cell transplantation. Eur J Pediatr. 2014;173(10):1399–403.
Chanoki M, Ishii M, Fukai K, Kobayashi H, Hamada T, Murakami K, Tanaka A. Farber's lipogranulomatosis in siblings: light and electron microscopic studies. Br J Dermatol. 1989;121(6):779–85.
Schmoeckel C. Subtle clues to diagnosis of skin diseases by electron microscopy:" Farber bodies" in disseminated lipogranulomatosis (Farber's disease). Am J Dermatopathol. 1980;2(2):153–4.
Navarro C, Fachal C, Rodriguez C, Padro LL, Dominguez C. Lipoid proteinosis. A biochemical and ultrastructural investigation of two new cases. Br J Dermatol. 1999;141(2):326–31.
Schafer A, Harzer K, Kattner E, Schafer HJ, Stoltenburg G, Lietz H. Disseminated lipogranulomatosis (Farber disease) with hydrops fetalis. Pathologe. 1996;17(2):145–9.
Jankovic J, Rivera V. Hereditary myoclonus and progressive muscular atrophy: a new syndrome. Trans Am Neurol Assoc. 1978;103:116–8.
Dyment DA, Sell E, Vanstone MR, Smith AC, Garandeau D, Garcia V, Carpentier S, Le Trionnaire E, Sabourdy F, Beaulieu CL, Schwartzentruber JA, McMillan HJ, FORGE Canada Consortium, Majewski J, Bulman DE, Levade T, Boycott KM. Evidence for clinical, genetic and biochemical variability in spinal muscular atrophy with progressive myoclonic epilepsy. Clin Genet. 2014;86(6):558–63.
Özkara B, Budak F. Spinal Muscular Atrophy with myoclonic epilepsy. Epilepsi: Journal of the Turkish Epilepsi Society. 2017;23(1):29–30.
Lance JW, Evans WA. Progressive myoclonic epilepsy, nerve deafness and spinal muscular atrophy. Clin Exp Neurol. 1984;20:141–51.
D'Ecclesia G, Scorrano V, Bernardini C, Poli V. Unusual familial association of epilepsy, myoclonus and muscular atrophy. Case report. Riv Neurol. 1985;55(5):313–6.
Taglioli M, Bartolini S, Volpi G, Alberti G, Ambrosetto G. Progressive familial myoclonic epilepsy with bulbo-spinal amyotrophy. Clinical, electrophysiological study, and biopsy of a case. Riv Neurol. 1990;60(5):201–6.
Ferlazzo E, Italiano D, An I, Calarese T, Laguitton V, Bramanti P, Di Bella P, Genton P. Description of a family with a novel progressive myoclonus epilepsy and cognitive impairment. Mov Disord. 2009;24(7):1016–22.
Haliloglu G, Chattopadhyay A, Skorodis L, Manzur A, Mercuri E, Talim B, Akçören Z, Renda Y, Muntoni F, Topaloğlu H. Spinal muscular atrophy with progressive myoclonic epilepsy: report of new cases and review of the literature. Neuropediatrics. 2002;33(06):314–9.
Dyment DA, Bennett SA, Medin JA, Levade T: ASAH1-related disorders. 2018.
Oguz Akarsu E, Tekturk P, Yapici Z, Tepgec F, Uyguner ZO, Baykan B. Eyelid myoclonic status epilepticus: a rare phenotype in spinal muscular atrophy with progressive myoclonic epilepsy associated with ASAH1 gene mutation. Seizure. 2016;42:49–51.
Li C, Park J, Simonaro CM, He X, Gordon RE, Friedman A, Ehleiter D, Paris F, Manova K, Hepbiloikler S. Insertional mutagenesis of the mouse acid ceramidase gene leads to early embryonic lethality in homozygotes and progressive lipid storage disease in heterozygotes. Genomics. 2002;79(2):218–24.
Eliyahu E, Park JH, Shtraizent N, He X, Schuchman EH. Acid ceramidase is a novel factor required for early embryo survival. FASEB J. 2007;21(7):1403–9.
Eliyahu E, Shtraizent N, Shalgi R, Schuchman EH. Construction of conditional acid ceramidase knockout mice and in vivo effects on oocyte development and fertility. Cell Physiol Biochem. 2012;30(3):735–48.
