
Abstract
Sneddon’s syndrome (SS) is a rare non-inflammatory thrombotic vasculopathy characterized by the combination of cerebrovascular disease with livedo racemosa(LR). The Orpha number for SS is ORPHA820. It has been estimated that the incidence of SS is 4 per 1 million per annum in general population and generally occurs in women between the ages of 20 and 42 years. LR may precede the onset of stroke by years and the trunk and/or buttocks are involved in nearly all patients. The cerebrovascular manifestations are mostly secondary to ischemia (transient ischemic attacks and cerebral infarct). Other neurological symptoms range from headache, cerebral hemorrhage, seizures, cognitive and psychiatric disturbances. The involved internal organs include heart, kidney, and eyes. Histological findings of skin are characteristic and the involved vessels are small to medium-sized arteries at the border of dermis to subcutis with a distinct histopathological time course. The main diagnostic criteria are general LR with typical histopathological findings on skin biopsy and focal neurological deficits. The pathogenesis is related to hypercoagulable state and intrinsic small-vessel vasculopathy. The optimal management remains an unsolved problem and long-term anticoagulation have been recommended for cerebral ischemic events based on the presumed pathogenesis. There are controversial results in treatment of SS with immunomodulatory agents. The aim of this review is to comprehensively discuss this disease.
Similar content being viewed by others


Introduction
Definition
Sneddon’s syndrome (SS) is a rare non-inflammatory thrombotic vasculopathy characterized by the combination of cerebrovascular disease with livedo racemosa (LR).
History
The association between LR and cerebrovascular manifestation was first described by Kimming in 1959 [1]. In 1960, Champion and Rook described a case of cutaneous condition, diffuse arterial occlusive disease and cerebral ischemia [2]. 5 years later, Sneddon reported six patients with severe and generalized bluish discoloration of the skin (also termed as livedo reticularis), “multiple cerebrovascular incidents of limited and benign nature” and no autopsy reported [3]. Since then the term SS has been used to describe the association of LR and multiple cerebral infarcts.
Classification
SS, which originally was a clinical diagnosis, is now regarded as a common clinical manifestation of different disease entities. Three forms of the syndrome were described by Schellong et al.: primary, where no causative factor could be identified, autoimmune with antiphospholipid antibodies (aPL) or coexisting systemic lupus erythematosus (SLE) or lupus-like disease and a thrombophilic form [4]. Francès et al. proposed that SS should be classified as idiopathic SS with neither aPL nor SLE, primary antiphospholipid syndrome (APS)-related SS, and SLE related SS with or without aPL [5]. Cases without any other pathological signs except LR should be historically labeled “idiopathic generalized LR” which may not exist as a separate entity but may represent a very early stage of SS.
Epidemiology
While a number of SS cases have been reported since 1965, there is no definite evidence of ethnic differences in incidence. It has been estimated that the incidence of SS is 4 per 1 million per annum in general population [6]. In hospital-based series of stroke patients, the frequency of SS is between 0.25% and 0.50% [7],[8]. A mortality rate of 9.5% is reported in a mean observation period of 6.2 years [6].
SS generally occurs in women between the ages of 20 and 42 years. For example, in the serial studies reported by Sneddon [3], 5 of 6 patients were women. Similarly, 34 of 46 patients were women reported by Francès and all cases were women in the report by Bolayir [5],[9]. However, SS does occur occasionally in girls as young as 10 years old and in women as advanced as 64 years old [10].
Genetic characteristics
The disorder mainly occurs sporadically, although a few familial cases have been reported [11]. Hademonos et al. [12] described in a review of the genetics of cerebrovascular disease and stroke that SS was inherited in an autosomal dominant pattern with unknown type of the gene and gene chromosome localization. In familial cases described by Rebollo et al. [13], either LR or full clinical SS was apparently transmitted in autosomal dominant pattern with complete or incomplete penetrance. The studies by Zhou et al. and Navon Elkan et al. have identified novel mutations in CECR1(cat eye syndrome chromosome region, candidate 1), encoding ADA2, as the cause of a syndrome including systemic vasculopathy and inflammation. Bras etal. extend the phenotypic spectrum of ADA2 deficiency to include a familial form of SS [14],[15].
Clinical description
Dermatologic manifestations
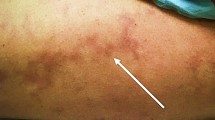
One of the diagnostic hallmarks of SS is LR (Figure 1a). LR is defined as a dusky erythematousto-violaceous, irregular, net-like pattern in the skin. LR may precede the onset of stroke by years and is located on limbs (100% each), trunk (84–89%), buttocks (68–74%), face (15–16%), or the hands or feet (53–59%) [5]. The trunk and/or buttocks are involved in nearly all patients. LR is noticed before cerebrovascular events in more than a half of patients. In some patients, the livedo is first detected at the time of stroke occurrence [16],[17]. In rare cases, LR appears after neurological symptoms [18]. In addition to LR, some patients have signs of Raynaud’s phenomenon involving hands and feet and some have widespread cutaneous, mottled-purple discoloration on the body which had been diagnosed as systemic angiomatosis [19],[20].

