
Abstract
Background
More than 70 % of the cases of congenital deafness are of genetic origin, of which approximately 80 % are non-syndromic and show autosomal recessive transmission (DFNB forms). To date, 60 DFNB genes have been identified, most of which cause congenital, severe to profound deafness, whereas a few cause delayed progressive deafness in childhood. We report the study of two Algerian siblings born to consanguineous parents, and affected by progressive hearing loss.
Method
After exclusion of GJB2 (the gene most frequently involved in non-syndromic deafness in Mediterranean countries), we performed whole-exome sequencing in one sibling.
Results
A frame-shift variant (c.1014delC; p.Ser339Alafs*15) was identified in EPS8L2, encoding Epidermal growth factor receptor Pathway Substrate 8 L2, a protein of hair cells’ stereocilia previously implicated in progressive deafness in the mouse. This variant predicts a truncated, inactive protein, or no protein at all owing to nonsense-mediated mRNA decay. It was detected at the homozygous state in the two clinically affected siblings, and at the heterozygous state in the unaffected parents and one unaffected sibling, whereas it was never found in a control population of 150 Algerians with normal hearing or in the Exome Variant Server database.
Conclusion
Whole-exome sequencing allowed us to identify a new gene responsible for childhood progressive hearing loss transmitted on the autosomal recessive mode.
Similar content being viewed by others


Background
Deafness is the most common sensory deficit in humans, with an incidence of 1 in 700 live births. It is estimated that about two thirds of prelingual severe to profound isolated (non-syndromic) deafness cases have a genetic cause in developed countries [1], and autosomal recessive inheritance (DFNB) accounts for 80 % of the genetic cases [2]. Cases with autosomal recessive non-syndromic hearing loss (ARNSHL) are more prevalent in populations where consanguineous marriages are common, including Maghrebi populations. To date, 60 DFNB genes have been identified (http://hereditaryhearingloss.org/). Recessive deafness is usually prelingual, severe or profound, and fully penetrant, but some DFNB genes cause progressive hearing loss with delayed onset in childhood [3, 4].
In Algeria, mutations in GJB2, encoding connexin 26 (Gap Junction Protein Beta 2), account for no less than 35 % of the cases [5–7]. Mutations in other DFNB genes have also been identified, which illustrates the ARNSHL genetic heterogeneity in this population [8]. However, the genetic bases of progressive deafness beginning in childhood have not been characterized yet. Here, we report the identification, by whole-exome sequencing (WES), of a new causal gene for recessively inherited progressive deafness in an Algerian family.
Patients and methods
Patients
The two affected siblings were recruited in deafness schools of Blida and Baraki in Algiers (Algeria), and clinically examined in the otorhinolaryngology department at Bab El Oued hospital in Algiers. Medical history and physical examination of the patients did not reveal any non-genetic cause for the deafness and confirmed its non-syndromic nature. Hearing thresholds were determined by pure tone audiometry between 125 Hz and 8000 Hz, using air-conduction and bone conduction of sound.
Methods
The study was approved by the local Ethics Committee and the Committee for the Protection of Individuals in Biochemical Research as required by French legislation. A written consent for genetic testing was obtained from every family member. DNA was extracted from peripheral blood lymphocytes using the Promega Wizard Genomic DNA Purification Kit (Promega, Madison, MI, USA, Cat. # A1120). Whole-exome sequencing and bioinformatic analysis were carried out as previously described [9]. To validate the mutation in EPS8L2, sequencing of exon 12 was performed by the Sanger technique with the following primers: EPS8L2-12 F 5′-GTCTGTGCTGAGGGGAGG-3′ and EPS8L2-12R 5′-CTCTCCAGAACTGGCCCAC-3′ (http://bioinfo.ut.ee/primer3-0.4.0/primer3/).
Results and discussion
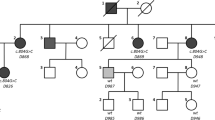
We studied an Algerian family with two siblings (IV.1 and IV.2) presenting with ARNSHI (Fig. 1a). The two affected children were born to normal hearing second-cousin parents, and reportedly had normal hearing for the first 3 years. The first signs of hearing impairment appeared around 4 years of age. By 6 years of age, they presented a bilateral elevation of the hearing threshold, starting at 30 dB at low frequencies (125 Hz) and increasing steadily up to 90 dB at high frequencies (8000 Hz) (Fig. 1b). At the age of 10, the hearing threshold increased further at all frequencies, ranging from 40 dB at 125 Hz, up to 100 dB at 8000 Hz (Fig. 1b and data not shown). Of note, audiograms showing parallel slopes at 6 and 10 years of age are rather unusual.