Alayoubi AM, Wang JC, Au BC, Carpentier S, Garcia V, Dworski S, El-Ghamrasni S, Kirouac KN, Exertier MJ, Xiong ZJ, Prive GG, Simonaro CM, Casas J, Fabrias G, Schuchman EH, Turner PV, Hakem R, Levade T, Medin JA. Systemic ceramide accumulation leads to severe and varied pathological consequences. EMBO Mol Med. 2013;5(6):827–42.
Lopez-Vasquez L, Dworski S, Pouliot R, Galbraith T, Kamani MA, Lacroix D, Auger FA, Medin JA. Acid ceramidase deficiency leads to multiple skin abnormalities in a mouse model of Farber disease. Mol Genet Metab. 2016;117(2):S76.
Yu FP, Islam D, Sikora J, Dworski S, Gurka' J, Lopez-Vasquez L, Liu M, Kuebler WM, Levade T, Zhang H, Medin JA: Chronic lung injury and impaired pulmonary function in a mouse model of acid ceramidase deficiency. Am J Physiol Lung Cell Mol Physiol. 2017;314(3):L406–20.
Sikora J, Dworski S, Jones EE, Kamani MA, Micsenyi MC, Sawada T, Le Faouder P, Bertrand-Michel J, Dupuy A, Dunn CK, Yang Xuan Ingrid C, Casas J, Fabrias G, Hampson DR, Levade T, Drake Richard R, Medin JA, Walkley SU. Acid ceramidase deficiency in mice results in a broad range of central nervous system abnormalities. Am J Pathol. 2017;187(4):864–83.
El-Darouti MA. A child with hoarse cry, subcutaneous nodules and joint contracture. In: Anonymous Springer, editor. Challenging Cases in Dermatology; 2013. p. 107–12.
Mitchell J, Solyom A, Makay B, Arslan N, Batu ED, Ozen S, Hügle B, Schuchman E, Magnusson B. Farber disease: implications of anti-inflammatory treatment. Mol Genet Metab. 2016;117(2):S82.
Haraoka G, Muraoka M, Yoshioka N, Wakami S, Hayashi I. First Case of Surgical Treatment of Farber's Diseas. Ann Plast Surg. 1997;39(4):405–10.
Lee SM, Oh C, Jung S, Kim H. Farber Disease Misdiagnosed as Hemangioendothelioma. J Korean Assoc Pediatr Surg. 2016;22(2):54–8.
Vormoor J, Ehlert K, Groll AH, Koch H, Frosch M, Roth J. Successful hematopoietic stem cell transplantation in Farber disease. J Pediatr. 2004;144(1):132–4.
Ehlert K, Frosch M, Fehse N, Zander A, Roth J, Vormoor J. Farber disease: clinical presentation, pathogenesis and a new approach to treatment. Pediatr Rheumatol. 2007;5(1):15.
Souillet G, Guiband P, Fensom AH, Maire I, Zabot MT. Outcome of displacement bone marrow transplantation in Farber’s disease: a report of a case. Correction of certain genetic diseases by transplantation, vol. 1989. London: COGENT; 1989. p. 137–41.
Yeager AM, Armfield Uhas K, Coles CD, Davis PC, Krause WL, Moser HW. Bone marrow transplantation for infantile ceramidase deficiency (Farber disease). Bone Marrow Transplant. 2000;26(3):357–63.
Ehlert K, Levade T, Di Rocco M, Lanino E, Albert MH, Führer M, Jarisch A, Güngör T, Ayuk F, Vormoor J. Allogeneic hematopoietic cell transplantation in Farber disease. J Inherit Metab Dis. 2018:1–8. https://doi.org/10.1007/s10545-018-0171-6
Sessa M, Lorioli L, Fumagalli F, Acquati S, Redaelli D, Baldoli C, Canale S, Lopez ID, Morena F, Calabria A, Fiori R, Silvani P, Vrancoita PM, Gabaldo M, Benedicenti F, Antonioli G, Assanelli A, Pia Cicalese M, del Carro U, Grazia MS, Natali MS, Quattrini A, Montini E, Di Serio C, Ciceri F, Roncarolo MG, Aiuti A, Naldini L, Biffi A. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet. 2016;388(10043):476–87.
Tardieu M, Zérah M, Gougeon M, Ausseil J, de Bournonville S, Husson B, Zafeiriou D, Parenti G, Bourget P, Poirier B, Furlan V, Artaud C, Baugnon T, Roujeau T, Crystal RG, Meyer C, Deiva K, Heard J. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: an uncontrolled phase 1/2 clinical trial. Lancet Neurol. 2017;16(9):712–20.