(two cases of SS ) a, diffused livedo racemosa (asterisks); b, FLAIR/T2 MRI brain scan showing multiple ischemic lesions (white arrows) and cortical atrophy (red arrows); c, skin biopsy showed intra-capillary (thin arrow) and parietal (thick arrow) widespread thrombosis; scale bar 100μm (Case1) and 50 μm (Case2). (courtesy of Dr. Alessandro Tessitore).
LR is similar to the familiar sign of livedo reticularis from which it differs by its shape (irregular, broken circular segments, resulting in a seemingly larger pattern), and persistence on warming [21],[22]. In the American literature, livedo reticularis is used interchangeably with LR despite their different pathophysiological basis. Livedo reticularis is caused by temporary vasoconstriction, whereas livedo racemosa results from persistent impairment of peripheral blood flow caused by occlusion of small or medium-sized arteries [23],[24].
Neurological manifestations
Stroke is another diagnostic hallmark of SS. This cerebrovascular disease mostly occurs due to ischemia (transient ischemic attacks and cerebral infarct) in the perfusion zones of middle cerebral artery or posterior cerebral artery [25],[26]. Hemiparesis, sensory disturbances, aphasia and visual field defects are the most common clinical signs. However, most patients are minor stroke. Headache represents the most frequent unspecific symptom [27],[28]. The 50% of these patients have migrainous headache [29]. The frequency of headache is not significantly higher in persons with positive aPL when compared to an aPL-negative cohort [29].
Cognitive impairment and psychiatric disturbances can occur in approximately 77% of SS patients and SS can also be a cause of dementia in the young [25],[30]. These patients develop cognitive dysfunction over years and then dementia due to the cumulative effects of multiple cerebral infarcts. Because of these features, SS may lead to psychiatric disturbances, including depression. Concentration, attention, memory, visual perception and visuospatial construction are the most commonly described cognitive dysfunctions [31]. However, patients with SS can develop topographic disorientation or dementia without antecedent clinical stroke [18]. Focal or secondary generalized seizures are commonly seen in SS, especially in patients with positive aPL [5],[32].
Intracerebral (ICH), subarachnoid or intraventricular hemorrhage are unusual in SS. Movement disorders including chorea and tremor were occasionally been described [33],[34].
Other manifestations
Hypertension occurs in a significant proportion (15%–65%) of SS patients [25],[26]. Heart valvulopathy was found in 41% of SS patients in one study and 61% in another study [35],[26]. Mitral valve thickening has been revealed more frequently in patients with aPL than in those without. Cases of SS with ischemic heart disease are described including patient associated with successful percutaneous coronary intervention [36]. The renal function is slowly deteriorated in SS. Zelger et al. reported a long-term follow-up of 21 patients and observed that 65% of patients showed decreased levels of creatinine clearance [6].
Ophthalmologic complications include central retinal artery occlusion, central retinal vein occlusion, retinal neovascularization, homonymous visual field defects, and internuclear ophthalmoloplegia [37]-[42]. Rehany et al. reported a young SS patient with internuclear ophtalmoplegia followed by ophthalmic artery occlusion [41]. Unilateral third cranial nerve palsy has been seen in a patient with SS and this is likely associated with microvascular complication [43].
Etiology
The precise etiology is unknown and around 50% of cases are idiopathic. Female reproductive hormones, oral contraceptives and hypertension are correlated with disease progression. The frequent presence of aPL suggest that SS is part of the clinical spectrum of the primary aPL syndrome. The proportion of aPL antibodies is widely different in various cohorts of SS [6],[35],[44]. The majority authors suggest that 40–50% of SS patients are aPL positive [5],[45]. However, some patients are aPL-negative which indicate that this syndrome may be a distinct entity or perhaps a group of different disorders.
Pathology
Dermatopathology
Skin biopsy is critically important for the early diagnosis of SS. Skin biopsy may reveal thrombosis of subcutaneous arterioles and compensatory capillary dilation with blood stagnation causing LR (Figure 1c). Several lines of evidence showed endarteritis obliterans from intimal endothelial proliferation and proliferation of medial smooth muscle cells [24],[46],[47], although skin biopsies were negative or non-specific in some cases [1],[48]. Pathogenesis of SS has been proposed a stepwise pattern of disease progression by Sepp et al. and Zelger et al. [49],[50], beginning as an inflammatory vascular process in small to medium sizes of arteries. After the initial phase (stage I) of endotheliitis, characterised by a plug, the formation of the plug of lymphohistiocytic cells and fibrin would occur in the early phase (II stage), followed by the intermediate phase (stage III) of subendothelial migration of smooth muscle cells, and completed by the late phase (stage IV) of vessel shrinkage and fibrosis. Skin biopsy may reveal different pathological phases in one patient. However, the severity of the clinical findings is not normally correlated with the histological findings. Deep skin biopsies increase sensitivity from 27% with one biopsy, to 53% with two biopsies, and to 80% with three biopsies taken from white areas in the most of cases [51].