Clinical and molecular data of the patients harboring a biallelic homozygous frameshift mutation in EPS8L2. a Pedigree showing the segregation of the mutation in the family. The + and – signs denote the wild type and mutant alleles, respectively. The two clinically affected siblings are indicated by black symbols. The double horizontal bar joining the parents in generation III indicates consanguinity. b Air-conduction audiometric curves for patient IV.2 at the ages of 6 (open symbols) and 10 (black symbols) years of age. For each pure tone frequency tested (from 0.25 to 8 kHz), the hearing thresholds (in dB HL) for the right and left ears are indicated with circles and diamonds, respectively. c DNA sequencing chromatograms showing the mutation (arrow). d Schematic representation of the human EPS8L2 protein. The protein (715 amino acids) contains a phosphotyrosine interaction domain (PID), an SRC Homology 3 (SH3) domain, and an effector region (46 % of amino acid sequence identity with F-actin binding domain of EPS8) including a sterile alpha motif/pointed domain (SAM/PNT)
First, we explored the possibility of mutations in GJB2 by Sanger sequencing of the non-coding exon and the single coding exon of this gene for each patient, and screened the patients for the common deletion reported in the promoter region [10]. Since no mutations were found, we then proceeded to sequence the whole exome of patient IV.2, and looked for homozygous, compound heterozygous, or heterozygous variants with a predicted pathogenicity. None of the genes known to cause autosomal recessive or autosomal dominant forms of deafness showed any such variants in coding and non-coding exons or splice sites. The STRC and OTOA genes, which were poorly covered by WES, were further analyzed by single nucleotide polymorphism (SNP) array and q-PCR. No evidence for genomic rearrangement was found. Variants with a prevalence superior to 0.01 % in the DBSNP132, 1000 genomes, HapMap, and Exome Variant Server databases were excluded. Given the parents' consanguinity, the causal mutation was searched preferentially among homozygous nonsense, frame-shift, splice site, and missense variants. From the originally-identified 75,753 SNPs and 5918 insertions/deletions, only 5 SNPs and 1 deletion (Table 1) appeared as good candidates. The four missense variants and the synonymous/splice exon variant were deemed non pathogenic by the prediction algorithms SIFT, PolyPhen2, Mutation Taster and Nnsplice. In contrast, the homozygous 1-bp deletion c.1014delC (p.Ser339Alafs*15), located in exon 12 of the EPS8L2 gene, was predicted to cause a frame shift leading to premature termination of translation. The causal role of this variant was further supported by prior work showing that inactivation of this gene in the mouse caused progressive deafness [11]. After Sanger sequencing of EPS8L2 exon 12 of the two affected children, the unaffected sister, and the parents, the segregation analysis confirmed that the mutation was homozygous only in the deaf children, whereas the parents and the unaffected sibling were heterozygous for this variant (Fig. 1a, c). Lastly, the variant was never detected in a control population of 150 Algerians with normal hearing or in the Exome Variant Server database. We concluded that this frame-shift mutation, predicted to result either in a truncated inactive protein, or in no protein at all due to nonsense-mediated mRNA decay, is responsible for the progressive hearing loss of the two Algerian patients. EPS8L2 can thus be added to the list of DFNB genes already reported to cause progressive ARNSHL in humans: SLC26A4 [12, 13], MYO3A [14], LOXHD1 [15], PJVK [16–20], SERPINB6 [21], TPRN [22, 23], TMPRSS3 [24, 25], GIPC3 [26], SYNE4 [27], GRXCR2 [28], CLIC5 [29], TMC1 [30], GRXCR1 [31], and LRTOMT [32]. Of note, about 25 % of the DFNB genes identified so far are responsible for progressive forms of deafness, and several of them (SLC26A4, LOXHD1, PJVK, TMPRSS3, GIPC3, TMC1, GRXCR1, LRTOMT) can cause either non-progressive or progressive forms of deafness.
EPS8L2 has 24 exons and codes for a 715 amino acid protein (Fig. 1d), which is a member of the actin-binding protein EPS8 (epidermal growth factor receptor pathway substrate 8) family. This family includes four members, EPS8 and the three EPS8-like proteins (EPS8L1, EPS8L2, and EPS8L3), with partially overlapping functions [33]. In the mouse ear, EPS8L2 was detected at the stereocilia tips of both cochlear and vestibular hair cells. Eps8L2 knockout mice undergo progressive hearing loss, as the result of the progressive disorganization of the hair bundles of both inner and outer hair cells. Scanning electron microscopy analysis of the hair bundles in the cochlea of these mice showed that stereocilia of the tall row are shorter and fewer than those of wild-type mice, whereas both the middle and small stereocilia rows seem to be preserved and unaffected [11]. By contrast, mutations in the EPS8 gene cause profound congenital deafness in mice and humans [34, 35]. In the mouse cochlea, Eps8 is essential for the initial elongation of stereocilia [34, 36], whereas Eps8L2 is required for their maintenance in mature hair cells [11].