Huang J, Khan A, Au BC, Barber DL, López-Vásquez L, Prokopishyn NL, Boutin M, Rothe M, Rip JW, Abaoui M, Nagree Murtaza S, Shaalee D, Axel S, Keating A, West Michael L, Klassen J, Turner PV, Sirrs S, Medin JA. Lentivector iterations and pre-clinical scale-up/toxicity testing: targeting mobilized CD34 cells for correction of Fabry disease. Mol Ther Methods Clin Dev. 2017;5:241–58.
Medin JA, Takenaka T, Carpentier S, Garcia V, Basile J, Segui B, Andrieu-Abadie N, Auge N, Salvayre R, Levade T. Retrovirus-mediated correction of the metabolic defect in cultured Farber disease cells. Hum Gene Ther. 1999;10(8):1321–9.
Ramsubir S, Nonaka T, Girbés CB, Carpentier S, Levade T, Medin JA. In vivo delivery of human acid ceramidase via cord blood transplantation and direct injection of lentivirus as novel treatment approaches for Farber disease. Mol Genet Metab. 2008;95(3):133–41.
Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nature Rev Genet. 2011;12(5):301–15.
Boelens JJ, Orchard PJ, Wynn RF. Transplantation in inborn errors of metabolism: current considerations and future perspectives. Br J Haematol. 2014;167(3):293–303.
Naldini L. Gene therapy returns to Centre stage. Nature. 2015;526(7573):351–60.
Morgan RA, Gray D, Lomova A, Kohn DB. Hematopoietic stem cell gene therapy: progress and lessons learned. Cell Stem Cell. 2017;21(5):574–90.
Scott CT, DeFrancesco L. Gene therapy's out-of-body experience. Nat Biotechnol. 2016;34(6):600–7.
Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S, Benedicenti F, Vallanti G, Biasco L, Leo S, Kabbara N, Zanetti G, Rizzo WB, Mehta NA, Cicalese MP, Casiraghi M, Boelens JJ, Del Carro U, Dow DJ, Schmidt M, Assanelli A, Neduva V, Di Serio C, Stupka E, Gardner J, von Kalle C, Bordignon C, Ciceri F, Rovelli A, Roncarolo MG, Aiuti A, Sessa M, Naldini L. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341(6148):1233158.
Walia JS, Neschadim A, Lopez-Perez O, Alayoubi A, Fan X, Carpentier S, Madden M, Lee C, Cheung F, Jaffray DA. Autologous transplantation of Lentivector/acid ceramidase–transduced hematopoietic cells in nonhuman Primates. Hum Gene Ther. 2011;22(6):679–87.
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L, Berry K, Church K. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713–22.
Mendell J, Al-Zaidy S, Shell R, Arnold W, Rodino-Klapac L, Prior T, Lowes L, Alfano L, Berry K, Church K, Kissel J, Nagendran S, L'Italien J, Sproule D, Wells C, Burghes A, Foust K, Kaspar B. AVXS-101 phase 1 gene therapy clinical trial in SMA type 1: end-of-study event free survival and achievement of developmental milestones. Neuromuscul Disord. 2017;27:S208.
Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, Mankin HJ, Murray GJ, Parker RI, Argoff CE, Grewal RP, Yu K. Replacement therapy for inherited enzyme deficiency—macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991;324(21):1464–70.
Schiffmann R, Kopp JB, Austin HA III, Sabnis S, Moore DF, Weibel T, Balow JE, Brady RO. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285(21):2743–9.
Kakkis ED, Muenzer J, Tiller GE, Waber L, Belmont J, Passage M, Izykowski B, Phillips J, Doroshow R, Walot I, Hoft R, Yu KT, Okazaki S, Lewis D, Lachman R, Thompson JN, Neufeld EF. Enzyme-replacement therapy in mucopolysaccharidosis I. N Engl J Med. 2001;344(3):182–8.
Thurberg BL, Maloney CL, Vaccaro C, Afonso K, Tsai AC, Bossen E, Kishnani PS, O'Callaghan M. Characterization of pre-and post-treatment pathology after enzyme replacement therapy for Pompe disease. Lab Investig. 2006;86(12):1208–20.
Harmatz P, Yu Z, Giugliani R, Schwartz IVD, Guffon N, Teles EL, Miranda MCS, Wraith JE, Beck M, Arash L, Scarpa M, Ketteridge D, Hopwood JJ. Enzyme replacement therapy for mucopolysaccharidosis VI: evaluation of long-term pulmonary function in patients treated with recombinant human N-acetylgalactosamine 4-sulfatase. J Inherit Metab Dis. 2010;33(1):51–60.