Brain pathology
Brain biopsy is rarely done in SS. The pathologic findings showed multiple vascular thrombosis without vasculitis in the first brain biopsy case [52]. There were cortical infarcts and leukoencephalopathy associated with intimal fibrosis of the basal vessels in a biopsy report [53]. The other autopsy report described a diffuse vasculopathy affecting small to medium sized arteries (including cerebral vessels) characterized by proliferation of the intima and media, with some vessels showing fibrotic occlusion and recanalization. Inflammation did not affect blood vessels [54]. The principal pathologic findings in the case reported by Hilton DA et al. were multiple small cortical infarcts associated with occlusion of medium sized arteries and prominent focal smooth muscle hyperplasia of smaller arterial vessels [55]. Although a granulomatous reaction in the leptomeninges has been reported [56], this reaction is not observed by others [54], suggesting that this abnormality does not always occur in SS.
Pathophysiology
Pathophysiology of SS remains incompletely understood to date. The existence of aPL antibodies suggests that symptoms are secondary to thrombotic process [57]. Vascular thrombosis and recanalization within skin and brain tissue support the pathophysiology of coagulopathy.
Although a basic thrombotic process may appear in SS, it is still not clear how it occurs in aPL-negative cases.Various abnormalities have been reported in isolated cases: activated protein C resistance [58], platelet aggregability [49], increased thromboglobulin levels [59], modifications of the ratio tissue plasminogen activator/inhibitor [60], familial deficiency in antithrombin III [46], and protein S deficiency [61]. None of these abnormalities have been confirmed in large series of studies. Protein Z, a downregulator of coagulation and linked to an increased risk of arterial thrombosis, is significantly lowered in patients with aPL-negative SS [62]. Heart valve abnormalities, either clinically audible or detected on echocardiogram, are frequently observed, and their total prevalence is similar in both groups. This raises the possibility that the embolic mechanism plays an important role in the occurrence of some neurologic and even skin manifestations of SS [63].
In some patients, no aPL or primary coagulation deficits are detected. Thus, the primary vasculopathy is considered as the pathophysiological change in SS. It is speculated that a nonvasculitic small and medium sized vessel arteriopathy causes both skin symptoms and cerebrovascular events [48]. However, the type and origin of the arteriopathy is largely unknown [64]. In some cases, the gene responsible for the arteriopathy may play a role [41],[65]. Besides, endothelial dysfunction may be secondary to acquired autoimmune or mediated by some unknown factors [66],[67].
Diagnosis
Any patient suspected of SS should undergo blood tests, thorough cardiovascular evaluation, cerebral MRI, cerebral angiography, and skin biopsy. Blood tests should screen for lupus anticoagulant, immunoglobulin IgG and possibly IgM anti-cardiolipinantibodies, anti-nuclear and anti-double-stranded DNA autoantibodies, thrombocytopenia, leukopenia, VDRL, cryoglobulins, circulating immune complexes, antithrombin-III, protein C, or protein S [3],[13],[68],[46]. The cerebrospinal fluid (CSF) is usually normal.
Lesions can be more clearly detected by MRI than by CT scan. These lesions are often small and multifocal, located predominantly in the periventricular deep white matter or pons (Figure 1b). Cerebral hemorrhages are rarely found in SS. Prominent microbleeds are found in familial SS, which might be a marker of impending haemorrhagic stroke [65],[69]. Cerebral angiography is abnormal up to 75% of patients with SS. The most common abnormality is an obliterating noninflammatory arteriopathy with stenosis and/or occlusion of intracranial vessels [70]-[73]. Other reported findings include: arteriovenous malformations involving meningeal branches, transdural anastomoses, granulomatous leptomeningeal infiltration, and a large network of fine collateral vessels [70]-[76]. Occurrence of an aneurysmatic dilatation of the ascending aorta is rare in SS [77].
The main criteria are general LR with typical histopathological findings on skin biopsy and focal neurological deficits. Supportive criteria are a history of transient ischemic attacks or stroke and evidence of small bright foci on T2-weighted MRI [68].
Differential diagnosis
Some diseases that can occur with generalized LR (such as SLE, polyarteritis nodosa, cryoglobulinemia, livedoid vasculitis, cold agglutinin disease, arteriosclerosis, cholesterin embolization and others [6],[22],[59]), must be excluded by appropriate clinical and laboratory tests. The differential diagnosis of neurological symptoms include multiple sclerosis, multiple embolization from cardiac source, infectious conditions (such as syphilis and lyme disease), and others [6],[43]. Central nervous system (CNS) vasculitis refers to a broad array of diseases that result in inflammation and destruction of the blood vessels of brain, spinal cord and the meninges. CNS vasculitis is classified into systemic vasculitis and isolated CNS angiitis. CNS vasculitis may present symptoms such as headache and stroke which mimic SS patients. However, in systemic vasculitis, CNS involvement always coexists with other clearly apparent systemic manifestations. Isolated CNS angiitis is twice as frequent in males as in females and onset most often occurs after 40 years of age. The sedimentation rate is moderately increased in about 30% patients and CSF inflammation is observed in about 90% of patients. Histological findings are characterized by infiltrations of the vascular walls with mononuclear cells including lymphocytes, macrophages, and histiocytes [78].