Conclusion
Deafness is a highly heterogeneous disorder, and many causative genes still remain to be identified. Here, despite several limitations (e.g. only a single family possibly analysed, uneven exon coverage at certain loci by WES), we were able to identify a new pathogenic variant responsible for ARNSHL in two Algerian siblings, by combining the powers of WES and genetics. This report is the first to incriminate EPS8L2, a gene formerly known to cause deafness in rodents, as a causal gene for progressive hearing loss in humans.
References
Petit C, Levilliers J, Hardelin J-P. Molecular genetics of hearing loss. Annu Rev Genet. 2001;35:589–646.
Morton NE. Genetic epidemiology of hearing impairment. Annu NY Acad Sci. 1991;630:16–31.
Bitner-Glindzicz M. Hereditary deafness and phenotyping in humans. Br Med Bull. 2002;63:73–94.
Piatto VB, Secches LV, Arroyo MAS, Lopes ACP, Maniglia JV. Nonsyndromic deafness - Molecular update. Open Biol J. 2009;2:80–90.
Denoyelle F, Marlin S, Weil D, Moatti L, Chauvin F, Garabedian EN, et al. Clinical features of the prevalent form of childhood deafness, DFNB1, due to a connexin-26 gene defect: implications for genetic counselling. Lancet. 1999;353:1298–303.
Frei K, Ramsebner R, Lucas T, Hamader G, Szuhai K, Weipoltshammer K, et al. GJB2 mutations in hearing impairment: identification of a broad clinical spectrum for improved genetic counseling. Laryngoscope. 2005;115:461–5.
Ammar-Khodja F, Makrelouf M, Malek R, Ibrahim H, Zenati A. Frequency of the 35 delG allele causing non-syndromic recessive deafness in the Algerian patients. Genet Couns. 2007;18:383–91.
Ammar-Khodja F, Bonnet C, Dahmani M, Ouhab S, Lefèvre GM, Ibrahim H, et al. Diversity of the causal genes in hearing impaired Algerian individuals identified by whole exome sequencing. Mol Genet Genom Med 2015;3:189-96.
Delmaghani S, Aghaie A, Michalski N, Bonnet C, Weil D, Petit C. Defect in the gene encoding the EAR/EPTP domain-containing protein TSPEAR causes DFNB98 profound deafness. Hum Mol Genet. 2012;21:3835–44.
del Castillo FJ, Rodriguez‐Ballesteros M, Alvarez A, Hutchin T, Leonardi E, de Oliveira CA, et al. A novel deletion involving the connexin‐30 gene, del(GJB6‐d13s1854), found in trans with mutations in the GJB2 gene (connexin‐26) in subjects with DFNB1 non‐syndromic hearing impairment. J Med Genet. 2005;42:588–94.
Furness DN, Johnson SL, Manor U, Ruttiger L, Tocchetti A, Offenhauser N, et al. Progressive hearing loss and gradual deterioration of sensory hair bundles in the ears of mice lacking the actin-binding protein Eps8L2. Proc Natl Acad Sci U S A. 2013;110:13898–903.
Abe S, Usami S, Hoover DM, Cohn E, Shinkawa H, Kimberling WJ. Fluctuating sensorineural hearing loss associated with enlarged vestibular aqueduct maps to 7q31, the region containing the Pendred syndrome gene. Am J Med Genet. 1999;82:322–8.
Bladwin CT, Weiss S, Farrer LA, De Stefano AL, Adair R, Franklyn B, et al. Linkage of congenital, recessive deafness (DFNB4) to chromosome 7q31 and evidence for genetic heterogeneity in the Middle Eastern Druze population. Hum Mol Genet. 1995;4:1637–42.
Walsh T, Walsh V, Vreugde S, Hertzano R, Shahin H, Haika S, et al. From flies' eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc Natl Acad Sci U S A. 2002;99:7518–23.
Grillet N, Schwander M, Hildebrand MS, Sczaniecka A, Kolatkar A, Velasco J, et al. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am J Hum Genet. 2009;85:328–37.
Delmaghani S, del Castillo FG, Michel V, Leibovici M, Aghaie A, Ron U, et al. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat Genet. 2006;38:770–8.
Schwander M, Sczaniecka A, Grillet N, Bailey JS, Avenarius M, Najmabadi H, et al. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J Neurosci. 2007;27:2163–75.
Ebermann I, Walger M, Scholl HPN, Issa PC, Luke C, Nurnberg G, et al. Truncating mutation of the DFNB59 gene causes cochlear hearing impairment and central vestibular dysfunction. Hum Mutat. 2007;28:571–7.
Chaleshtori MH, Simpson MA, Farrokhi E, Dolati M, Rad LH, Geshnigani SA, et al. Novel mutations in the pejvakin gene are associated with autosomal recessive non-syndromic hearing loss in Iranian families. Clin Genet. 2007;72:261–3.