Wasserstein MP, Jones SA, Soran H, Diaz GA, Lippa N, Thurberg BL, Culm-Merdek K, Shamiyeh E, Inguilizian H, Cox GF. Successful within-patient dose escalation of olipudase alfa in acid sphingomyelinase deficiency. Mol Genet Metab. 2015;116(1):88–97.
Wasserstein MP, Diaz GA, Lachmann RH, Jouvin M, Nandy I, Ji AJ, Puga AC. Olipudase alfa for treatment of acid sphingomyelinase deficiency (ASMD): safety and efficacy in adults treated for 30 months. J Inherit Metab Dis. 2018:1–10. https://doi.org/10.1007/s10545-017-0123-6.
Schuchman EH, Clancy J, Dimango E, Petrache I, Gulbins E, He X. Novel use of the lysosomal enzyme acid ceramidase for the treatment of inflammatory lung diseases, including cystic fibrosis. Mol Genet Metab. 2015;114(2):S105.
Pewzner-Jung Y, Tavakoli Tabazavareh S, Grassme H, Becker KA, Japtok L, Steinmann J, Joseph T, Lang S, Tuemmler B, Schuchman EH, Lentsch AB, Kleuser B, Edwards MJ, Futerman AH, Gulbins E. Sphingoid long chain bases prevent lung infection by Pseudomonas aeruginosa. EMBO Mol Med. 2014;6(9):1205–14.
He X, Dworski S, Zhu C, DeAngelis V, Solyom A, Medin JA, Simonaro CM, Schuchman EH. Enzyme replacement therapy for Farber disease: proof-of-concept studies in cells and mice. BBA Clinc. 2017;7:85–96.
Boado RJ, Lu JZ, Hui EK, Lin H, Pardridge WM. Insulin receptor antibody− α-N-Acetylglucosaminidase fusion protein penetrates the primate blood–brain barrier and reduces Glycosoaminoglycans in Sanfilippo type B fibroblasts. Mol Pharm. 2016;13(4):1385–92.
Zhang Z, Mandal AK, Mital A, Popescu N, Zimonjic D, Moser A, Moser H, Mukherjee AB. Human acid ceramidase gene: novel mutations in Farber disease. Mol Genet Metab. 2000;70(4):301–9.
Bär J, Linke T, Ferlinz K, Neumann U, Schuchman EH, Sandhoff K. Molecular analysis of acid ceramidase deficiency in patients with Farber disease. Hum Mutat. 2001;17(3):199–209.
Alves MQ, Le Trionnaire E, Ribeiro I, Carpentier S, Harzer K, Levade T, Ribeiro MG. Molecular basis of acid ceramidase deficiency in a neonatal form of Farber disease: identification of the first large deletion in ASAH1 gene. Mol Genet Metab. 2013;109(3):276–81.
Chikova IA, Buchinskaya NV, Kostik ММ, Avramenko VV, Krasnogorskaya OL, Nasirov RA, Levade T, Chasnyk VG. Farber disease- disease description with case reports. Voprosy Sovremennoj Pediatrii. 2014;13(6):78–84.
Muramatsu T, Sakai N, Yanagihara I, Yamada M, Nishigaki T, Kokubu C, Tsukamoto H, Ito M, Inui K. Mutation analysis of the acid ceramidase gene in Japanese patients with Farber disease. J Inherit Metab Dis. 2002;25(7):585–92.
Behin A, Nelson I, Bonne G, Romero N, Froissart R. Mutations in ASAH1 may cause spinal muscular atrophy. Neuromuscul Disord. 2015;25:S225.
Yildiz EP, Yesil G, Bektas G, Caliskan M, Tatlı B, Aydinli N, Ozmen M. Spinal muscular atrophy with progressive myoclonic epilepsy linked to mutations in ASAH1. Clin Neurol Neurosurg. 2018;164:47–9.
Acknowledgments
We thank Dr. Monty Mckillop and Dr. Jamie Harshman for input and critical assessment of this review.
Availability of data and materials
Data collected in this review are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
FPSY conducted the literature review, summarized the findings, and drafted the complete manuscript. SA, TL, and JAM, reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable, literature review only.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
A description of the literature search method used to identify FD and SMA-PME patients from research articles and case reports. (DOCX 20 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yu, F.P.S., Amintas, S., Levade, T. et al. Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J Rare Dis 13, 121 (2018). https://doi.org/10.1186/s13023-018-0845-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-018-0845-z