Treatment
The optimal management of patients with SS remains an unsolved issue and controlled trials have not yet been performed. Although nifedipine may reduce skin symptoms, it does not prevent cerebrovascular complications [6]. Based on the presumed pathogenesis of SS, some researchers have recommended long-term anticoagulation for cerebral ischemic events [79],[80]. Khamashta et al. reported in a retrospective study of 183 patients with APS syndrome that the number of events per year of follow up was 0.18 with aspirin, 0.23 with low dose warfarin (INR < 3), and 0.015 with high-dose warfarin (INR ≥ 3). In a retrospective study, Francès et al. found that antiplatelet therapy was less effective in aPL-positive than in aPL-negative patients. Among the former, the number of recurrent cerebral events per year of follow-up was 0.5 with antiplatelet therapy and 0.06 with anticoagulation. The three aPL-positive patients who experienced a new cerebral ischemic event under anticoagulation had an INR less than 3 at the time of recurrence [5]. The benefits of warfarin in the APS are greater than the risks of bleeding [79].
In SS patients with acute ischemic stroke, thrombolytic therapy might be safe and effective [81]. Angiotensin converting enzyme inhibitors (ACEI) and prostaglandin E1 (PGE1) have also been used in the treatment of SS [82]. The prevention of smoking and prevention of usage of estrogen oral contraceptive may prevent or decrease the severity of neurological symptoms [6]. Moreover, cardiovascular risk factors should also be treated.
Corticosteroids, cyclophosphamide, and azathioprine have been reported ineffective in treatment of SS [83]. However, one study demonstrated improvements in neurological and cognitive symptoms in a patient after 8 months of monthly intravenous cyclophosphamide therapy [73]. Future research should focus on identifying potential subgroups of patients who may be good responders.
Conclusion
SS is a clinically syndrome that probably stems from a number of acquired or congenital hemostatic abnormalities which preferentially involves the cerebral and cutaneous vascular beds. To a certain extent, the small number of cases may reflect the unfamiliarity with the syndrome rather than a true reflection of its incidence. Further research exploring the pathogenesis of SS is needed to get more etiological subgroups. Future therapy should identify different treatment modalities for different etiological subgroups.
Abbreviations
- SS:
-
Sneddon’s syndrome
- LR:
-
Livedo racemosa
- aPL:
-
Antiphospholipid antibodies
- SLE:
-
Systemic lupus erythematosus
- APS:
-
Antiphospholipid syndrome
- ACEI:
-
Angiotensin converting enzyme inhibitors
- PGE1:
-
Prostaglandin E1
References
Kimming J: Arteriolopathie:livedo rasemosa. Dermatol Wochenschr. 1959, 139: 211
Champion RH, Rook A: Cutaneous arteriolitis. Proc R Soc Med. 1960, 53: 568
Sneddon IB: Cerebrovascular lesions and livedo reticularis. Br J Dermatol. 1965, 77: 180-185. 10.1111/j.1365-2133.1965.tb14628.x.
Schellong SM, Weissenborn K, Niedermeyer J, Wollenhaupt J, Sosada M, Ehrenheim C, Lubach D: Classification of Sneddon’s syndrome. Vasa. 1997, 26: 215-221.
Francès C, Papo T, Wechsler B, Laporte JL, Biousse V, Piette JC: Sneddon syndrome with or without antiphospholipid antibodies. A comparative study in 46 patients. Medicine (Baltimore). 1999, 78: 209-219. 10.1097/00005792-199907000-00001.
Zelger B, Sepp N, Stockhammer G, Dosch E, Hilty E, Ofner D, Aichner F, Fritsch PO: Sneddon’s syndrome. A long-term follow-up of 21 patients. Arch Dermatol. 1993, 129: 437-447. 10.1001/archderm.1993.01680250049006.
Berciano J: Sneddon syndrome: another mendelian etiology of stroke. Ann Neurol. 1988, 24: 586-587. 10.1002/ana.410240422.
De Reuck J, De Reus R, De Koninck J: Sneddon’s syndrome. A not unusual cause of stroke in young women. Cerebral Vascular Disease 6. Proceedings of the World Federation of Neurology 13th International Salzburg Conference: 25–27 September, 1986. Edited by: Meyer JS, Lechner H, Reivich M, Ott EO. 1987, Excerpta Medica, Amsterdam, 171-174.
Bolayir E, Yilmaz A, Kugu N, Erdogan H, Akyol M, Akyuz A: Sneddon’s syndrome: clinical and laboratory analysis of 10 cases. Acta Med Okayama. 2004, 58: 59-65.