Collin RWJ, Kalay E, Oostrik J, Caylan R, Wollnik B, Arslan S, et al. Involvement of DFNB59 mutations in autosomal recessive nonsyndromic hearing impairment. Hum Mutat. 2007;28:718–23.
Huang M, Duman D, Cengiz FB, Bademci G, Tokgöz-Yilmaz S, Hişmi B, et al. A truncating mutation in SERPINB6 is associated with autosomal recessive nonsyndromic sensorineural hearing loss. Am J Hum Genet. 2010;86:797–804.
Rehman AU, Morell RJ, Belyantseva IA, Khan SY, Boger ET, Shahzad M, et al. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am J Hum Genet. 2010;86:378–88.
Li Y, Pohl E, Boulouiz R, Schraders M, Nurnberg G, Charif M, et al. Mutations in TPRN cause a progressive form of autosomal-recessive nonsyndromic hearing loss. Am J Hum Genet. 2010;86:479–84.
Veske A, Oehlmann R, Younus F, Mohyuddin A, Müller-Myhsok B, Qasim Mehdi S, et al. Autosomal recessive non-syndromic deafness locus (DFNB8) maps on chromosome 21q22 in a large consanguineous kindred from Pakistan. Hum Mol Genet. 1996;5:165–8.
Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, Chrast R, et al. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat Genet. 2001;27:59–63.
Charizopoulou N, Lelli A, Schraders M, Ray K, Hildebrand MS, Ramesh A, et al. Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human. Nat Commun. 2011;2:201–12.
Horn HF, Brownstein Z, Lenz DR, Shivatzki S, Dror AA, Dagan-Rosenfeld O, et al. The LINC complex is essential for hearing. J Clin Invest. 2013;123:740–50.
Imtiaz A, Kohrman DC, Naz SA. Frameshift mutation in GRXCR2 causes recessively inherited hearing loss. Hum Mutat. 2014;35:618–24.
Seco CZ, Oonk AM, Domınguez-Ruiz M, Draaisma JMT, Gandi M, Oostrik J, et al. Progressive hearing loss and vestibular dysfunction caused by a homozygous nonsense mutation in CLIC5. Eur J Hum Genet. 2014;23:189–94.
de Heer AM, Collin RW, Huygen PL, Schraders M, Oostrik J, Rouwette M, et al. Progressive sensorineural hearing loss and normal vestibular function in a Dutch DFNB7/11 family with a novel mutation in TMC1. Audiol Neurootol. 2011;16:93–105.
Mori K, Miyanohara I, Moteki H, Nishio SY, Kurono Y, Usami SI. Novel Mutations in GRXCR1 at DFNB25 lead to progressive hearing loss and dizziness. Ann Otol Rhinol Laryngol. 2015;0003489415575061.
Ichinose A, Moteki H, Hattori M, Nishio SY, Usami SI. Novel mutations in LRTOMT associated with moderate progressive hearing loss in autosomal recessive inheritance. Ann Otol Rhinol Laryngol. 2015;0003489415575043.
Offenhäuser N, Borgonovo A, Disanza A, Romano P, Ponzanelli I, Iannolo G, et al. The eps8 family of proteins links growth factor stimulation to actin reorganization generating functional redundancy in the Ras/Rac pathway. Mol Biol Cell. 2004;15:91–8.
Zampini V, Ruttiger L, Johnson SL, Franz C, Furness DN, Waldhaus J, et al. Eps8 regulates hair bundle length and functional maturation of mammalian auditory hair cells. PLoS Biol. 2011;9, e1001048.
Behlouli A, Bonnet C, Abdi S, Bouaita A, Lelli A, Hardelin J-P, et al. EPS8, encoding an actin-binding protein of cochlear hair cell stereocilia, is a new causal gene for autosomal recessive profound deafness. Orph J Rar Dis. 2014;9:55.
Manor U, Disanza A, Grati MH, Andrade L, Lin H, Di Fiore PP, et al. Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr Biol. 2011;21:167–72.
Acknowledgments
We thank all the family members for their participation in the study. This work was supported by grants from the Algerian government, the BNP Paribas Foundation, the French LABEX LIFESENSES grant [reference ANR-10-LABX-65], and the Pasteur-Weizmann program.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
FAK and CP designed the project. MD and CB analysed WES and Sanger sequencing results. HI and ZM contributed to clinical evaluation of the patients. MD, FAK, CB, GML, JPH, and CP wrote the article. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dahmani, M., Ammar-Khodja, F., Bonnet, C. et al. EPS8L2 is a new causal gene for childhood onset autosomal recessive progressive hearing loss. Orphanet J Rare Dis 10, 96 (2015). https://doi.org/10.1186/s13023-015-0316-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-015-0316-8