Villaizán C, Ferrada MJ, Legarda I, Iriarte J, Narbona J, Martínez-Lage JM: Sneddon’s syndrome vasculopathy with antiphospholipid antibodies in a child. Neurologia. 1995, 10: 41-45.
Szmyrka-Kaczmarek M, Daikeler T, Benz D, Koetter I: Familial inflammatory Sneddon’s syndrome-case report and review of the literature. Clin Rheumatol. 2005, 24: 79-82. 10.1007/s10067-004-0981-9.
Hademenos GJ, Alberts MJ, Awad I, Mayberg M, Shepard T, Jagoda A, Latchaw RE, Todd HW, Viste K, Starke R, Girgus MS, Marler J, Emr M, Hart N: Advances in the genetics of cerebrovascular disease and stroke. Neurology. 2001, 56: 997-1008. 10.1212/WNL.56.8.997.
Rebollo M, Val JF, Garijo F, Quintana F, Berciano J: Brain. Livedo reticularis and cerebrovascular lesions (Sneddon’s syndrome). Clinical, radiological and pathological features in eight cases. Brain. 1983, 106: 965-979. 10.1093/brain/106.4.965.
Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, Stone DL, Chae JJ, Rosenzweig SD, Bishop K, Barron KS, Kuehn HS, Hoffmann P, Negro A, Tsai WL, Cowen EW, Pei W, Milner JD, Silvin C, Heller T, Chin DT, Patronas NJ, Barber JS, Lee CC, Wood GM, Ling A, Kelly SJ, Kleiner DE, Mullikin JC, Ganson NJ, et al: Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014, 370: 911-920. 10.1056/NEJMoa1307361.
Bras J, Guerreiro R, Santo GC: Mutant ADA2 in vasculopathies. N Engl J Med. 2014, 371: 478-480. 10.1056/NEJMc1405506.
Cavestro C, Richetta L, Pedemonte E, Asteggiano G: Sneddon’s syndrome presenting with severe disabling bilateral headache. J Headache Pain. 2009, 10: 211-213. 10.1007/s10194-009-0109-3.
Killeen T, Wanke I, Mangiardi J, Cesnulis E: Ruptured, fusiform, distal lenticulostriate aneurysm causing intraventricular haemorrhage in a patient with antiphospholipid-negative Sneddon’s syndrome. Clin Neurol Neurosurg. 2014, 116: 80-82. 10.1016/j.clineuro.2013.11.009.
Marianetti M, Mina C, Marchione P, Giacomini P: Sneddon’s Syndrome presenting with topographic disorientation. J Clin Neurosci. 2011, 18: 980-981. 10.1016/j.jocn.2010.11.027.
Schlez A, Lischka G, Schaumburg-Lever G, Ganten T, Jünger M: Raynaud symptoms as principal signs in a case of Sneddon’s syndrome. J Eur Acad Dermatol Venereol. 2001, 15: 365-366.
Heckmann JG, Lüfti M: Images in clinical medicine. Angiomatosis associated with Sneddon’s syndrome. N Engl J Med. 2004, 350: e11-10.1056/ENEJMicm020035.
Burton JL: Livedo reticularis, porcelain-white scars, and cerebral thromboses. Lancet. 1988, 1: 1263-1265. 10.1016/S0140-6736(88)92080-6.
Bruyn RP, van der Veen JP, Donker AJ, Valk J, Wolters EC: Sneddon’s syndrome. Case report and literature review. J Neurol Sci. 1987, 79: 243-253. 10.1016/0022-510X(87)90232-2.
Copeman PW: Livedo reticularis:Signs in the skin of disturbance of blood viscosity and of blood flow. Br J Dermatol. 1975, 93: 519-529. 10.1111/j.1365-2133.1975.tb02244.x.
Marsch WC, Muckelmann R: Generalized racemose livedo with cerebrovascular lesions (Sneddon syndrome): an occlusive arteriolopathy due to proliferation and migration of medial smooth muscle cells. Br J Dermatol. 1985, 112: 703-708. 10.1111/j.1365-2133.1985.tb02341.x.
Boesch SM, Plörer AL, Auer AJ, Poewe W, Aichner FT, Felber SR, Sepp NT: The natural course of Sneddon syndrome: clinical and magnetic resonance imaging findings in a prospective six year observation study. J Neurol Neurosurg Psychiatry. 2003, 74: 542-544. 10.1136/jnnp.74.4.542.
Tourbah A, Piette JC, Iba-Zizen MT, Lyon-Caen O, Godeau P, Francès C: The natural course of cerebral lesions in Sneddon syndrome. Arch Neurol. 1997, 54: 53-60. 10.1001/archneur.1997.00550130037013.
Tietjen GE, Al-Qasmi MM, Shukairy MS: Livedo reticularis and migraine: a marker for stroke risk?. Headache. 2002, 42: 352-355. 10.1046/j.1526-4610.2002.02106.x.
Tietjen GE, Gottwald L, Al-Qasmi MM, Gunda P, Khuder SA: Migraine is associated with livedo reticularis: a prospective study. Headache. 2002, 42: 263-267. 10.1046/j.1526-4610.2002.02078.x.
Tietjen GE, Al-Qasmi MM, Gunda P, Herial NA: Sneddon’s syndrome: another migraine-stroke association?. Cephalalgia. 2006, 26: 225-232. 10.1111/j.1468-2982.2005.01032.x.
Adair JC, Digre KB, Swanda RM, Hartshorne MF, Lee RR, Constantino TM, Knoefel JE: Sneddon’s syndrome: cause of cognitive decline in young adults. Neuro Psychiatry Neuropsychol Behav Neurol. 2001, 14: 197-204.
Weissenborn K, Rückert N, Ehrenheim C, Schellong S, Goetz C, Lubach D: Neuropsychological deficits in patients with Sneddon’s syndrome. J Neurol. 1996, 243: 357-363. 10.1007/BF00868412.
Kalashnikova LA, Nasonov EL, Stoyanovich LZ, Kovalyov VU, Kosheleva NM, Reshetnyak TM: Sneddon’s syndrome and the primary antiphospholipid syndrome. Ter Arkh. 1993, 65: 64-70.
Kalashnikova LA, Aleksandrova EN, Novikov AA, Dobrynina LA, Nasonov EL, Sergeeva EV, Berkovskiĭ AL: Anti-phosphatidylethanolamine antibodies in patients with Sneddon’s syndrome. Klin Med (Mosk). 2005, 83: 46-49.
Da Silva AM, Rocha N, Pinto M, Alves V, Farinha F, Correia AP, Coelho T, Magalhães M: Tremor as the first neurological manifestation of Sneddon’s syndrome. Mov Disord. 2005, 20: 248-251. 10.1002/mds.20270.
Kalashnikova LA, Nasonov EL, Borisenko VV, Usman VB, Prudnikova LZ, Kovaljov VU, Kushekbaeva AF: Sneddon’s syndrome: cardiac pathology and antiphospholipid antibodies. Clin Exp Rheumatol. 1991, 9: 357-361.
Scheuermann S, Schlundt C: STEMI of the anterior wall associated with Sneddon’s syndrome. Herz. 2014, 39: 352-353. 10.1007/s00059-013-3834-y.
Pauranik A, Parwani S, Jain S: Simultaneous bilateral central retinal arterial occlusion in a patient with sneddon syndrome: case history. Angiology. 1987, 38: 158-163. 10.1177/000331978703800211.
Aggermann T, Haas P, Binder S: Central retinal vein occlusion as a possible presenting manifestation of sneddon syndrome. J Neuroophthalmol. 2007, 27: 240-241. 10.1097/WNO.0b013e31814b259f.
Gobert A: Sneddons syndrome with bilateral peripheral retinal neovascularization. Bull Soc Belge Ophtalmol. 1995, 255: 85-90.
Narbay G: Sneddon’s syndrome in a patient with homonymous hemianopia with macular sparing. Bull Soc Belge Ophtalmol. 1996, 263: 103-107.
Rehany U, Kassif Y, Rumelt S: Sneddon’s syndrome: neuro-ophthalmologic manifestations in a possible autosomal recessive pattern. Neurology. 1998, 51: 1185-1187. 10.1212/WNL.51.4.1185.
Song HB, Woo SJ, Jung CK, Lee YJ, Ahn J, Park KH, Kwon OK: Acute central retinal artery occlusion associated with livedoid vasculopathy: a variant of Sneddon’s syndrome. Korean J Ophthalmol. 2013, 27: 376-380. 10.3341/kjo.2013.27.5.376.
Jiménez-Gallo D, Albarrán-Planelles C, Linares-Barrios M, González-Fernández JA, Espinosa-Rosso R, Báez-Perea JM: Sneddon syndrome presenting with unilateral third cranial nerve palsy. J Neuroophthalmol. 2014, 34: 50-52. 10.1097/WNO.0b013e3182a3060d.
Francès C, Piette JC, Viard JP, Tourbah A: Anti-beta 2-glycoprotein I antibodies in Sneddon’s syndrome. Dermatology. 1993, 186: 273-10.1159/000247371.
Kalashnikova LA, Nasonov EL, Kushekbaeva AE, Gracheva LA: Anticardiolipin antibodies in Sneddon’s syndrome. Neurology. 1990, 40: 464-467. 10.1212/WNL.40.3_Part_1.464.
Quimby SR, Perry HO: Livedo reticularis and cerebrovascular accidents. J Am Acad Dermatol. 1980, 3: 377-383. 10.1016/S0190-9622(80)80331-8.
Baleva M, Chauchev A, Dikova C, Stamenov B, Nikoevski N, Tzankov N, Nikovov K: Sneddon’s syndrome: echocardiographic, neurological, and immunologic findings. Stroke. 1995, 26: 1303-1304.
Rumpl E, Rumpl H: Recurrent transient global amnesia in a case with cerebrovascular lesions and livedo reticularis (sneddon syndrome). J Neurol. 1979, 221: 127-131. 10.1007/BF00313110.
Sepp N, Zelger B, Schuler G, Romani N, Fritsch P: Sneddon’s syndrome—an inflammatory disorder of small arteries followed by smooth muscle proliferation. Immunohistochemical and ultrastructural evidence. Am J Surg Pathol. 1995, 19: 448-453. 10.1097/00000478-199504000-00006.
Zelger B, Sepp N, Schmid KW, Hintner H, Klein G, Fritsch PO: Life history of cutaneous vascular lesions in Sneddon’s syndrome. Hum Pathol. 1992, 23: 668-675. 10.1016/0046-8177(92)90323-U.
Wohlrab J, Fischer M, Wolter M, Marsch WC: Diagnostic impact and sensitivity of skin biopsies in Sneddon’s syndrome. A report of 15 cases. Br J Dermatol. 2001, 145: 285-288. 10.1046/j.1365-2133.2001.04348.x.
Geschwind DH, FitzPatrick M, Mischel PS, Cummings JL: Sneddon’s syndrome is a thrombotic vasculopathy: neuropathologic and neuroradiologic evidence. Neurology. 1995, 45: 557-560. 10.1212/WNL.45.3.557.
Muerza FM, González G, Ortiz E, Saracibar N: Cerebral hemorrhage in sneddon syndrome. Rev Neurol. 1998, 27: 74-76.
Pinol Aguade J, Ferrandiz C, Ferrer Roca O, Ingelmo M: Livedo reticularis y accidentes cerebrovasculares. Med Cutan Ibero Lat Am. 1975, 3: 257-265.
Hilton DA, Footitt D: Neuropathological findings in Sneddon’s syndrome. Neurology. 2003, 60: 1181-1182. 10.1212/01.WNL.0000055931.66009.D3.
Boortz-Marx RL, Clark HB, Taylor S, Wesa KM, Anderson DC: Sneddon’s syndrome with granulomatous leptomeningeal infiltration. Stroke. 1995, 26: 492-495. 10.1161/01.STR.26.3.492.
Köner O, Günay I, Cetin G, Celebi S: Mitral valve replacement in a patient with sneddon syndrome. J Cardiothorac Vasc Anesth. 2005, 19: 661-664. 10.1053/j.jvca.2005.07.011.
Montalbán J, Ordi J, Barquinero J, Vilardell M: Sneddon’s syndrome and anticardiolipin antibodies. Stroke. 1988, 19: 785-786.
Rumpl E, Neuhofer J, Pallua A, Willeit J, Vogl G, Stampfel G, Platz T: Cerebrovascular lesions and livedo reticularis (Sneddon’s syndrome)–a progressive cerebrovascular disorder?. J Neurol. 1985, 231: 324-330. 10.1007/BF00313710.
Sumi Y, Ozaki Y, Itoh S, Katayama H, Tanaka S: Cerebral blood flow-SPECT in a patient with Sneddon’s syndrome. Ann Nucl Med. 1999, 13: 109-112. 10.1007/BF03164886.
Sayin R, Bilgili SG, Karadag AS, Tombul T: Sneddon syndrome associated with protein S deficiency. Indian J Dermatol Venereol Leprol. 2012, 78: 407-
Ayoub N, Esposito G, Barete S, Soria C, Piette JC, Francès C: Protein Z deficiency in antiphospholipid-negative Sneddon’s syndrome. Stroke. 2004, 35: 1329-1332. 10.1161/01.STR.0000127534.54538.15.
Asherson RA, Khamashta MA, Ordi-Ros J, Derksen RH, Machin SJ, Barquinero J, Outt HH, Harris EN, Vilardell-Torres M, Hughes GR: The “primary” antiphospholipid syndrome: major clinical and serological features. Medicine (Baltimore). 1989, 68: 366-374. 10.1097/00005792-198911000-00004.
Cirillo G, Tessitore A, Cirillo M, Salemi F, Liguori S, Esposito S, Tedeschi G: Livedo and ischemic strokes: diagnostic hints of a rare condition. Neurol Sci. 2013, 34: 2073-2075. 10.1007/s10072-013-1452-3.
Llufriu S, Cervera A, Capurro S, Chamorro A: Familial Sneddon’s syndrome with microbleeds in MRI. J Neurol Neurosurg Psychiatry. 2008, 79: 962-10.1136/jnnp.2007.131912.
Moral A, Vidal JM, Moreau I, D’Olhaberriague L, Montalbán J: Sneddon’s syndrome with antiphospholipid antibodies and arteriopathy. Stroke. 1991, 22: 1327-1328. 10.1161/01.STR.22.10.1327.
Duval A, Darnige L, Glowacki F, Copin MC, Martin De Lassalle E, Delaporte E, Auxenfants E: Livedo, dementia, thrombocytopenia, and endotheliitis without antiphos- pholipid antibodies: seronegative antiphospholipid-like syndrome. J Am Acad Dermatol. 2009, 61: 1076-1078. 10.1016/j.jaad.2008.12.046.
Stockhammer G, Felber SR, Zelger B, Sepp N, Birbamer GG, Fritsch PO, Aichner FT: Sneddon’s syndrome: diagnosis by skin biopsy and MRI in 17 patients. Stroke. 1993, 24: 685-690. 10.1161/01.STR.24.5.685.
Koennecke HC: Cerebral microbleeds on MRI: prevalence, associations, and potential clinical implications. Neurology. 2006, 66: 165-171. 10.1212/01.wnl.0000194266.55694.1e.
Ellie E, Julien J, Henry P, Vital C, Ferrer X: Divry-Van bogaert cortico-meningeal angiomatosis and Sneddon’s syndrome. Nosological study. Apropos of 4 cases. Rev Neurol (Paris). 1987, 143: 798-805.
Antiphospholipid Antibodies in Stroke Study Group: Clinical and laboratory findings in patients with antiphospholipid antibodies and cerebral ischemia. Stroke. 1990, 21: 1268-1273. 10.1161/01.STR.21.9.1268.
Antoine JC, Michel D, Garnier P, Absi L, Boucheron S, Barral FG, Laurent B: Sneddon syndrome: 9 cases. Rev Neurol (Paris). 1994, 150: 435-443.
Hannon PM, Kuo SH, Strutt AM, York MK, Kass JS: Improvement of neuro- logical symptoms and memory and emotional status in a case of Seno- negative sneddon syndrome with cyclophosphamide. Clin Neurol Neurosurg. 2010, 112: 544-547. 10.1016/j.clineuro.2010.04.002.
Levine SR, Welch KM: The spectrum of neurologic disease associated with anti- phospholipid antibodies. Lupus anticoagulants and anticardiolip in antibodies. Arch Neurol. 1987, 44: 876-883. 10.1001/archneur.1987.00520200078024.
Blom RJ: Sneddon syndrome: CT, arteriography, and MR imaging. Familial Sneddon’s syndrome: clinical, hematologic, and radiographic findings in two brothers. J Comput Assist Tomogr. 1989, 13: 119-122. 10.1097/00004728-198901000-00026.
Pettee AD, Wasserman BA, Adams NL, McMullen W, Smith HR, Woods SL, Ratnoff OD: Familial Sneddon’s syndrome: clinical, hematologic, and radiographic findings in two brothers. Neurology. 1994, 44: 399-405. 10.1212/WNL.44.3_Part_1.399.
Kraemer M, Baumgaertel MW, Berlit P: Miscarriage, peripheral thromboses and aortic aneurysm in antiphospholipid-antibody-negative Sneddon’s syndrome. J Neurol. 2007, 254: 1599-1600. 10.1007/s00415-007-0568-x.
Alba MA, Espígol-Frigolé G, Prieto-González S, Tavera-Bahillo I, García-Martínez A, Butjosa M, Hernández-Rodríguez J, Cid MC: Central nervous system vasculitis: still more questions than answers. Curr Neuropharmacol. 2011, 9: 437-448. 10.2174/157015911796557920.
Khamashta MA, Cuadrado MJ, Mujic F, Taub NA, Hunt BJ, Hughes GR: The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med. 1995, 332: 993-997. 10.1056/NEJM199504133321504.
Rosove MH, Brewer PM: Antiphospholipid thrombosis: clinical course after the first thrombotic event in 70 patients. Ann Intern Med. 1992, 117: 303-308. 10.7326/0003-4819-117-4-303.
Sun J, Zhang F, Gao F, Wang J, Selim M, Lou M: Intravenous thrombolysis in Sneddon’s syndrome. J Clin Neurosci. 2012, 19: 326-328. 10.1016/j.jocn.2011.05.024.
Flöel A, Imai T, Lohmann H, Bethke F, Sunderkötter C, Droste DW: Therapy of Sneddon syndrome. Eur Neurol. 2002, 48: 126-132. 10.1159/000065510.
Akbal A, Kurtaran A, Selçuk B, Ersöz M, Akyüz M: A rarely seen syndrome in rehabilitation of hemiplegia: antiphospholipid antibody-negative Sneddon’s syndrome. Rheumatol Int. 2010, 30: 1489-1491. 10.1007/s00296-009-1093-2.
Acknowledgements
We thank Dr. Alessandro Tessitore for providing us with clinical radiographies and histological image to complete our review. We also thank Dr. Chu Chen from School of Medicine, Louisiana State University, USA, for the critical revision of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SW, ZX and HL participated in the process of the literature review and in the drafting the final manuscript. In addition, HL supervised the project. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Wu, S., Xu, Z. & Liang, H. Sneddon’s syndrome: a comprehensive review of the literature. Orphanet J Rare Dis 9, 215 (2014). https://doi.org/10.1186/s13023-014-0215-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-014-0215-4